

Abstract
Diabetes Mellitus is a metabolic cum vascular syndrome with resultant abnormalities in both micro- and macrovasculature. The adverse long-term effects of diabetes mellitus have been described to involve many organ systems. Apart from hyperglycemia, abnormalities of angiogenesis may cause or contribute toward many of the clinical manifestations of diabetes. These are implicated in the pathogenesis of vascular abnormalities of the retina, kidneys, and fetus, impaired wound healing, increased risk of rejection of transplanted organs, and impaired formation of coronary collaterals. A perplexing feature of the aberrant angiogenesis is that excessive and insufficient angiogenesis can occur in different organs in the same individual. The current article hereby reviews the molecular mechanisms including abnormalities in growth factors, cytokines, and metabolic derangements, clinical implications, and therapeutic options of dealing with abnormal angiogenesis in diabetes.
Keywords: Angiogenesis, diabetes mellitus, growth factor, neovascularization, vascular endothelial cell
INTRODUCTION
Diabetes Mellitus (DM) is a metabolic cum vascular syndrome of multiple etiology characterized by chronic hyperglycemia with disturbances of carbohydrate, fat, and protein metabolism, resulting from defects in insulin secretion, insulin action, or both leading to changes in both small blood vessels (microangiopathy) and large blood vessels (macroangiopathy).
Micro- and macrovascular beds are altered in diabetes by various changes in neovascular mechanism.[1] However, from the vascular point of view, diabetes is a paradoxical disease.[2] Excessive angiogenesis plays a role in diabetic retinopathy (DR),[3] nephropathy,[4] and in the vessel wall, potentially producing atherosclerotic plaque destabilization. Insufficient angiogenesis contributes to impaired wound healing and skin ulcers,[5] impaired coronary collateral vessel (CV) development, embryonic vasculopathy in pregnancies complicated by maternal diabetes, and transplant rejection in diabetic recipients. Furthermore, diabetic neuropathy is a complication linked with reduced nutritive blood flow secondary to diabetes. Defective arteriogenesis, a process of formation or remodeling of arterioles and arteries, has also been reported in diabetic patients.[6] Impaired release of endothelial progenitor cells (EPCs) from the bone marrow and defective function of these cells are the other features of diabetes that further contribute to abnormal neovascularization and increased cardiovascular risk.[7]
Angiogenesis is the process of formation of new capillary network (microvascular) in response to hypoxia or other stimuli.[8] The process of angiogenesis involves the local secretion of angiogenic factors from both hypoxic endothelium and supporting pericytes that induce endothelial proliferation and sprouting of neovessels. This is different from arteriogenesis, a process of growth and expansion of existing arteriole networks in response to acute arterial occlusion (e.g. arterial occlusion) or physical forces (e.g. exercise-induced shear stress). In 1987, Osterby and Nyberg[4] described abnormal blood vessels in glomeruli of patients with long-term type 1 diabetes, and later these findings were shown to occur in type 2 diabetic patients.[9]
MECHANISM OF ABNORMAL ANGIOGENESIS IN DIABETES
Angiogenesis is a complex multisequential process involving interaction between a number of pro- and anti-angiogenic mediators, growth factors, and cytokines, endothelium, and the extracellular matrix (ECM). The angiogenesis is characterized by ECM degeneration, endothelial cell (EC) proliferation, EC survival, EC migration, EC morphology changes, and EC anastomoses. Following enzymatic degradation of the basement membrane (BM), chemotactic and mitogenic factors cause ECs to extend through the gaps, elongate, align, and proliferate to form a capillary sprout.[10] The joining of two sprouts initiates blood flow in the newly formed loop, and subsequent interactions between EC and pericytes trigger the construction of a new BM and lead to vessel maturation and stabilization.[10] Normal angiogenesis depends on the delicate balance of stimulatory and inhibitory factors; up- or downregulation of any of these factors leads to aberrant vessel formation with pathological consequences.
Various mechanisms contribute toward causation of stimulation or inhibition of angiogenesis. They involve an array of growth factors, cytokines, hematopoietic factors, and metabolic disturbances induced changes. The factors involved in the dysregulation of angiogenesis are summarized in Table 1.
Table 1.
Molecular Mechanisms of abnormal Angiogenesis / Arteriogenesis in Diabetes

Stimulation of angiogenesis
Vascular endothelial cell growth factor signaling pathway
The vascular endothelial cell growth factor (VEGF) family includes VEGF (A-D) and placental growth factor. VEGF interacts with different tyrosine kinase receptors (Flt-1, Flk-1, Flt-4). They act as mitogens for vascular ECs and also stimulate EPC mobilization from the bone marrow. The VEGF-A family has a role in the development, maintenance, and remodeling of the vasculature, acting through the receptor tyrosine kinases VEGFR-1 and VEGFR-2.[11] Diabetic patients have reduced number of circulating EPCs, with the extent of reduction directly proportional to HbA1c levels.[12] Myocardium of diabetic patients expresses reduced VEGF and VEGF receptors,[13] as well as increased production of an angiogenesis inhibitor angiostatin induced by hyperglycemia.[14] In diabetes, the VEGF-A164 and VEGF-A188 isoforms are increased and can be reduced by insulin treatment.[13] Cooper et al.[15] have demonstrated that short-term diabetes led to elevated VEGF-A and VEGF receptor- 2 (VEGFR-2) mRNA, whereas in long-term diabetic animals, VEGF-A remained elevated and VEGFR-2 was unaltered.
Diabetes results in defective VEGF signaling leading to impaired Flk-1 activation that affects the various processes of angiogenesis including EC growth and migration, monocyte and EPC recruitment, and EPC release by bone marrow. At the same time, decreased VEGF sensing due to impaired Flk-1 activation results in increased serum VEGF levels that lead to pathologic angiogenesis. Waltenberger et al.[16] noted that monocytes from diabetic patients failed to respond to VEGF despite activation of the Flt- 1 receptor. Sasso et al.[17] have demonstrated increased VEGF expression in the myocardium of diabetic patients compared with that in non-diabetic patients, whereas expression levels of VEGF receptors 1 and 2 (Flt-1 and Flk-1, respectively) were reduced. Most importantly, the extent of Flk-1 phosphorylation/activation was severely reduced in diabetic patients. This was associated with a reduced activation of serine-threonine protein kinase Akt-1 and endothelial nitric oxide synthase (eNOS), the principal effectors of the VEGF signaling pathway. These two studies suggest that whereas Flt- 1 activation under diabetic conditions is normal, Flk-1 activation is not. The role of Flt-1 in VEGF signaling remains controversial. Unlike Flk-1, which is expressed in the endothelium and in certain bone marrow cell populations, including EPCs, Flt-1 is expressed in endothelium and mononuclear cells, including monocytes. It is involved in the regulation of cell migration either via an independent signaling pathway or secondary to Flk-1 activation via an intracellular cross-talk or direct receptor heterodimerization.
Flk-1 is the principal receptor involved in transmitting VEGF signaling [Figure 1]. It regulates cell proliferation via activation of the extracellular receptor kinase (Erk-1/2) and Akt-1, a master regulator of cell function. Two most crucial activities of Akt-1 include: firstly, activation of eNOS stimulating nitric oxide (NO) production required for EC proliferation, and inhibition of apoptosis; and secondly, for the maintenance of the intact vasculature in adult tissues. Simons et al. has proposed the sequence of events to explain diabetic angiogenic abnormalities [Figure 2].[18] The abnormally activated Flk-1 leads to increased levels of VEGF to compensate for the deficiency of VEGF signaling. High circulating VEGF levels lead to increased permeability of vascular structures throughout the body. In the retina, this results in the formation of protein-rich exudates containing VEGF that induces a local inflammatory response resulting in capillary sprouting.[19] A similar process in the arterial wall promotes capillary sprouting and plaque destabilization. Simultaneously, the lack of Flk-1 activation in ECs and abnormal VEGF dependent activation of monocytes impair the arteriogenic response that requires monocyte recruitment and monocyte and EC migration and proliferation. In addition, VEGF/ Flk-1 signaling is required for bone marrow release of circulating EPCs that plays a role in arteriogenesis. The abnormal release of EPCs will further reduce arteriogenic response.
Figure 1.
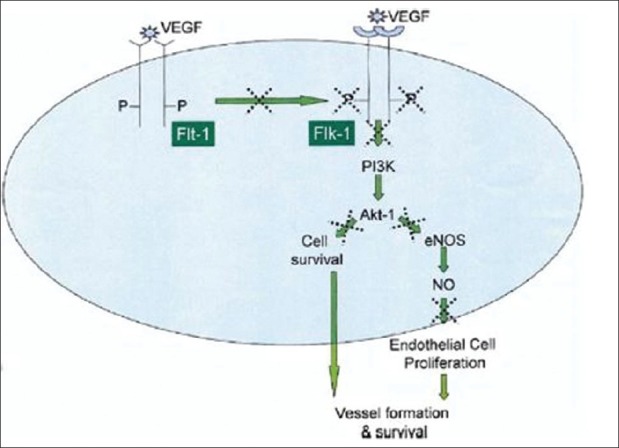
Schematic representation of vascular endothelial growth factor (VEGF) signaling in endothelial cells is presented with putative signaling abnormalities observed in diabetes, indicated by the dotted X symbol. See text for details. eNOS: endothelial nitric oxide synthase; Flk-1: VEGF sreceptor-2; Flt-1: VEGF receptor
Figure 2.
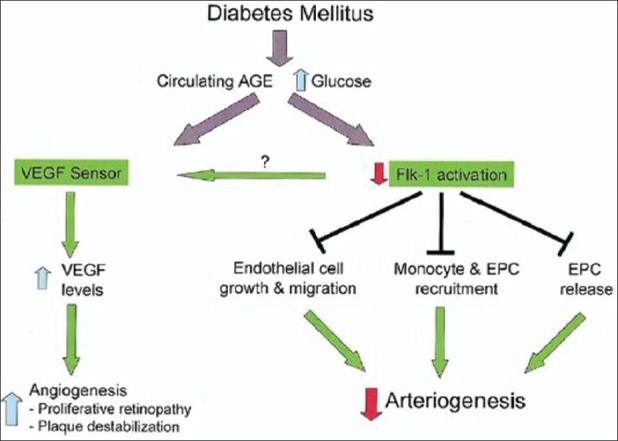
Graphical representation of the proposed paradigm of neovascularization abnormalities in diabetes mellitus. Defective VEGF signaling results in impaired Flk-1 activation that affects endothelial cell growth and migration, monocyte and endothelial progenitor cell (EPC) recruitment, and EPC release by the bone marrow. As a result, arteriogenesis is impaired. At the same time, decreased VEGF sensing, due to impaired Flk-1 activation, results in increased serum VEGF levels that lead to pathologic angiogenesis (retina, atheroma). AGE: advanced glycosylated end-products.
Endothelium-derived NO plays an important role in the angiogenic actions of VEGF,[20] transforming growth factor (TGF)-β1,[21] and basic fibroblast growth factor (bFGF).[22] The induction of angiogenesis by these growth factors can be blocked by inhibitors of NO synthase.[22,23]
Hypoxia
Hypoxia is one of the major inducers of angiogenesis.[24] Hypoxic conditions lead to the upregulation of hypoxia- inducible factor, a transcription factor known to bind to the hypoxia response element in the promotor region of the VEGF gene.[25] The presence of hypoxic environment triggers cells to upregulate VEGF, stromal- derived factor-1 (SDF-1), platelet-derived growth factor (PDGF), or angiopoietin-2. Hypoxia, hyperglycemia, vasopressor hormones (angiotensin II and arginine vasopressin), and various cytokines (TGF-β and IL1) and growth factors [tumor necrosis factor (TNF), fibroblast growth factor (FGF), and PDGF] have been shown to increase VEGF transcription and stability.[26]
Chronic inflammation
DM is characterized by a chronic inflammatory state with concomitant cytokine and growth factor secretion. In fact, inflammation-induced release of a huge amount of factors (e.g. IL1, IL6, TNF, CCL5, TGF-β) leads to angiogenic stimulation. Hyperglycemia itself is an angiogenic enhancer and negatively impacts many aspects of neovascularization. Although genetically diabetic (db/db) mice with elevated TGF-β mRNA levels also showed a twofold increase in transcripts for VEGF, treatment with anti-TGF-β antibodies only slightly reduced VEGF levels despite completely neutralizing TGF-β expression.[27] This evidence suggests that TGF-β is only one of the several factors capable of inducing VEGF in diabetes.
Oxidative stress
Diabetes is characterized by the presence of oxidative and nitrosative stress. There is evidence which indicates that reactive oxygen species (ROS) activate signaling pathways that promote angiogenesis.[7]
Hyperglycemia and AGE products
Diabetic patients presenting with poor glycemic control have high levels of advanced glycation end products (AGEs), which are known to promote tissue fibrosis in organs with end-stage damage. AGEs and activation of AGE receptors in diabetes contribute to impaired angiogenic potential[28] in vitro, while in vivo inhibition of AGE formation in diabetic mice can restore ischemia-induced angiogenesis in peripheral limbs.[29] Neutralization of the receptor for AGEs (RAGE) can restore angiogenic potential during wound healing in diabetic mice.[30] AGE modification of vasogenic growth factors impairs their angiogenic potential both in vitro[31] and in vivo.[32] However, the angiogenic role of AGEs remains somewhat controversial, with several studies reporting that these adducts can promote aspects of the angiogenic process in vitro, including stimulation of EC proliferation[33] and tube formation,[34] perhaps through the induction of the angiogenic peptide VEGF.[34]
This results in extensive reduction of tissue perfusion and consequently ischemia-induced angiogenesis. In addition, AGE activates synthesis of various pro-fibrotic and pro-angiogenic proteins, such as insulin-like growth factor binding protein-related protein-2 (IGFPB-rP2)/connective tissue growth factor (CTGF) in skin fibroblasts and in renal mesangial cells.[35]
Advanced lipoxidation end products
Advanced lipoxidation end (ALE) increase the expression of a wide range of inflammatory factors, such as CXCL10, CCL2, COX-2, integrins, IL6, IL8, and inducible NOS (iNOS), in monocytes. Most of these molecules are established angiogenic activators.[36] Abnormalities in the arachidonic acid cascade involving both the cyclooxygenase and lipoxygenase pathways create a pro-angiogenic environment. Antonipillai et al. reported a deficiency in the cyclooxygenase product prostacyclin (PGI2) accompanied by elevated levels in the alternate lipoxygenase product 12-hydroxyeicosatetraenoic acid (12-HETE) in both human cadavers and animal models of diabetes.[36] Subnormal levels of PGI2 are found in umbilical vessels of diabetic mothers and in vascular tissue from type 1 DM patients, and 12-HETE has been shown to stimulate angiogenesis and mitogenesis, possibly by inhibiting renin secretion and preventing the generation of superoxide ion that accompanies vasoconstriction. Renal production of HETE and the HETE/PGI2 ratio are elevated early in the course of type 2 DM and continue to increase as diabetic nephropathy advances.[36] Long-standing diabetes causes fixed activity of the cyclooxygenase pathway, which can be neither stimulated nor inhibited by pharmacological means beyond a certain point.
Others
Fibroblast growth factor
Like VEGF, FGF is a heparin-binding protein with mitogenic, chemotactic, and pro-angiogenic properties.[37] FGF exists in acidic and basic forms (aFGF and bFGF, also known as FGF-1 and FGF-2, respectively) and is primarily triggered by endothelial injury by hypoxia and hemodynamic stress.[38] FGF is localized in capillary BM in a heparin-bound inactive state. The release of FGF in response to injury stimulates the initiation of neovascularization.[37] FGF induces EC proliferation, survival, differentiation, and migration of ECs, smooth muscle cells, macrophages, and fibroblasts.[37] These effects are mediated through the tyrosine kinase receptor FGFR1, leading to activation of protein kinase C (PKC). Elevated bFGF levels occur in pregnant diabetic females, with maximum expression in diabetic pregnancies complicated by retinopathy and correlate positively with glycemic control at birth[38] and with fetal and placental size.[39] Macrosomia, organomegaly, excess subcutaneous adiposity, and modest increases in skeletal length are well-documented characteristics of children born to diabetic mothers and may be due to the actions of bFGF.[39]
Upregulation of integrins
Integrins mediate cell attachment to ECM components [fibronectin (Fn), vitronectin, laminin, collagen], thereby promoting cellular migration and the mobilization of growth factors necessary for angiogenesis.[40] Casaroli et al. have reported upregulation of integrin α5b1 as well as co-distribution of αvβ3and its ligand vitronectin in tissues from humans with proliferative DR.[41] Stromblad et al. have also used vitronectin receptor antagonists to inhibit neovascularization via induction of EC apoptosis.[42]
Upregulation of Fn and Fn-f
Fn is a high-molecular weight ECM glycoprotein component normally found in the internal limiting membrane of the adult retina and in vessel walls, with greatest concentration at pericyte–endothelial barriers.[36] Certain Fn fragments (Fn-f) have been shown to be potent pro-angiogenic factors with the capability to stimulate EC proliferation, migration, and the expression of BM-degrading matrix metalloproteinases (MMPs).[36] Furthermore, Fn-f have been shown to stimulate tissue plasminogen activator (tPA) catalyzed plasminogen activation; the plasmin produced then activates proforms of MMPs. Plasmin and MMPs degrade Fn into Fn-f, thus perpetuating the cycle[36] [Figure 3].
Figure 3.

The vascular ECM expansion, i.e., characteristic of diabetic retinopathy involves overexpression of fibronectin and, therefore, of fibronectin fragments from the enzymatic degradation of fibronectin. Fobronectin fragments catalyze the conversion of plasminogen to plasmin via tissue plasminogen activator. Plasmin activates matrix metalloproteinases, which are necessary to generate the active formof the fibronectin-cleaving enzymes, thereby increasing the fibronectinfragment pool ina vicious-cycle-like manner.
Hyperglycemia-induced Fn overexpression leads to increased degradation of Fn to pro-angiogenic Fn-f. This ensues aberrant angiogenesis characteristic of DR.[36] Subsequent insulin therapy and re-establishment of normoglycemia failed to return Fn levels to normal, and Fn overexpression continued through reproductive cycles after the cells were removed from the hyperglycemic environment.[36] The irreversibility of glucose-mediated derangement is explained by permanent modification of long-lived ECM proteins and can create a bad metabolic memory.
Angiotensin
The diabetic state is associated with increased plasma renin activity and elevated levels of angiotensin converting enzyme (ACE) and angiotensin II (AII), the most potent vasopressor. Vasoconstriction results in increased contact between circulating blood elements and vessel walls, leading to generation of superoxide ion by the endothelium.[43] Lipid peroxidation by the oxygen radical destroys ECs, impairs collagen metabolism, and disrupts the ECM.[44] AII stimulates VEGF expression in human mesangial cells, ECs, retinal cells, pericytes, and smooth muscle cells.[45] AII mediates bFGF-induced vascular smooth muscle cell hyperplasia.[46]
Excessive glucose metabolism via polyol pathway
Polyol metabolic pathway reduces glucose [Figure 4] to sorbitol by aldose reductase. Sorbitol accumulation leads to decrease in myoinositol content, abnormal phosphoinositide metabolism, decreased [Na+K+]-ATPase activity,[9] and increased collagen cross-linking and vascular permeability.[38,47] The latter permits extravasation of proteinases and plasma adhesion proteins from vessels, thereby hastening neovascularization.
Figure 4.
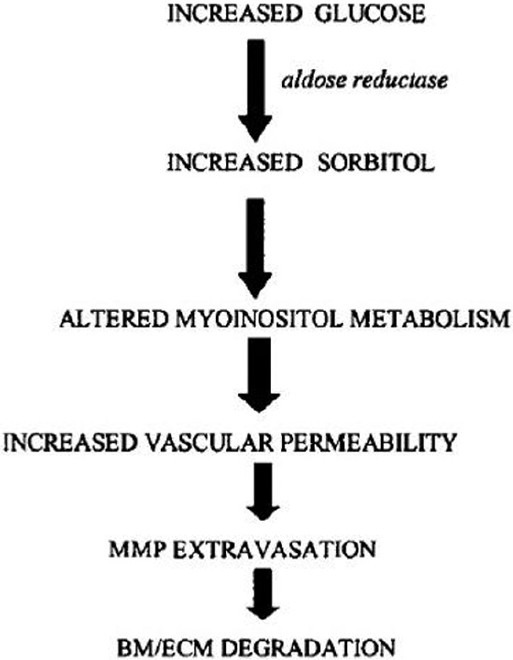
Hyperglycemiais associated with increased production of sorbitol viathe enzyme aldosereductase, resulting in deranged myoinositol metabolism which may increase vascular permeability, thereby increasing the rate of matrix metalloproteinase extravasation. The resulting degradation of the basement membrane/ extracellular matrix contributes to the increased rate of angiogenesis associated with diabetes.
f. Excess tissue factor
Elevated expression of TF mRNA in diabetes causes thrombotic episodes that result in retinal non-perfusion induced ischemia and the release of pro-angiogenic factors responsible for aberrant angiogenesis in DR.[48] Insulin and TNF-α and -β may potentiate the overexpression of TF mRNA.[48]
Metabolic derangement
Diabetes is associated with enhanced lipolysis [Figure 5] leading to elevated levels of monobutyrin (1-butyryl- glycerol). Initially, monobutyrin induces an increase in retinal blood flow rate.[49] However, in longer term, retinal blood vessels develop resistance to vasodilatation by monobutyrin, suggesting that monobutyrin downregulates specific receptors.[49] The resultant retinal non-perfusion and ischemia may trigger the release of various pro- angiogenic growth factors.
Figure 5.
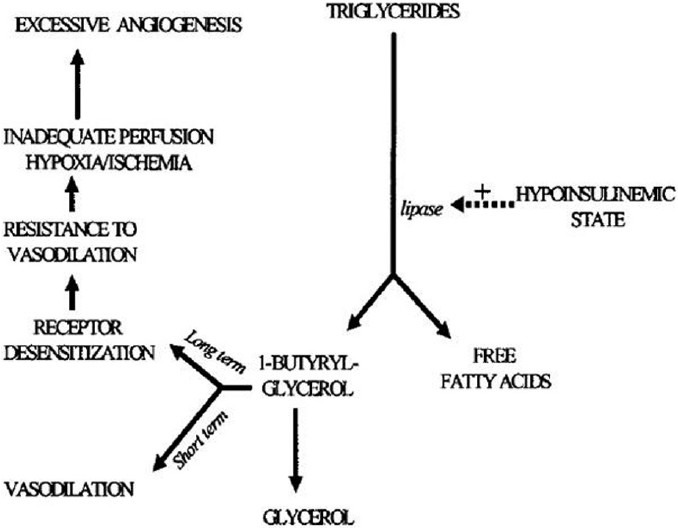
The lipolytic nature of diabetes mellitus may promote excessive angiogenesis. In view of deficient insulin, hormone sensitive lipase actively breaks down triglycerides into free fatty acids and monoacyl glycerol moieties, 1-butyryl glycerol among them. Elevation in the level of 1-butyryl glycerol has vasodilatory effects in short term. In long term, 1-butyryl glycerol receptor becomes desensitized, decreasing the ability of the vessels to dilate and creating ischemia and suboptimal perfusion, which in turn triggers proangiogenic factors (VEGF and others).
Deficiencies in serum 1,25-dihydroxy vitamin D[1,25(OH)2D3], a known inhibitor of angiogenesis, may have a role in excessive angiogenesis in diabetes.[50] There is an inverse relationship between the severity of diabetic retinal neovascularization and serum concentrations of 1,25 (OH)2D3.[50]
Inhibition of angiogenesis
Inadequate ECM/BM degradation
Decreased levels of urokinase plasminogen activator (uPA) contribute to the impaired degradation of the BM/ECM. uPA converts plasminogen to plasmin, which promotes angiogenesis by degrading Fn, laminin, and the proteoglycan protein core, by activating MMPs and by mobilizing bFGF from the ECM pool.[26] Plasmin and uPA contribute to fibrinolysis and anticoagulatory effect.[48] The decreased uPA levels and supranormal levels of plasminogen activator inhibitor-1 (PAI-1) associated with diabetes creates an antifibriolytic state which impedes nutritive blood flow, and impairs CV formation by hindering ECM degradation. This prevents new capillary outgrowth and puts the ischemic diabetic at a greater risk of atherosclerosis, coronary artery disease (CAD), or peripheral arterial disease (PAD).[51]
Upregulation of the TGF-β leads to glomerular and tubular hypertrophy and sclerosis.[27] TGF-β1 suppresses MMPs and increases the expression of protease inhibitors such as PAI-1, thereby impairing matrix degradation.[27] TGF-β is implicated in the pathogenesis of both excessive and deficient angiogenesis. Increased levels of TGF-β promote matrix expansion, which encroaches upon vascular beds and impedes nutritive flow. The resulting ischemia upregulates pro-angiogenic substances’ expression. However, in situations with deficient angiogenesis, TGF-β induced matrix expansion was not extensive enough to create ischemia of the severity required to trigger angiogenesis.
Growth factor and cytokine imbalance
Decreased VEGF may contribute to inadequate angiogenesis in diabetes and insulin-resistant states.[52] There are reports of markedly decreased expression of VEGF and subnormal concentrations of TGF-β1 in diabetic dermal wounds.[53,54] Insulin activates the P13- kinase/Akt pathway, which in turn leads to upregulation of VEGF.[52]
NG-dimethyl-arginine [asymmetric dimethyl arginine (ADMA)] is an endogenous competitive inhibitor of NO synthase.[55] ADMAs are elevated in patients with uremia,[55] hypercholesterolemia, hyperglycemia, and atherosclerosis.[56,57] The major metabolic pathway for ADMA is dimethylarginine dimethylaminohydrolase (DDAH).[58] DDAH activity is reduced in the presence of hypercholesterolemia and hyperglycemia.[58] A reduction in DDAH activity leads to increased levels of ADMA. ADMA inhibits bFGF-induced angiogenesis. The impaired angiogenesis can be reversed by oral l-arginine, consistent with a role for ADMA as an endogenous inhibitor of angiogenesis.[59] Diabetes with endothelial dysfunction is accompanied by lowered eNOS activity. ADMA levels may be higher due to reduced DDAH activity and/or renal insufficiency.
The angiopoietins are a family of endothelium-specific growth factors involved in the maturation, stabilization, and remodeling of vessels.[53] Tie-2 is the receptor tyrosine kinase for all four Angs identified thus far; the Ang-1/ Tie-2 system acts in coordination with VEGF at later stages of vascular development.[53] The ligand for the Tie-1 receptor tyrosine kinase (RTK) controls vascular EC integrity.[53] Furthermore, Ang-2 is a known Tie-2 antagonist and is induced at sites of vascular remodeling in order to promote a more plastic vascular state. Diabetic wound healing is associated with increased Ang-2 protein expression and Ang-2 levels remain elevated longer post-wounding in diabetics. Tie-2 protein disappears completely upon wounding in the diabetic, and VEGF protein levels are markedly decreased.[53]
PKC inhibits neovascularization at low concentrations, but promotes it at higher concentrations. The mechanism of PKC-induced angiogenesis antagonism involves non-enzymatic glycosylation, inadequate BM degradation, and ECM expansion.[52] Amadori-glycated albumin secondary to hyperglycemia activates mesangial cell PKCα[60] and β,[61] which in turn activate TGF-β, ultimately leading to hypertrophy of the ECM and diffuse intercapillary sclerosis.[51]
Signal transduction problems
VEGF-mediated monocyte infiltration of arterioles triggers the release of pro-arteriogenic cytokines and growth factors, which trigger further monocyte migration and additional VEGF secretion during CV formation.[62] VEGF induces monocyte migration under normoglycemic conditions, but fails to do so in diabetes. In diabetics, VEGF binds to its receptor in diabetes, but the downstream signal transduction pathway is problematic.[62]
ANGIOGENESIS AND SPECIFIC COMPLICATIONS
Diabetic retinopathy
Proliferative DR is characterized by retinal vessel microaneurysms, hemorrhages, exudates, and edema. One of the primary changes in DR involves loss of pericytes in retinal capillaries, which may lead to vascular failure and chronic hypoxia. Hypoxia-inducible factor (HIF) transcription factors then promote the rapid formation of neovessels, ultimately resulting in exacerbated angiogenesis. The sudden establishment of angiogenic vessels leads to leaky and malfunctioning vascular structures accompanied by delicate BM.
In the retina, the primary sources of VEGF-A are ganglion cells, Muller cells, and retinal pigment epithelium cells.[63] High-affinity VEGF receptors have been identified on retinal ECs and pericytes.[64] VEGF-A increases vascular permeability mediated by leukocyte-mediated endothelial injury, fenestrae formation, dissolution of tight junctions, and transcellular bulk flow, and leads to macular edema. Hypoxia is a key regulator of VEGF-induced ocular neovascularization via the production of HIF-1. HIF-1 is composed of two subunits: HIF1a and HIF1b. Under normoxic conditions, HIF1a is rapidly degraded and undetectable. Conversely, under hypoxic conditions, HIF1a expression increases due to suppressed hydroxylation and decreased degradation.[65]
Funatsu and colleagues simultaneously measured vitreous plasma levels of VEGF and endostatin, and platelet factor 4 (PF-4) (angiogenic inhibitors) in diabetic patients. Results from these studies demonstrated that vitreous levels of VEGF and endostatin significantly correlated with the severity of DR. These findings suggest that the balance between VEGF and angiogenic inhibitors may determine the proliferation of angiogenesis in DR.[66]
Pigment epithelium derived factor (PEDF), a glycoprotein secreted by retinal pigment epithelium and originally described as a neurotrophic factor, is a potent antiangiogenic agent.[67] In ischemia-induced retinal neovascularization, intraocular concentration of VEGF is high, while that of PEDF is low. This factor is known to prevent VEGF and erythropoietin in retinal ECs. Recombinant PEDF (or PEDF agonists) could prove to be of major value in limiting or preventing proliferative retinopathy; furthermore, PEDF neurotrophic properties could be of value in diabetic neuropathy. Enhanced Fn and subsequent Fn-F do have a role to play.
Diabetic nephropathy
Podocytes secrete VEGF, which along with low NO bioavailability in diabetes, contribute to the increased vascular permeability in the glomerulus and cause glomerular injury in diabetic nephropathy[68] [Figure 6]. VEGF-A expression can stimulate EC proliferation and inhibit apoptosis.[69] The upregulation of VEGF-A in early stages of diabetic nephropathy results in the initial progression of the disease and excessive blood vessel formation. The decline of VEGF-A in the later phase of diabetic nephropathy may reflect a loss of endogenous VEGF-A due to the disruption of podocytes and tubular cells in chronic kidney damage.[70]
Figure 6.
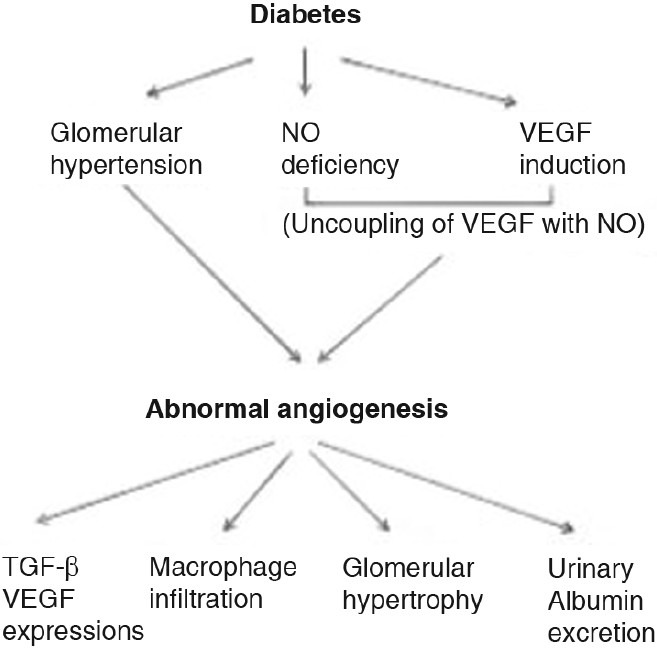
Mechanism and pathogenic role of abnormal angiogenesis in diabetic nephropathy
Hyperglycemia and hypertension induced glomerular hyperfiltration leads to progression of abnormal angiogenesis in diabetes. The beneficial effect of lowering blood pressure could be mediated by VEGF-A inhibition. It is postulated that these vessels function as a by-pass to reduce intraglomerular pressure given that abnormal vessels were found to connect intraglomerular capillaries to peritubular capillaries.[24] Hence, reducing intraglomerular pressure as a consequence of lowering systemic blood pressure might reduce the need for the development of by-pass vessels.
Angiopoietins are critical for the normal vascular differentiation, maintenance, and turnover of blood vessels.[71] Angiopoietin-1 and -2 are ligands for the Tie-2 receptor tyrosine kinase, expressed mainly by endothelia; angiopoietin-1 stimulates receptor activation, leading to promotion of endothelial survival and stabilization. Angiopoietin-2 is considered a natural antagonist of angiopoietin-1.[72] Alterations in the expression of the angiopoietins have been implicated in the progression of diabetic nephropathy. A decreased ratio of angiopoietin-1 to angiopoietin-2 might play a role alongside VEGF-A in the pathobiology of diabetic nephrology.
Another relevant angiogenic marker present in diabetic nephropathy is TGF and its type II receptor. This factor is markedly excreted in the urine in diabetic patients. This emphasizes the angiogenic enhancement in diabetic patients.
Impaired wound healing
The normal wound healing process proceeds through five phases: hemostasis, inflammation and debridement, proliferation, epithelialization, and remodeling. The non-healing nature of the diabetic wound has been attributed to disturbances in both the inflammation/debridement phase and the proliferation phase.[72] A generalized microangiopathy could also prevent the adequate transfer of nutrients to the wounded tissue, thereby interfering with the normal healing process.[73] This is characterized by reduced angiogenesis, decreased arteriolar number and density, loss of vascular tone, and a reduction in the cross- sectional area of new vessel walls, delayed formation of granulation tissue, decreased collagen content, and low breaking strength, as compared with normal littermates.[45] The presence of small abnormal blood vessels – often cuffed with collagen, laminin, Fn, and fibrin – has been reported at the wound edge of diabetic ulcers.[74] Fibroblasts isolated from diabetic ulcers exhibit diminished proliferative capacity. These diabetic wound fibroblasts show characteristically abnormal morphological features such as multiple lamellar and vesicular bodies, an absence of microtubular structures, and enlarged, dilated endoplasmic reticuli, indicative of a hypertrophic phenotype.[72] The lack of microtubules is noteworthy; given the well-established role of microtubules in the regulation of cell migration and the plane of cell division, the absence of mictotubular structures is immediately suggestive of a mechanism, whereby aggregation of lymphocytes, granulocytes, and macrophages, and subsequent cell proliferation are impeded.[72] Prolonged expression of certain ECM molecules, including Fn, has been observed in tissue from chronic diabetic ulcers of duration greater than 12 months, whereas these matrix molecules disappear early in the course of normal wound healing.[72]
Impaired CV formation
CV growth is a compensatory mechanism in response to the ischemia created by advanced CAD, PAD, and atherosclerosis in other vascular beds.[62] A biochemical signal produced by the ischemic myocardium initiates the DNA synthesis and mitotic events leading to growth of collaterals.[7] Increased morbidity and mortality from atherosclerosis and the ensuing CAD and PAD in diabetes is due to an impaired ability to form CV in the diabetic milieu.[27] Compared with age-matched non-diabetics, these patients often present with more widespread vascular disease and a greater number of vascular occlusions with lower capillary density in diabetics with myocardial infarction.[10] Diabetics had a greater frequency of total occlusions of the proximal RCA and LAD.[75]
Embryonic vasculopathy
Embryonic vasculopathy is a well-documented phenomenon in gestational DM, leading to congenital cardiac malformations.[76,77] In normal pregnancies, conceptuses show narrow vessels with flattened mesenchymal and mesodermal cells firmly attached to the abluminal endothelial surface.[77] In contrast, conceptuses exposed to hyperglycemia show capillaries with wider diameters and mesenchymal and mesodermal cells that are plumper and only loosely attached to the abluminal endothelial surface. Abnormal placental angiogenesis is the link between maternal diabetes and embryonic vasculopathy.[78] However, altered expression of angiogenic growth factor in diabetic placenta correlates with reduced fetal capillary branching, maldevelopment of the villous tree, and impaired maternal vascular adaptation to pregnancy, and may provide a mechanistic explanation for the decreased success rate of diabetic pregnancies.
Transplant failure
There is a greater incidence of transplant rejection associated with tissues/organs grafted into a diabetic recipient.[79] This is attributed to impaired angiogenesis caused by the delayed expression of pro-angiogenic factors in grafts. Kawakami et al. report that therapeutic induction of angiogenesis around the time of islet cell transplantation in diabetic recipients greatly increases the survival and functionality of the graft.[80]
Diabetic neuropathy
Pathological changes in the nerves of diabetics include increased resistance to ischemic conduction block and reduced conduction velocity.[81] Endoneural hypoxia of sensory nerve fibers secondary to reduction of nutritive blood flow is responsible for neuropathy. This subsequently leads to imbalances in sorbitol–inositol metabolism as a result of excess glucose metabolism via the polyol pathway.[46]
Malignancies
Diabetes has been associated with an increased prevalence of certain cancers, such as endometrial, liver, biliary, pancreatic, and renal.[82] These findings could be due to an enhanced propensity for neovascularization of tumors in diabetic patients.
THERAPEUTIC IMPLICATION OF ANGIOGENESIS
As the understanding of the mechanisms underlying the diabetic neovascularization has increased, novel therapeutic strategies have been proposed.
VEGF inhibitors
The implication of VEGF in vasculoproliferative DR led to the intravitreal use of anti-VEGF agents in the management of these patients.
Diabetic retinopathy
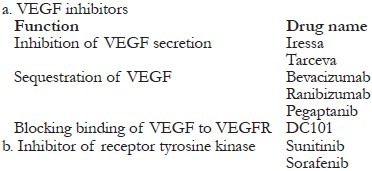
VEGF inhibition might trigger undesirable side effects including impaired wound healing, abrogation of CV growth, hypertension, and proteinuria, which are the prevalent features of diabetic patients. Therefore, a careful evaluation of the molecular vascular events and their association with other diabetic processes like chronic oxidative stress, inflammation, etc. must be accomplished before addressing these pathways toward treatment.
Angiopoietins
Administration of angiopoietin-1 has been shown to suppress DR by preventing leukocyte adhesion, EC injury, and blood retinal barrier breakdown.[83] Systemic adenoviral delivery of COMP-Ang-1 (a modified form of angiopoietin-1) reduced renal fibrosis in db/db mice.[84] This strategy also caused significant improvement in hyperglycemia, an event possibly related to the systemic administration of angiopoietin-1, which could itself account for the amelioration of diabetic nephropathy.
Wound healing
Vascularization in wound healing can be promoted by several interventions. These include therapeutic agents involving growth factors, bioactive matrices, mechanical systems, tissue engineering biomaterials, and hyperbaric oxygen therapy (HBO). HBO, for instance, is already in use and stimulates wound healing by promoting growth factor synthesis at the wound bed and by mobilizing EPCs.
Wound healing
-
-
Growth factors
-
-
PDGF
-
-
EGF
-
-
Bioactive matrices
-
-
Tissue engineering biomaterials
-
-
Placental extract
-
-
Hyperbaric oxygen
-
-
Nicotine (angiogenic promoter)
Pro-angiogenic factors cause hypotension and edema in the case of VEGF and anemia, thrombocytopenia, and renal toxicity in the case of FGF. Hypotension is believed to be mediated through an increase in NO synthesis.[85] Induction of angiogenesis in certain tissues, where angiogenesis is deficient, may cause the unwanted increase in angiogenesis in tissues which already experience a pathological excess of angiogenesis. For example, improvements in wound healing and neuropathy by therapeutic induction of angiogenesis in the extremities may create or exacerbate pre-existing retinopathy or nephropathy.[25]
Others
Disruption of RAAS system
Vasodilation by using angiotensin converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) corrects the hypoxia-induced endoneural angiogenesis.[86] They also block the AgII mediated activation of VEGF and TGF-β.
Aldose reductase inhibitors
The aldose reductase inhibitor, sorbinil, has been used to counteract the increased permeability of vessels created by aberrant angiogenesis in the diabetic milieu.[22]
PKC inhibition
Tranilast, an inhibitor of the PKC-dependent angiogenesis signaling pathway that works downstream of VEGF binding, inhibits phorbolmyristate (PMA)-dependent stimulation of [3H]-thymidine incorporation and VEGF- and PMA-induced gene expression of integrin aV in bovine retinal ECs.[87] Oral administration of LY333531, a competitive inhibitor of PKCb, lowered the rates of glomerular filtration and albumin excretion in diabetic rats in a dose-dependent manner.[88] The thiazolidinedione (TZD) class of insulin sensitizers has also been shown to activate diacyl glycerol (DAG) kinase and inhibit PKC activity.[89]
Inhibition of non-enzymatic glycosylation
Aminoguanidine's protection against the anti-angiogenic effects of hyperglycemia appears to be mediated through its ability to inhibit AGE modification of ECM proteins essential for proper wound healing and angiogenesis.[28]
Dietary arginine supplementation
Arginine is a conditionally essential amino acid, vasorelaxor, angiogenesis promoter, antiatherogenic and antithrombotic factor, It serves as the substrate for synthesis of NO.[90] It regulates vascular tone, hemodynamics, wound healing, and microcirculation. Dietary arginine supplementation reverses or ameliorates EC dysfunction secondary to diabetes.[90] Dietary arginine supplementation induces eNOS and increases blood flow in the femoral artery and calf muscle of patients with PAD and reduces the occurrence of myocardial ischemia in patients with CAD.
Antioxidant therapy
Raxofelast, a synthetic analogue of vitamin E, inhibits lipid peroxidation and scavenges radical oxide species, including superoxide ion.[44] Raxofelast also increases VEGF expression in dermal wounds with increased wound capillary density, breaking strength, and collagen content.[44]
Statins
Statins have demonstrated improvement in the endothelial release of pro-angiogenic cytokines, increase the number and function of circulating EPCs, and upregulate the Akt pathway to promote angiogenesis.[91]
Correction of angioblast deficiencies
Hematopoietic stem cells play an important role in angiogenesis. Exogenous CD34+ cells injected into animals with hind limb ischemia become incorporated into the neovasculature.[10] CD34+ cells from insulin-dependent DM (IDDM) mice produced fewer ECs as compared to CD34+ cells from healthy controls, supposedly because hypoinsulinemia associated with the diabetic state impaired angioblast differentiation.[10] Han et al. have shown that inhibition of TGF-β gene expression by antisense oligodeoxynucleotides (ODNs) successfully reduces kidney weight and mRNA expression of the matrix molecules, Fn and collagen type IV, in mouse models of diabetic nephropathy.[92] The decrease in kidney weight has been attributed to reversal of high-glucose-induced proximal tubular epithelial cell hypertrophy, a hallmark of diabetic nephropathy in both humans and animal models.[92]
CONCLUSION
Vascular complications contribute to the bulk of morbidity and mortality linked with diabetes. Angiogenesis mediates these complications in different ways. It makes sense therefore to target this process by pharmacological strategies to optimize vascular health. However, many questions still remain regarding the use of angiogenesis as a therapeutic approach. More detailed studies are required to elucidate the inherent molecular mechanisms that hold the angiogenic paradox and to predict which patients could benefit from each therapeutic approach.
ACKNOWLEDGMENTS
All the authors would extend their heartfelt thanks to Dr Jagadeesh Tangudu, M Tech, MS, PhD, and Mrs. Sowmya Jammula, M Tech, for their immense and selfless contribution toward manuscript preparation, language editing, and final approval of text.
Footnotes
Source of Support: Nil
Conflict of Interest: No.
REFERENCES
- 1.Simons M. Angiogenesis, arteriogenesis, and diabetes, paradigm reassessed? J Am Coll Cardiol. 2005;46:835–7. doi: 10.1016/j.jacc.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 2.Scares R. Angiogenesis in diabetes.Unraveling the angiogenic paradox. Open Circ Vasc J. 2010;3:3–9. [Google Scholar]
- 3.Wilkinson-Berka JL. Vasoactive factors and diabetic retinopathy: Vascular endothelial growth factor, cycoloxygenase-2 and nitric oxide. Curr Pharm Des. 2004;10:3331–48. doi: 10.2174/1381612043383142. [DOI] [PubMed] [Google Scholar]
- 4.Osterby R, Nyberg G. New vessel formation in the renal corpuscles in advanced diabetic glomerulopathy. J Diabet Complications. 1987;1:122–7. doi: 10.1016/s0891-6632(87)80069-7. [DOI] [PubMed] [Google Scholar]
- 5.Galiano RD, Tepper OM, Pelo CR, Bhatt KA, Callaghan M, Bastidas N, et al. Topical vascular endothelial growth factor accelerates diabetic wound healing through increased angiogenesis and by mobilizing and recruiting bone marrow-derived cells. Am J Pathol. 2004;164:1935–47. doi: 10.1016/S0002-9440(10)63754-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abaci A, Oguzhan A, Kahraman S, Eryol NK, Unal S, Arinç H, et al. Effect of diabetes mellitus on formation of coronary collateral vessels. Circulation. 1999;99:2239–42. doi: 10.1161/01.cir.99.17.2239. [DOI] [PubMed] [Google Scholar]
- 7.Tepper OM, Galiano RD, Capla JM, Kalka C, Gagne PJ, Jacobowitz GR, et al. Human endothelial progenitor cells from type II diabetics exhibit impaired proliferation, adhesion, and incorporation into vascular structures. Circulation. 2002;106:2781–6. doi: 10.1161/01.cir.0000039526.42991.93. [DOI] [PubMed] [Google Scholar]
- 8.Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931–4. [PubMed] [Google Scholar]
- 9.Hohenstein B, Hausknecht B, Boehmer K, Riess R, Brekken RA, Hugo CP. Local VEGF activity but not VEGF expression is tightly regulated during diabetic nephropathy in man. Kidney Int. 2006;69:1654–61. doi: 10.1038/sj.ki.5000294. [DOI] [PubMed] [Google Scholar]
- 10.Barzilai JI, Kronmall RA, Bittner V, Eaker E, Evans C, Foster ED. Coronary artery disease and coronary artery bypass grafting in diabetic patients age >65 years (Report from Coronary Artery Surgery Study (CASS) Registry) Am J Cardiol. 1994;74:334–9. doi: 10.1016/0002-9149(94)90399-9. [DOI] [PubMed] [Google Scholar]
- 11.Harper SJ, Bates DO. VEGF-A splicing: The key to anti-angiogenic therapeutics? Nat Rev Cancer. 2008;8:880–7. doi: 10.1038/nrc2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Loomans CJ, de Koning EJ, Staal FJ, Rookmaaker MB, Verseyden C, de Boer HC, et al. Endothelial progenitor cell dysfunction: A novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes. 2004;53:195–9. doi: 10.2337/diabetes.53.1.195. [DOI] [PubMed] [Google Scholar]
- 13.Chou E, Suzuma I, Way KJ, Opland D, Clermont AC, Naruse K, et al. Decreased cardiac expression of vascular endothelial growth factor and its receptors in insulin-resistant and diabetic states: A possible explanation for impaired collateral formation in cardiac tissue. Circulation. 2002;105:373–9. doi: 10.1161/hc0302.102143. [DOI] [PubMed] [Google Scholar]
- 14.Weihrauch D, Lohr NL, Mraovic B, Ludwig LM, Chilian WM, Pagel PS, et al. Chronic hyperglycemia attenuates coronary collateral development and impairs proliferative properties of myocardial interstitial fluid by production of angiostatin. Circulation. 2004;109:2343–8. doi: 10.1161/01.CIR.0000129225.67353.1F. [DOI] [PubMed] [Google Scholar]
- 15.Cooper ME, Vranes D, Youssef S, Stacker SA, Cox AJ, Rizkalla B, et al. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes. 1999;48:2229–39. doi: 10.2337/diabetes.48.11.2229. [DOI] [PubMed] [Google Scholar]
- 16.Waltenberger J, Lange J, Kranz A. Vascular endothelial growth factor-A-induced chemotaxis of monocytes is attenuated in patients with diabetes mellitus: A potential predictor for the individual capacity to develop collaterals. Circulation. 2000;102:185–90. doi: 10.1161/01.cir.102.2.185. [DOI] [PubMed] [Google Scholar]
- 17.Sasso FC, Torella D, Carbonara O, Ellison GM, Torella M, Scardone M, et al. Increased vascular endothelial growth factor expression but impaired vascular endothelial growth factor receptor signaling in the myocardium of type 2 diabetic patients with chronic coronary heart disease. J Am Coll Cardiol. 2005;46:827–34. doi: 10.1016/j.jacc.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 18.Simons M. Angiogenesis: Where do we stand now? Circulation. 2005;111:1556–66. doi: 10.1161/01.CIR.0000159345.00591.8F. [DOI] [PubMed] [Google Scholar]
- 19.Adamis AP, Aiello LP, D’Amato RA. Angiogenesis and ophthalmic disease. Angiogenesis. 1999;3:9–14. doi: 10.1023/a:1009071601454. [DOI] [PubMed] [Google Scholar]
- 20.van der Zee R, Murohara T, Luo Z, Zollmann F, Passeri J, Lekutat C, et al. Vascular endothelial growth factor/vascular permeability factor augments nitric oxide release form quiescent rabbit and humanvascular endothelium. Circulation. 1997;95:1030–7. doi: 10.1161/01.cir.95.4.1030. [DOI] [PubMed] [Google Scholar]
- 21.Inoue N, Venema RC, Sayegh HS, Ohara Y, Murphy TJ, Harrison DG. Molecular regulation of the bovine endothelial cell nitric oxide synthase by transforming growth factor-beta 1. Arterioscler Thromb Vasc Biol. 1995;15:1255–61. doi: 10.1161/01.atv.15.8.1255. [DOI] [PubMed] [Google Scholar]
- 22.Babaei S, Teichert-Kuliszewska K, Monge JC, Mohamed F, Bendeck MP, Stewart DJ. Role of nitric oxide in the angiogenic response in vitro to basic fibroblast growth factor. Circ Res. 1998;82:1007–15. doi: 10.1161/01.res.82.9.1007. [DOI] [PubMed] [Google Scholar]
- 23.Papapetropoulos A, Garcia-Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest. 1997;100:3131–9. doi: 10.1172/JCI119868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;6:843–5. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- 25.Freedman SB, Isner JM. Therapeutic angiogenesis for coronary artery disease. Ann Intern Med. 2002;136:54–71. doi: 10.7326/0003-4819-136-1-200201010-00011. [DOI] [PubMed] [Google Scholar]
- 26.Williams B. A potential role for angiotensin II-induced vascular endothelial growth factor expression in the pathogenesis of diabetic nephropathy? Miner Electrolyte Metab. 1998;24:400–5. doi: 10.1159/000057401. [DOI] [PubMed] [Google Scholar]
- 27.Ziyadeh FN, Hoffman BB, Han DC, Iglesias-de la Cruz MC, Hong SW, Isono M, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db mice. Proc Natl Acad Sci U S A. 2000;97:8015–20. doi: 10.1073/pnas.120055097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teixeira AS, Andrade SP. Glucose-induced inhibition of angiogenesis in the rat sponge granuloma is prevented by aminoguanidine. Life Sci. 1999;64:655–62. doi: 10.1016/s0024-3205(98)00607-9. [DOI] [PubMed] [Google Scholar]
- 29.Tamarat R, Silvestre JS, Huijberts M, Benessiano J, Ebrahimian TG, Duriez M, et al. Blockade of advanced glycation end-product formation restores ischemia-induced angiogenesis in diabetic mice. Proc Natl Acad Sci U S A. 2003;100:8555–60. doi: 10.1073/pnas.1236929100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goova MT, Li J, Kislinger T, Qu W, Lu Y, Bucciarelli LG, et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am J Pathol. 2001;159:513–25. doi: 10.1016/S0002-9440(10)61723-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Giardino I, Edelstein D, Brownlee M. Nonenzymatic glycosylation in vitro and in bovine endothelial cells alters basic fibroblast growth factor activity: A model for intracellular glycosylation in diabetes. J Clin Invest. 1994;94:110–7. doi: 10.1172/JCI117296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capogrossi MC. Sugar-induced modification of fibroblast growth factor 2 reduces its angiogenic activity in vivo. Am J Pathol. 2002;161:531–41. doi: 10.1016/S0002-9440(10)64209-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffmann S, Friedrichs U, Eichler W, Rosenthal A, Wiedemann P. Advanced glycation end products induce choroidal endothelial cell proliferation, matrix metalloproteinase-2 and VEGF upregulation in vitro. Graefes Arch Clin Exp Ophthalmol. 2002;240:996–1002. doi: 10.1007/s00417-002-0568-6. [DOI] [PubMed] [Google Scholar]
- 34.Yamagishi S, Yonekura H, Yamamoto Y, Katsuno K, Sato F, Mita I, et al. Advanced glycation end products-driven angiogenesis in vitro: Induction of the growth and tube formation of human microvascular endothelial cells through autocrine vascular endothelial growth factor. J Biol Chem. 1997;272:8723–30. doi: 10.1074/jbc.272.13.8723. [DOI] [PubMed] [Google Scholar]
- 35.Twigg SM, Chen MM, Joly AH, Chakrapani SD, Tsubaki J, Kim HS, et al. Advanced glycosylation end products up-regulate connective tissue growth factor (insulin-like growth factor-binding protein- related protein 2) in human fibroblasts: A potential mechanism for expansion of extracellular matrix in diabetes mellitus. Endocrinology. 2001;142:1760–9. doi: 10.1210/endo.142.5.8141. [DOI] [PubMed] [Google Scholar]
- 36.Braunwald E, Isselbacher KJ, Petersdorf RG, Wilson JD, Martin JB, Fauci AS. Harrison's Principles of Internal Medicine. 11th ed. New York: McGraw-Hill; 1987. [Google Scholar]
- 37.Grant MB, Caballero S, Bush DM, Spoerri PE. Fibronectin fragments modulate human retinal capillary cell proliferation and migration. Diabetes. 1998;47:1335–40. doi: 10.2337/diab.47.8.1335. [DOI] [PubMed] [Google Scholar]
- 38.Zimering MB, Eng J. Increased basic fibroblast growth factor-like substance in plasma from a subset of middle-aged or elderly male diabetic patients with microalbuminuria or proteinuria. J Clin Endocrinol Metab. 1996;81:4446–52. doi: 10.1210/jcem.81.12.8954057. [DOI] [PubMed] [Google Scholar]
- 39.Hill DJ, Petrik J, Arany E. Growth factors and the regulation of fetal growth. Diabetes Care. 1998;21:B60–9. [PubMed] [Google Scholar]
- 40.Tomanek RJ, Schatteman GC. Angiogenesis: New insights and therapeutic potential. Anat Rec. 2000;261:126–35. doi: 10.1002/1097-0185(20000615)261:3<126::AID-AR7>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 41.Casaroli-Marano RP, Preissner KT, Vilaro S. Fibronectin, laminin, vitronectin, and their receptors at newly-formed capillaries in proliferative diabetic retinopathy. Exp Eye Res. 1995;60:5–17. doi: 10.1016/s0014-4835(05)80079-x. [DOI] [PubMed] [Google Scholar]
- 42.Stromblad S, Cheresh DA. Cell adhesion and angiogenesis. Trends Cell Biol. 1996;6:462–7. doi: 10.1016/0962-8924(96)84942-7. [DOI] [PubMed] [Google Scholar]
- 43.Cooke JP. The pathophysiology of peripheral arterial disease: Rational targets for drug intervention. Vasc Med. 1997;2:227–30. doi: 10.1177/1358863X9700200311. [DOI] [PubMed] [Google Scholar]
- 44.Galeano M, Torre V, Deodato B, Campo GM, Colonna M, Sturiale A, et al. Raxofelast, a hydrophilic vitamin E-like antioxidant, stimulates wound healing in genetically diabetic mice. Surgery. 2001;129:467–77. doi: 10.1067/msy.2001.112072. [DOI] [PubMed] [Google Scholar]
- 45.Gilbert RE, Kelly DJ, Cox AJ, Wilkinson-Berka JL, Rumble JR, Osicka T, et al. Angiotensin converting enzyme inhibition reduced retinal overexpression of vascular endothelial growth factor and hyperpermeability in experimental diabetes. Diabetologia. 2000;43:1360–7. doi: 10.1007/s001250051539. [DOI] [PubMed] [Google Scholar]
- 46.Cameron NE, Cotter MA, Robertson S. Angiotensin converting enzyme inhibition prevents development of muscle and nerve dysfunction and stimulates angiogenesis in streptozotocin-diabetic rats. Diabetologia. 1992;35:12–8. doi: 10.1007/BF00400846. [DOI] [PubMed] [Google Scholar]
- 47.Williamson JR, Chang K, Rowold E, Marvel J, Tomlinson M, Sherman WR, et al. Sorbinil prevents diabetes-induced increases in vascular permeability but does not alter collagen crosslinking. Diabetes. 1985;34:703–5. doi: 10.2337/diab.34.7.703. [DOI] [PubMed] [Google Scholar]
- 48.Samad F, Pandey M, Loskutoff DJ. Tissue factor gene expression in the adipose tissue of obese mice. Proc Natl Acad Sci U S A. 1998;95:7591–6. doi: 10.1073/pnas.95.13.7591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Halvoresen YD, Bursell SE, Wilkison WO, Clermont AC, Brittis M, McGovern TJ, et al. Vasodilation of rat retinal microvessels induced by monobutyrin. Dysregulation in diabetes. J Clin Invest. 1993;92:2872–6. doi: 10.1172/JCI116908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aksoy H, Akcay F, Kurtul N, Baykal O, Avci B. Serum 1,25-dihydroxy vitamin D (1,25(OH)2D3), 25-hydroxy vitamin D (25(OH)D) and parathormone levels in diabetic retinopathy. Clin Biochem. 2000;33:47–51. doi: 10.1016/s0009-9120(99)00085-5. [DOI] [PubMed] [Google Scholar]
- 51.Yarom R, Zirkin H, Stammler G, Rose AG. Human coronary microvessels in diabetes and ischemia.Morphometric study of autopsy material. J Pathol. 1992;166:265–70. doi: 10.1002/path.1711660308. [DOI] [PubMed] [Google Scholar]
- 52.Nyengaard JR, Rasch R. The impact of experimental diabetes mellitus in rats on glomerular capillary number and sizes. Diabetologia. 1993;36:189–94. doi: 10.1007/BF00399948. [DOI] [PubMed] [Google Scholar]
- 53.Kampfer H, Pfeilschifter J, Frank S. Expressional regulation of angiopoietin-1 and -2 and the tie-1 and -2 receptor tyrosine kinases during cutaneous wound healing: A comparative study of normal and impaired repair. Lab Invest. 2001;81:361–73. doi: 10.1038/labinvest.3780244. [DOI] [PubMed] [Google Scholar]
- 54.Bitar MS, Labbad ZN. Transforming growth factor-beta and insulin- like growth factor-1 in relation to diabetes-induced impairment of wound healing. J Surg Res. 1996;61:113–9. doi: 10.1006/jsre.1996.0090. [DOI] [PubMed] [Google Scholar]
- 55.Vallance P, Leone A, Claver A, Collier J, Moncada S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet. 1992;339:572–5. doi: 10.1016/0140-6736(92)90865-z. [DOI] [PubMed] [Google Scholar]
- 56.Bode-Boger SM, Boger RH, Kienke S, Junker W, Frolich JC. Elevated L-arginine/dimethylarginine ratio contributes to enhanced systemic NO production by dietary L-arginine in hypercholesterolemic rabbits. Biochem Biophys Res Commun. 1996;219:598–603. doi: 10.1006/bbrc.1996.0279. [DOI] [PubMed] [Google Scholar]
- 57.Boger RH, Bode-Boger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, et al. Asymmetric dimethylarginine (ADMA): A novel risk factor for endothelial dysfunction: Its role in hypercholesterolemia. Circulation. 1998;98:1842–7. doi: 10.1161/01.cir.98.18.1842. [DOI] [PubMed] [Google Scholar]
- 58.Cooke JP. Does ADMA cause endothelial dysfunction? Arterioscler Thromb Vasc Biol. 2000;20:2032–7. doi: 10.1161/01.atv.20.9.2032. [DOI] [PubMed] [Google Scholar]
- 59.Jang JJ, Ho HK, Kwan HH, Fajardo LF, Cooke JP. Angiogenesis is impaired by hypercholesterolemia: Role of asymmetric dimethylarginine. Circulation. 2000;102:1414–9. doi: 10.1161/01.cir.102.12.1414. [DOI] [PubMed] [Google Scholar]
- 60.Wang A, Nomura M, Patan S, Ware JA. Inhibition of protein kinase C alpha prevents endotheilial cell migration and vascular tube formation in vitro and myocardial neovascularization in vivo. Circ Res. 2002;90:609–16. doi: 10.1161/01.res.0000012503.30315.e8. [DOI] [PubMed] [Google Scholar]
- 61.Ido Y, Chang KC, Lejeune WS, Bjercke RJ, Reiser KM, Williamson JR, et al. Vascular dysfunction induced by AGE is mediated by VEGF via mechanisms involving reactive oxygen species, guanylate cyclase, and protein kinase C. Microcirculation. 2001;8:251–63. doi: 10.1038/sj/mn/7800079. [DOI] [PubMed] [Google Scholar]
- 62.Waltenberger J. Impaired collateral vessel development in diabetes: Potential cellular mechanisms and therapeutic implications. Cardiovasc Res. 2001;49:554–60. doi: 10.1016/s0008-6363(00)00228-5. [DOI] [PubMed] [Google Scholar]
- 63.Mizutani M, Kern TS, Lorenzi M. Accelerated death of retinal microvascular cells in human and experimental diabetic retinopathy. J Clin Invest. 1996;97:2883–90. doi: 10.1172/JCI118746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burgos R, Simo R, Audi L, Mateo C, Mesa J, Garcia-Ramirez M, et al. Vitreous levels of vascular endothelial growth factor are not influenced by its serum concentrations in diabetic retinopathy. Diabetologia. 1997;40:1107–9. doi: 10.1007/s001250050794. [DOI] [PubMed] [Google Scholar]
- 65.Sell DR, Lapolla A, Odetti P, Fogarty J, Monnier VM. Pentosidine formation in skin correlates with severity of complications in individuals with long-standing IDDM. Diabetes. 1992;41:1286–92. doi: 10.2337/diab.41.10.1286. [DOI] [PubMed] [Google Scholar]
- 66.Okamoto T, Yamagishi S, Inagaki Y, Amano S, Takeuchi M, Kikuchi S, et al. Incadronate disodium inhibits advanced glycation end products- induced angiogenesis in vitro. Biochem Biophys Res Commun. 2002;297:419–24. doi: 10.1016/s0006-291x(02)02218-0. [DOI] [PubMed] [Google Scholar]
- 67.Filleur S, Nelius T, de Riese W, Kennedy RC. Characterization of PEDF: A multi-functional serpin family protein. J Cell Biochem. 2009;106:769–75. doi: 10.1002/jcb.22072. [DOI] [PubMed] [Google Scholar]
- 68.Nakagawa T, Sato W, Glushakova O, Heinig M, Clarke T, Campbell-Thompson M, et al. Diabetic eNOS knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol. 2007;18:539–50. doi: 10.1681/ASN.2006050459. [DOI] [PubMed] [Google Scholar]
- 69.McGinn S, Saad S, Poronnik P, Pollock CA. High glucose-mediated effects on endothelial cell proliferation occur via p38 MAP kinase. Am J Physiol Endocrinol Metab. 2003;285:E708–17. doi: 10.1152/ajpendo.00572.2002. [DOI] [PubMed] [Google Scholar]
- 70.Yuan HT, Li XZ, Pitera JE, Long DA, Woolf AS. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia-inducible factor-1 alpha. Am J Pathol. 2003;163:2289–301. doi: 10.1016/s0002-9440(10)63586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Woolf AS, Gnudi L, Long DA. Roles of angiopoietins in kidney development and disease. J Am Soc Nephrol. 2009;20:239–44. doi: 10.1681/ASN.2008020243. [DOI] [PubMed] [Google Scholar]
- 72.Loots MA, Lamme EN, Zeegelaar J, Mekkes JR, Bos JD, Middlekoop E. Differences in cellular infiltrate and extracellular matrix of chronic diabetes and venous ulcers venous acute wounds. J Invest Dermatol. 1998;111:850–7. doi: 10.1046/j.1523-1747.1998.00381.x. [DOI] [PubMed] [Google Scholar]
- 73.Shyng YC, Devlin H, Sloan P. The effect of streptozotocin-induced experimental diabetes mellitus on calvarial defect healing and bone turnover in the rat. Int J Oral Maxillofac Surg. 2001;30:70–4. doi: 10.1054/ijom.2000.0004. [DOI] [PubMed] [Google Scholar]
- 74.Ferguson MW, Herrick SE, Spencer MJ, Shaw JE, Boulton AJ, Sloan P. The histology of diabetic foot ulcers. Diabet Med. 1996;13:S30–3. [PubMed] [Google Scholar]
- 75.Melidonis A, Tournis S, Kouvaras G, Baltaretsou E, Hadanis S, Hajissavas I, et al. Comparison of coronary collateral circulation in diabetic nondiabetic patients suffering from coronary artery disease. Clin Cardiol. 1999;22:465–71. doi: 10.1002/clc.4960220706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reece EA, Homko CJ. Multifactorial basis of the syndrome of diabetic embryopathy. Teratology. 1996;54:171–82. doi: 10.1002/(SICI)1096-9926(199610)54:4<171::AID-TERA1>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 77.Pinter E, Mahooti S, Wang Y, Imhof BA, Madri JA. Hyperglycemia-induced vasculopathy in the murine vitelline vasculature: Correlation with PECAM 1/CD31 tyrosine phosphorylation state. Am J Pathol. 1999;154:1367–79. doi: 10.1016/S0002-9440(10)65391-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Helske S, Vuorela P, Carpen O, Hornig C, Weich H, Halmesmaki E. Expression of vascular endothelial growth factor receptors 1, 2 and 3 in placentas from normal and complicated pregnancies. Mol Hum Reprod. 2001;7:205–10. doi: 10.1093/molehr/7.2.205. [DOI] [PubMed] [Google Scholar]
- 79.Vasir B, Reitz P, Xu G, Sharma A, Bonner-Weir S, Weir GC. Effects of diabetes and hypoxia on gene markers of angiogenesis (HGF, cMET, uPA and uPAR, TGF-alpha, TGF-beta, bFGF and Vimentin) in cultured and transplanted rat islets. Diabetologia. 2000;43:763–72. doi: 10.1007/s001250051374. [DOI] [PubMed] [Google Scholar]
- 80.Kawakami Y, Iwata H, Gu YJ, Miyamoto M, Murakami Y, Balamurugan AN, et al. Successful subcutaneous pancreatic islet transplantation using an angiogenic growth factor releasing device. Pancreas. 2001;23:375–81. doi: 10.1097/00006676-200111000-00007. [DOI] [PubMed] [Google Scholar]
- 81.Cameron NE, Cotter MA, Archibald V, Dines KC, Maxfield EK. Anti-oxidant and pro- oxidant effects on nerve conduction velocity, endoneurial blood flow and oxygen tension in non-diabetic andbstreptozotocindiabetic rats. Diabetologia. 1994;37:449–59. doi: 10.1007/s001250050131. [DOI] [PubMed] [Google Scholar]
- 82.Wideroff L, Gridley G, Mellemkjaer L, Chow WH, Linet M, Keehn S, et al. Cancer incidence in a population-based cohort of patients hospitalized with diabetes mellitus in Denmark. J Natl Cancer Inst. 1997;89:1360–5. doi: 10.1093/jnci/89.18.1360. [DOI] [PubMed] [Google Scholar]
- 83.Joussen AM, Poulaki V, Tsujikawa A, Qin W, Qaum T, Xu Q, et al. Suppression of diabetic retinopathy with angiopoietin-1. Am J Pathol. 2002;160:1683–93. doi: 10.1016/S0002-9440(10)61115-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee S, Kim W, Moon SO, Sung MJ, Kim DH, Kang KP, et al. Renoprotective effect of COMP-angiopoietin- 1 in db/db mice with type 2 diabetes. Nephrol Dial Transplant. 2007;22:396–408. doi: 10.1093/ndt/gfl598. [DOI] [PubMed] [Google Scholar]
- 85.Lutty GA, McLeod DS, Merges C, Diggs A, Plouet J. Localization of vascular endothelial growth factor in human retina and choroid. Arch Ophthalmol. 1996;114:971–7. doi: 10.1001/archopht.1996.01100140179011. [DOI] [PubMed] [Google Scholar]
- 86.Maxfield EK, Cameron NE, Cotter MA, Dines KC. Angiotensin II receptor blockade improves nerve function, modulates nerve blood flow and stimulates endoneurial angiogenesis in streptozotocin-diabetic rats and nerve finction. Diabetologia. 1993;36:1230–7. doi: 10.1007/BF00400799. [DOI] [PubMed] [Google Scholar]
- 87.Zygmunt M. Placental circulation: Clinical significance. Early Pregnancy. 2001;5:72–3. [PubMed] [Google Scholar]
- 88.Ishii H, Jirousek MR, Koya D, Takagi C, Xia P, Clermont A, et al. Amelioration of vascular dysfunction in diabetic rats by an oral PKC beta inhibitor. Science. 1996;272:728–31. doi: 10.1126/science.272.5262.728. [DOI] [PubMed] [Google Scholar]
- 89.Haneda M, Koya D, Kikkawa R. Cellular mechanisms in the development and progression of diabetic nephropathy: Activation of the DAG-PKC-ERK pathway. Am J Kidney Dis. 2001;38:S178–81. doi: 10.1053/ajkd.2001.27438. [DOI] [PubMed] [Google Scholar]
- 90.Wu G, Meininger CJ. Arginine nutrition and cardiovascular function. J Nutr. 2000;130:2626–9. doi: 10.1093/jn/130.11.2626. [DOI] [PubMed] [Google Scholar]
- 91.Dulak J, Jozkowicz A. Anti-angiogenic and Anti-Inflammatory effects of statin: Relevance to Anti-Cancer Therapy. Curr Cancer Drug Targets. 2005;5:79–94. doi: 10.2174/156800905774932824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Han DC, Hoffman BB, Hong SW, Guo J, Ziyadeh FN. Therapy with antisense TGF-beta-1 oligodeoxynucleotides reduces kidney weight and matrix mRNAs in diabetic mice. Am J Physiol Renal Physiol. 2000;278:F628–34. doi: 10.1152/ajprenal.2000.278.4.F628. [DOI] [PubMed] [Google Scholar]