

Background: Versatile peroxidases oxidize different substrates, including low redox potential aromatics.
Results: To investigate this activity, 13 variants were characterized, and five x-ray structures obtained.
Conclusion: Phenols and dyes are oxidized in a low efficiency site in the heme channel and a high efficiency site at an exposed tryptophan.
Significance: This study supplies new structural-functional information on versatile peroxidase, and provides variants with improved activity on phenols.
Keywords: Crystal Structure, Enzyme Kinetics, Enzyme Mechanisms, Fungi, Lignin Degradation, Peroxidase, Pre-steady-state Kinetics, Protein Engineering, Site-directed Mutagenesis, Versatile Peroxidase
Abstract
Versatile peroxidase shares with manganese peroxidase and lignin peroxidase the ability to oxidize Mn2+ and high redox potential aromatic compounds, respectively. Moreover, it is also able to oxidize phenols (and low redox potential dyes) at two catalytic sites, as shown by biphasic kinetics. A high efficiency site (with 2,6-dimethoxyphenol and p-hydroquinone catalytic efficiencies of ∼70 and ∼700 s−1 mm−1, respectively) was localized at the same exposed Trp-164 responsible for high redox potential substrate oxidation (as shown by activity loss in the W164S variant). The second site, characterized by low catalytic efficiency (∼3 and ∼50 s−1 mm−1 for 2,6-dimethoxyphenol and p-hydroquinone, respectively) was localized at the main heme access channel. Steady-state and transient-state kinetics for oxidation of phenols and dyes at the latter site were improved when side chains of residues forming the heme channel edge were removed in single and multiple variants. Among them, the E140G/K176G, E140G/P141G/K176G, and E140G/W164S/K176G variants attained catalytic efficiencies for oxidation of 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonate) at the heme channel similar to those of the exposed tryptophan site. The heme channel enlargement shown by x-ray diffraction of the E140G, P141G, K176G, and E140G/K176G variants would allow a better substrate accommodation near the heme, as revealed by the up to 26-fold lower Km values (compared with native VP). The resulting interactions were shown by the x-ray structure of the E140G-guaiacol complex, which includes two H-bonds of the substrate with Arg-43 and Pro-139 in the distal heme pocket (at the end of the heme channel) and several hydrophobic interactions with other residues and the heme cofactor.
Introduction
Ligninolytic peroxidases are enzymes of biotechnological interest playing a central role in lignin biodegradation by white-rot fungi (1). Three ligninolytic peroxidase families, differing in substrate specificity, have been described and widely characterized (2, 3). Classical manganese peroxidase (MnP)5 (EC 1.11.1.13), first described in Phanerochaete chrysosporium, requires Mn2+ to complete its catalytic cycle, and generates Mn3+ that acts as a diffusible oxidizer of low redox potential compounds (including lignin phenolic units and simple phenols) (4). A new MnP type, characterized by its ability to directly oxidize not only Mn2+ but also phenols and other low redox potential compounds has been suggested in Phlebia radiata (5), and similar genes identified in recently sequenced fungal genomes (6–8). Unlike MnP, lignin peroxidase (LiP) (EC 1.11.1.14), first described also from P. chrysosporium, is characterized by its capacity to oxidize high redox potential nonphenolic aromatic substrates, including lignin model compounds and polycyclic aromatic hydrocarbons, among others (9, 10). Finally, versatile peroxidase (VP) (EC 1.11.1.16), thoroughly investigated in Pleurotus eryngii, combines catalytic properties of MnP, LiP, and generic peroxidases (low redox-potential peroxidases of plant and fungal origin) being able to oxidize Mn2+, as well as phenolic and nonphenolic aromatic compounds and dyes (11). This promiscuity is precisely what makes VP interesting for different environmental and industrial applications, including processing of plant feedstocks in the sustainable production of fuels, chemicals, and other products (3).
VP combines features of the catalytic cycles of the other peroxidases mentioned above, as described by Pérez-Boada et al. (12) (Fig. 1). At least seven steps can be described in this cycle, depending on the nature of the substrate to be oxidized. It starts when the resting enzyme (VP, containing Fe3+) is two-electron oxidized by peroxide yielding compound I, which contains a ferryl oxo iron (Fe4+ = O) and a porphyrin cation radical (P+·) (VP-IA). VP-IA catalyzes one-electron substrate oxidation in direct contact with heme, as in Mn2+ oxidation (13), or through the formation of a tryptophanyl radical (resulting in VP-IB) (14) responsible for the oxidation of high redox potential substrates (e.g. veratryl alcohol) (12). In both ways, typical compound II containing Fe4+ = O (VP-IIA) is simultaneously formed. VP-IIA can also one-electron oxidize substrates directly in contact with the heme or through the tryptophanyl radical characteristic of VP-IIB (15), restoring the resting state of the enzyme.
FIGURE 1.
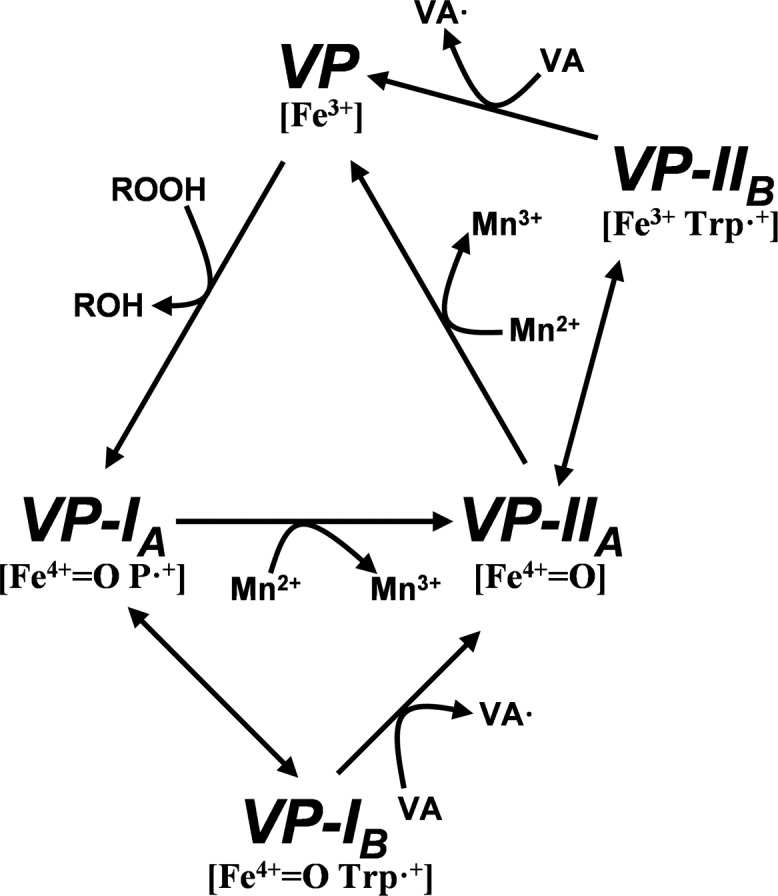
VP catalytic cycle proposed by Pérez-Boada et al. (12). Resting state peroxidase (VP, containing Fe3+) is two-electron oxidized by hydroperoxide, yielding compound I (VP-IA, containing Fe4+-oxo and porphyrin cation radical, P+·). VP-IA catalyzes one-electron oxidation of substrates in direct contact with heme (e.g. Mn2+) or through the formation of an alternative compound I (VP-IB) containing a tryptophanyl radical, which is responsible for the oxidation of high redox potential aromatic compounds (e.g. veratryl alcohol, shown as VA). In both ways VP-IIA is formed. This transient state of the enzyme bears only one oxidation equivalent (on the Fe4+-oxo) and can oxidize another substrate molecule, interacting directly with the heme group or through the tryptophanyl radical (VP-IIB) to recover the resting state.
According to the above catalytic properties, VP combines structural features of the other ligninolytic peroxidases, although some peculiarities make it not a mere MnP/LiP hybrid (16). In this respect, the existence of two different substrate oxidation sites is well established in VP. The Mn2+ oxidation site is formed by three acidic residues at a small channel giving access to the internal heme propionate. This catalytic site has higher plasticity than in MnP because VP is able to efficiently oxidize Mn2+ in the absence of one of the three acidic residues (13). On the other hand, high redox-potential substrates are oxidized by both VP and LiP at an exposed tryptophanyl radical, followed by long-range electron transfer to heme (pyrrolic ring-C) (12, 17). However, the two enzymes show different kinetic constants oxidizing substrates at this site, due to differences in the amino acid residues forming the catalytic tryptophan environment (18). VP, like all other heme peroxidases, presents an access channel to the distal heme pocket enabling the entrance of H2O2 for activation of the cofactor (19). In several peroxidases, such as horseradish peroxidase (HRP) (EC 1.11.1.7) and Coprinopsis cinerea peroxidase (CiP) (EC 1.11.1.7), it is assumed that phenolic compounds are oxidized through this channel in direct contact with the heme (20), and the same site has been suggested for LiP oxidation of anionic dyes (21). To determine whether the main heme access channel is the oxidation site for phenolic substrates and dyes in VP, directed mutagenesis was performed removing bulky and charged residues at the channel entrance. Two of the variants produced also included mutations at the exposed Trp-164, which has been described as responsible for high redox potential substrate oxidation by VP (12). The steady-state and transient-state kinetics of the different variants were analyzed and the most interesting ones were crystallized. Some of the heme-channel mutations improved VP oxidation of phenolic compounds, providing variants with a potential biotechnological interest (22).
EXPERIMENTAL PROCEDURES
Chemicals
Catechol (purity ≥99%), dithiothreitol (purity ≥98%), 2,6-dimethoxyphenol (DMP) (purity 99%), ferrocyanide (purity ≥99.99%), guaiacol (purity ≥98%), hemin (purity ≥98%), p-hydroquinone (HQ) (purity ≥99%), isopropyl β-d-thiogalactopyranoside (purity ≥99%), manganese(II) sulfate (purity ≥99.99%), and oxidized glutathione (purity ≥98%) were from Sigma; H2O2, sodium tartrate (purity ≥99.5%), and urea (purity ≥99.5%) were from Merck; and ABTS (purity >98%) was from Roche Applied Science. None of the above chemicals was further purified.
Heterologous Expression
Nonmutated native (wild-type) recombinant VP and different directed variants were obtained by Escherichia coli expression (23). The cDNA encoding the sequence of mature isoenzyme VPL of P. eryngii (allelic variant VPL2; GenBankTM AF007222) (24) was cloned in the pFLAG1 vector (International Biotechnologies Inc.) yielding pFLAG1-VPL2. E. coli DH5α was selected for plasmid propagation, whereas E. coli W3110 was used for native and mutated VP expression. The VP proteins accumulated in inclusion bodies, and were activated in vitro and purified as indicated below.
Site-directed Mutagenesis
Mutations were introduced by polymerase chain reaction (PCR) using the pFLAG1-VPL2 plasmid as template, and the QuikChangeTM kit from Stratagene. For each mutation, both a direct and a reverse primer were designed complementary to opposite strands of the same DNA region, but only the direct constructions with indication of the changed triplets (underlined) and the mutations introduced (bold) are included below: (i) P76G, 5′-CGACACCATTGAGACTAATTTCGGCGCCAATGCTGGCATCG-3′; (ii) F142G, 5′-GGACCACCTCGTGCCAGAGCCTGGTGATTCTGTTGACTC-3′; (iii) K176D, 5′-GCCGCTGCCGACGACGTTGACCCATCGATTCC-3′; (iv) K176G, 5′-GCCGCTGCCGACGGAGTTGACCCATCGATTCCTGG-3′; (v) K215Q, 5′-CCCAGGCACTGCTGACAACCAGGGAGAAGCCCAATCTCC-3′; (vi) K215G, 5′-CCCAGGCACTGCTGACAACGGCGGAGAAGCCCAATCTCC-3′; (vii) E140G, 5′-GGACCACCTCGTGCCAGGACCTTTTGATTCTGTTG-3′; (viii) P141G, 5′-CCACCTCGTGCCAGAGGGTTTTGATTCTGTTGACTCC-3′; (ix) E140G/P141G, 5′-CCGGACCACCTCGTGCCAGGCGGTTTTGATTCTGTTGACTCC-3′; (x) W164S, 5′-CCCGTCGAGGTTGTTTCGCTCCTGGCTTCGC-3′; (xi) the double variant E140G/K176G was obtained using plasmid pFLAG1-VPL2-E140G as template and the K176G primers; (xii) the triple variant E140G/P141G/K176G was obtained using plasmid pFLAG1-VPL2-E140G/P141G as template and the K176G primers; and (xiii) the triple variant E140G/W164S/K176G was obtained using plasmid pFLAG1-VPL2-E140G/K176G as template and the W164S primers. The mutated genes were sequenced using an ABI 3730 DNA Analyzer (Applied Biosystem) to assure that only the desired mutations occurred.
PCR (50 μl final volume) were carried out in a PerkinElmer Gene Amp PCR System 240 using 10 ng of template DNA, 500 μm each dNTP, 125 ng of direct and reverse primers, 2.5 units of Pfu Turbo polymerase (Stratagene), and the manufacturer's buffer. Reaction conditions were as follows: (i) a “hot start” at 95 °C for 1 min; (ii) 18 cycles at 95 °C for 50 s, 55 °C for 50 s, and 68 °C for 10 min; and (iii) a final cycle at 68 °C for 10 min.
Enzyme Production, Activation, and Purification
Native VP and its directed variants were produced in E. coli W3110 after transformation with the corresponding plasmids. Cells were grown for 3 h in Terrific Broth (25), induced with 1 mm isopropyl β-d-thiogalactopyranoside, and grown for a further 4 h. The apoenzyme accumulated in inclusion bodies, as observed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and was recovered by solubilization in 50 mm Tris-HCl (pH 8.0) containing 8 m urea, 1 mm EDTA, and 1 mm dithiothreitol for 30 min at room temperature. Subsequent in vitro folding was performed using 0.16 m urea, 5 mm CaCl2, 20 μm hemin, 0.5 mm oxidized glutathione, 0.1 mm dithiothreitol, and 0.1 mg/ml of protein in 50 mm Tris-HCl (pH 9.5) (23). Active enzyme was purified by Resource-Q chromatography using a 0–0.3 m NaCl gradient (2 ml/min, 20 min) in 10 mm sodium tartrate (pH 5.5) containing 1 mm CaCl2.
Spectroscopic Analyses
Electronic absorption spectra were recorded at 25 °C using a Shimadzu UV-1800 spectrophotometer. The concentrations of native VP and directed variants in 10 mm sodium tartrate (pH 5.0) were calculated from the absorption at 407 nm using an extinction coefficient of 150 mm−1 cm−1 (24). For spectroscopic characterization of the transient states in the VP catalytic cycle, 1 eq of H2O2 was added to the resting enzyme in 10 mm sodium tartrate (pH 5.0) yielding VP-I. Addition of 1 eq of ferrocyanide to VP-I yielded VP-II.
Steady-state Kinetics
Oxidation of ABTS (cation radical ϵ436 29,300 m−1 cm−1), DMP (coerulignone dimeric product ϵ469 55,000 m−1 cm−1), HQ (p-benzoquinone ϵ247 21,000 m−1 cm−1), catechol (o-benzoquinone ϵ392 1,456 m−1 cm−1), and guaiacol (3,3′-dimethoxy-4,4′-byphenylquinone ϵ470 26,600 m−1 cm−1) were estimated at pH 3.5, and that of Mn2+ (Mn3+-tartrate complex ϵ238 6,500 m−1 cm−1) at pH 5.0. The chemical structures of the above substrates are shown in Fig. 2. All enzymatic activities were measured as initial velocities from linear increments due to appearance of the reaction product, at 25 °C in 100 mm sodium tartrate (of different pH values) in the presence of 0.1 mm H2O2. Steady-state kinetic constants were calculated from oxidation of increasing substrate concentrations. Mean values ± S.E. for affinity constant (Michaelis constant, Km) and enzyme turnover (catalytic constant, kcat) were obtained by nonlinear least-squares fitting of the experimental measurements to the Michaelis-Menten model. Fitting of these constants to the normalized equation: v = (kcat/Km)[S]/(1 + [S]/Km) yielded the catalytic efficiency values (kcat/Km) with their corresponding standard errors.
FIGURE 2.

Chemical structures of the VP substrates used in this study. A, guaiacol; B, DMP; C, cathecol; D, HQ; and E, ABTS.
Transient-state Kinetics
Transient-state kinetic constants were measured at 25 °C using stopped-flow equipment (Bio-Logic) including a three-syringe module (SFM300) synchronized with a diode array detector (J&M), and Bio-Kine software. VP-I formation was investigated by mixing the resting enzyme with increasing concentrations of H2O2 in 100 mm sodium tartrate (pH 3.0) under pseudo first-order conditions (excess of substrate) and followed at 397 nm (the isosbestic point of VP-I and VP-II). To investigate VP-II formation, VP-I was first prepared by mixing 4 μm resting enzyme with 1 eq of H2O2 in 10 mm sodium tartrate (pH 5.0). After 0.6 s aging in a delay line, an excess of HQ in 100 mm (final concentration) sodium tartrate (pH 3.5) was added, and VP-II formation was followed at 416 nm (the isosbestic point of VP-II and resting enzyme). The first step to investigate VP-II reduction consisted of production and reduction of VP-I by premixing a solution of 4 μm enzyme and 4 μm ferrocyanide with 1 eq of H2O2 in 10 mm sodium tartrate (pH 5.0). The mixture was incubated for 6 s in the delay line, and VP-II reduction was followed at 406 nm (the Soret maximum of resting enzyme) after mixing with different concentrations of HQ in 100 mm (final concentration) sodium tartrate (pH 3.5). In all cases, the final enzyme concentration was 1 μm. All kinetic traces exhibited single-exponential character from which pseudo first-order rate constants were calculated.
Crystallization and X-ray Structure
A screening for optimal crystallization conditions by both the sitting- and hanging-drop vapor diffusion methods was performed using the commercial Crystal Screens I and II, Index Screen, SaltRx, and Additive Screen from Hampton Research. These conditions were further refined by varying buffer type, temperature, sample size, precipitant agent, and additives. Finally, crystals of three single (E140G, P141G, and K176G) and one double (E140G/K176G) variant, as well as of the E140G-guaiacol complex were obtained by the sitting-drop vapor diffusion method. Crystals of the different variants (obtained in the absence of any substrate) were soaked with the reducing substrates mentioned above with the aim of obtaining additional enzyme-substrate complexes.
Final crystallization conditions are provided below. The protein (10 mg/ml in 10 mm sodium tartrate, pH 5.0), or the protein plus substrate (1:1 molar ratio) in the case of E140G-guaiacol co-crystallization, were mixed in a 1:1 ratio (v/v) with a solution containing 1.4 m ammonium sulfate and 100 mm sodium cacodylate (pH 5.0). Different additives were necessary for crystallization of each specific variant: 2% 1,3-propanediol (E140G and K176G), 1.5% methanol (P141G), 20 mm hexamine cobalt (III) chloride (E140G/K176G), and 2.4% inositol (E140G-guaiacol) (final concentrations). Crystals appeared after 8–15 days at 22 °C, and were flash cooled in a cryosolution containing the crystallization solution plus 25% glycerol.
Complete data sets were collected at the beamline ID14.2 of the ESRF (Grenoble, France). Data processing was done using MOSFLM and reduced with SCALA, from the CCP4 package (26). The structures were solved by molecular replacement with MOLREP using the native VP (allelic variant VPL2) crystal structure (PDB entry 2BOQ) as a search probe. Consecutive cycles of refinement and manual rebuilding were done using REFMAC (27) and COOT (28). All the refined structures were validated using MolProbity (29). The statistics of data collection, processing, and refinement are shown in Table 1. The atomic coordinates for the E140G, P141G, K176G, and E140G/K176G variants and the E140G-guaiacol complex were deposited in the Protein Data Bank (with accession numbers 4FDQ, 4FEF, 4FCS, 4FCN and 4G05, respectively). Although crystals of the E140G/P141G/K176G variant could not be obtained, a structural model for this triple variant was prepared by in silico mutagenesis at Pro-141 of the E140G/K176G crystal structure using the mutagenesis tool of the PyMOL Molecular Graphics System (version 1.4.1; Schrödinger, LCC).
TABLE 1.
Data collection and refinement statistics
| E140G | P141G | K176G | E140G/K176G | E140G-guaiacol | |
|---|---|---|---|---|---|
| Space group | I41 | I41 | I41 | I41 | I41 |
| Unit cell (Å) | a = b = 96.23 c = 98.73 | a = b = 96.58 c = 98.06 | a = b = 96.27 c = 98.91 | a = b = 96.34 c = 98.94 | a = b = 96.43 c = 98.78 |
| Resolution range (Å) | 48–1.6 (1.69–1.6)a | 68–2.0 (2.11–2.0) | 48–1.5 (1.58–1.50) | 48.2–1.7 (1.79–1.70) | 69–2.35 (2.48–2.35) |
| Rmerge (%)b | 7.7 (43.3) | 11.0 (41.1) | 6.2 (45.4) | 8.3 (49.5) | 18.0 (45.4) |
| Multiplicity | 4.3 (4.3) | 4.3 (4.3) | 5.0 (4.9) | 4.2 (4.2) | 4.1 (4.1) |
| I/σ (I) | 13.9 (3.2) | 11.5 (3.7) | 17.2 (3.3) | 12.9 (3.0) | 3.9 (1.6) |
| Completeness (%) | 98.3 (97.1) | 100.0 (100.0) | 99.9 (99.5) | 99.9 (99.7) | 100.0 (99.9) |
| Refinement | |||||
| Rc/Rfree | 15.4/17.9 | 13.9/17.6 | 15.1/17.6 | 14.8/17.4 | 17.4/21.8 |
| Bond root mean square deviation (Å) | 0.030 | 0.034 | 0.034 | 0.035 | 0.021 |
| Angle root mean square deviation (°) | 2.342 | 2.100 | 2.757 | 2.519 | 2.467 |
| Average B factor (Å2) | 15.32 | 15.02 | 13.41 | 15.45 | 8.46 |
| PDB ID | 4FDQ | 4FEF | 4FCS | 4FCN | 4G05 |
a Data in parentheses are for the highest resolution shell.
b Rmerge = ΣhklΣi|Ii (hkl) − 〈I (hkl) 〉|/ΣhklΣi|Ii (hkl)|, where Ii(hkl) is the intensity of the ith measurement of reflection (hkl) and 〈I(hkl)〉 is the mean intensity.
c r = Σhkl|Fobs − Fcalc|/Σhkl|Fobs |, where Fobs and Fcalc are the observed and calculated structure factors.
RESULTS
Directed Mutagenesis at the VP Heme Channel
Several bulky residues at the main heme access channel of P. eryngii VP (Fig. 3A) were substituted by glycines in single (P76G, E140G, P141G, F142G, K176G, and K215G) and multiple (E140G/P141G, E140G/K176G, and E140G/P141G/K176G) variants with the aim of facilitating the access of substrates to the heme cofactor in the peroxide-activated enzyme. In this way we attempted to make VP more similar to HRP and CiP (Fig. 3, C and D), which are able to oxidize low redox potential substrates (e.g. simple phenols) in direct contact with the heme. In the same way, two basic residues were replaced by those amino acids present in P. chrysosporium LiP (Fig. 3B) (K176D and K215Q variants) to determine the importance of the local charge in a hypothetical substrate oxidation site at the heme channel, as suggested for anionic dye oxidation by LiP. Two more variants were produced (W164S and E140G/W164S/K176G) lacking the catalytic Trp-164 responsible for high redox potential substrate oxidation by VP, which is exposed to the solvent at ∼25 Å from the channel edge (supplemental Fig. S1A).
FIGURE 3.

Comparison of the heme access channel in different peroxidase families. A, VP (isoenzyme VPL) from P. eryngii; B, LiP (isoenzyme H8) from P. chrysosporium; C, HRP (isoenzyme C) from Armoracia rusticana; and D, CiP from C. cinerea (also reported as Arthromyces ramosus “nomen nudum” peroxidase, ARP) (PDB entries 2BOQ, 1B82, 1ATJ, and 1ARP, respectively). Those residues forming the channel opening are indicated.
All mutated variants were expressed in E. coli W3110, folded in vitro, and purified. The electronic absorption spectra of the resting and transient states (VP-I and VP-II) of these variants exhibited the same maxima as for the native enzyme, indicating that the mutations did not cause any substantial change in the heme environment. The x-ray diffraction study described below demonstrated that the mutations did not affect the overall protein fold, and hence the changes were basically located in the mutated residues.
Steady-state Kinetics of Mutated Variants
The steady-state kinetic constants for oxidation of ABTS (Fig. 2E) to a stable cation radical, and of different simple phenols (Fig. 2, A–D) to the corresponding quinones, by native VP and 13 single and multiple variants are provided below.
ABTS Oxidation
Double-hyperbolic curves were obtained for oxidation of different concentrations of ABTS by native VP, as shown in Fig. 4A using a semilogarithmic x axis scale. Nonlinear fitting to data for each of the curve regions enabled calculation of two sets of kinetic constants (in the μm and mm ranges), which revealed the existence of two oxidation sites characterized by high and low efficiency reactions (Table 2). Substitution of Trp-164 completely suppressed the high efficiency oxidation of ABTS in the W164S and E140G/W164S/K176G variants (Table 2) confirming the location of the high efficiency oxidation site at this exposed tryptophan residue. By contrast, the single mutations enlarging the heme access channel did not cause significant changes at the high efficiency site, but affected the low efficiency oxidation of this substrate (Table 2). So, the P76G variant experienced a 3-fold decrease, whereas the K176G and E140G variants exhibited a 4-fold increase in their catalytic efficiency (a rebound effect was observed for the E140G variant, with the high efficiency oxidation of ABTS showing a simultaneous 3-fold decrease). On the other hand, substitution of the positively charged residues at the heme channel in the K176D and K215Q variants did not significantly affect ABTS oxidation at the low efficiency site. This showed that the channel charge density does not affect the oxidation of this bulky anionic compound by VP.
FIGURE 4.

Biphasic kinetics for ABTS (A), DMP (B), and HQ (C) oxidation by native VP. Double hyperbolic curves are shown with the x axis in logarithmic scale.
TABLE 2.
Steady-state kinetic constants (Km (μm), kcat(s−1), and kcat/Km (s−1 mm−1)) of native VP, and single and multiple variants for oxidation of ABTS at high and low efficiency sitesa
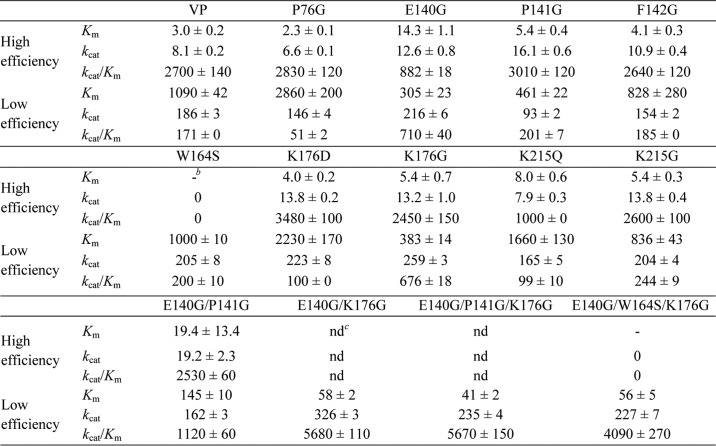
a Reactions at 25 °C in 0.1 m tartrate (pH 3.5).
b –, not determined because of a lack of activity.
c ND, not determined because of interference with the low efficiency oxidation site. Means and 95% confidence limits.
Regarding the multiple variants: (i) the E140G/P141G double mutation improved the affinity of the enzyme for ABTS at the low efficiency oxidation site with respect to the already improved E140G and P141G single variants by 2- and 3-fold, respectively (although it reverted the kcat increase observed in the E140G variant); (ii) the E140G/K176G double mutation increased the catalytic efficiency of the corresponding single variants oxidizing ABTS at the low efficiency site (up to 33-fold compared with native VP); (iii) the triple variant E140G/W164S/K176G, designed to avoid interferences from the catalytic Trp-164, retained the kinetic constants of the E140G/K176G variant confirming the improvement of low efficiency oxidation of ABTS by enlarging the heme channel; and (iv) further channel enlargement by incorporating the P141G mutation in the E140G/P141G/K176G variant did not confer an additional improvement in ABTS oxidation compared with the E140G/K176G variant (Table 2). When the increased catalytic efficiency of these multiple variants at the low efficiency site was analyzed, it was found to be basically due to the improvement in affinity for ABTS (Km decrease from 1090 μm in native VP to 145, 58, 56, and 41 μm in the E140G/P141G, E140G/K176G, E140G/W164S/K176G, and E140G/P141G/K176G variants, respectively) that reached a value similar to that of the high efficiency oxidation site in native VP (∼3 μm; Table 2). This affinity improvement, together with the characteristic high kcat values of the heme catalytic site (around 200–300 s−1) compared with the high efficiency site (only 8 s−1), turned this site into an additional high efficiency site for ABTS oxidation in the better variants.
Oxidation of Phenols
The steady-state kinetic constants for oxidation of four different phenols (HQ, catechol, guaiacol, and DMP) are shown in Tables 3 and 4. Native VP exhibited double hyperbolic kinetics for HQ and DMP oxidation (Fig. 4, B and C). As in the case of ABTS, two sets of kinetic constants could be measured by nonlinear fitting to data in each of the curve regions. These sets correspond to low (49 and 2.8 s−1 mm−1 for HQ and DMP, respectively) and high (656 and 71 s−1 mm−1 for HQ and DMP, respectively) efficiency oxidation sites. By contrast, catechol and guaiacol oxidation apparently showed a single hyperbolic behavior, and only one set of catalytic constants, the Km values in the millimolar range, was obtained. This result initially suggested the existence of a single oxidation site in native VP for these two phenols.
TABLE 3.
Steady-state kinetic constants of native VP, and single variants ([Km (μm), kcat(s−1), and kcat/Km (s−1 mm−1)]) for oxidation of catechol, guaiacol, HQ, and DMP (the last two at high and low efficiency sites)a
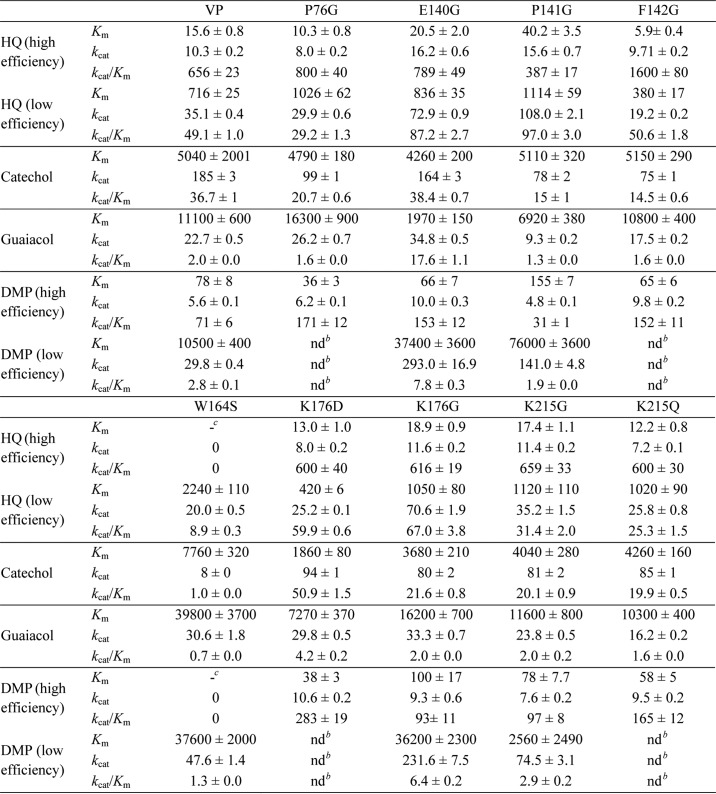
a Reactions at 25 °C in 0.1 M tartrate (pH 3.5).
b Not determined because saturation was not reached during reaction.
c Not determined because of a lack of activity. Means and 95% confidence limits.
TABLE 4.
Steady-state kinetic constants of native VP, and multiple variants (Km (μm), kcat (s−1), and kcat/Km (s−1 mm−1)) for oxidation of catechol, guaiacol, HQ, and DMP (the last two ones at high and low efficiency sites)
Reactions were at 25 °C in 0.1 m tartrate (pH 3.5).
| VP | E140G/P141G | E140G/K176G | E140G/P141G/K176G | E140G/W164S/K176G | |
|---|---|---|---|---|---|
| HQ (high efficiency) | |||||
| Km | 15.6 ± 0.8 | 39.7 ± 2.5 | 25.1 ± 2.4 | 36.2 ± 1.6 | –a |
| kcat | 10.3 ± 0.2 | 14.6 ± 0.4 | 20.0 ± 0.9 | 20.4 ± 0.5 | 0 |
| kcat/Km | 656 ± 23 | 368 ± 15 | 800 ± 40 | 565 ± 13 | 0 |
| HQ (low efficiency) | |||||
| Km | 716 ± 25 | 1180 ± 100 | 618 ± 36 | 410 ± 38 | 884 ± 71 |
| kcat | 35.1 ± 0.4 | 65.5 ± 1.9 | 82.1 ± 1.3 | 82.2 ± 2.5 | 85.6 ± 2.3 |
| kcat/Km | 49.1 ± 1.0 | 55.6 ± 3.3 | 132.8 ± 1.0 | 203.2 ± 14.2 | 96.8 ± 5.8 |
| Catechol | |||||
| Km | 5040 ± 2001 | 10,500 ± 800 | 2,630 ± 70 | 4,720 ± 320 | 34,10 ± 140 |
| kcat | 185 ± 3 | 105.6 ± 4.0 | 185.6 ± 1.3 | 135.7 ± 3.7 | 164.8 ± 2.8 |
| kcat/Km | 36.7 ± 1 | 10.1 ± 0.4 | 70.7 ± 1.5 | 28.8 ± 1.3 | 48.4 ± 1.2 |
| Guaiacol | |||||
| Km | 11100 ± 600 | 5,100 ± 460 | 2,730 ± 250 | 14,200 ± 900 | 5,850 ± 570 |
| kcat | 22.7 ± 0.5 | 3.1 ± 0.2 | 46.7 ± 1.2 | 19.3 ± 0.9 | 54.2 ± 2.5 |
| kcat/Km | 2.0 ± 0.0 | 0.6 ± 0.0 | 17.1 ± 1.2 | 1.4 ± 0.0 | 9.3 ± 0.5 |
| DMP (high efficiency) | |||||
| Km | 78 ± 8 | 104 ± 10 | 189 ± 14 | 119 ± 9 | –a |
| kcat | 5.6 ± 0.1 | 4.6 ± 0.1 | 17.3 ± 0.4 | 20.4 ± 0.5 | 0 |
| kcat/Km | 71 ± 6 | 44.6 ± 3.4 | 91.3 ± 4.5 | 171.0 ± 9.7 | 0 |
| DMP (low efficiency) | |||||
| Km | 10500 ± 400 | 16,000 ± 800 | 2,970 ± 130 | 2,380 ± 200 | 3,330 ± 260 |
| kcat | 29.8 ± 0.4 | 61.0 ± 1.5 | 67.0 ± 0.9 | 56.6 ± 1.3 | 50.2 ± 1.7 |
| kcat/Km | 2.8 ± 0.1 | 3.8 ± 0.1 | 22.6 ± 0.8 | 23.8 ± 1.6 | 15.1 ± 0.7 |
a Not determined because of a lack of activity. Means and 95% confidence limits.
When the W164S and E140G/W164S/K176G variants were analyzed, it was observed that they had lost the ability to oxidize HQ and DMP at the high efficiency site (Tables 3 and 4) proving the key role of Trp-164 in the high efficiency oxidation of these phenols, as previously found for ABTS. In contrast, HQ and DMP oxidation at the low efficiency site was maintained in both variants. The W164S variant also showed changes in its catalytic behavior with the other two phenols assayed. A small change in guaiacol Km (3.6-fold increase) and a strong effect on catechol kcat (23-fold decrease), in both cases reducing the catalytic efficiency, were observed when this single variant was compared with native VP. These results revealed that Trp-164 is also involved in the high efficiency oxidation of these two phenols, although it was not shown by the native enzyme kinetics described above.
The enlargement of the heme channel improved the ability of the enzyme to oxidize the four phenols at the low efficiency site. In particular, the P141G, K176G, E140G, and E140G/K176G mutations increased the catalytic efficiency on one (HQ), two (HQ and DMP), three (HQ, DMP and guaiacol), or the four (HQ, DMP, guaiacol, and catechol) phenols, respectively (Tables 3 and 4). The observed improvements in the three single variants with respect to native VP were due to increases (2–10-fold) in the kcat values (except for oxidation of guaiacol that was mainly due to the 6-fold Km decrease) (Table 3). The improvement of the E140G/K176G double variant oxidizing the four phenols was a result of both the 2-fold kcat increase (with the only exception of catechol oxidation) and the up to 4-fold decrease of Km (Table 4). Although the single E140G and P141G substitutions increased the catalytic efficiency toward HQ at the low efficiency site, as described above, the double E140G/P141G variant reversed this improvement (Table 4). By contrast, the E140G/P141G/K176G triple variant provided an additional improvement with respect to the E140G/K176G variant due to the 1.5-fold decrease of Km (Table 4).
Three additional single mutations (P76G, F142G, and K215G) enlarging the heme channel entrance were designed, and the resulting variants were characterized for high and low efficiency oxidation of phenols (Table 3). The K215G variant did not exhibit significant kinetic changes. Surprisingly, the other two variants showed increased efficiencies for DMP (P76G) and for DMP and HQ (F142G) at the high efficiency oxidation site. The increase in DMP oxidation efficiency at the high efficiency site was even more pronounced for the K176D variant (Table 3). This is the only variant with a change in the local charge of the main heme access that resulted in a positive catalytic effect, although affecting a VP site (Trp-164) located at a distant region of the enzyme.
Transient-state Kinetics of VP-directed Variants
Kinetic constants for formation and reduction of the transient states of the VP catalytic cycle (VP-I and VP-II) were determined using HQ as reducing substrate, which is oxidized to the p-semiquinone radical (HQ•) (Equations 1–3). With this purpose, stopped-flow spectrophotometry was performed with native VP and the most interesting variants after the steady-state kinetic studies (namely W164S, E140G, P141G, K176G, E140G/K176G, E140G/P141G/K176G, and E140G/W164S/K176G). HQ was used because its oxidation does not interfere with the absorbance of the enzyme in the 380–450 nm range.
VP-I Formation
The observed pseudo first-order rate constants (k1obs) for VP-I formation (Equation 1) exhibited a linear dependence of H2O2 concentration passing through the origin (data not shown). Fitting k1obs versus H2O2 concentration to a straight line yielded slope values corresponding to the apparent second-order rate constant for VP-I formation (k1app). The similar k1app values (∼3000 s−1 mm−1) obtained for native VP and the directed variants (Table 5) indicated that mutations did not affect formation of VP-I by H2O2.
TABLE 5.
Transient-state kinetic constants of variants compared with native VP
Apparent second-order rate constants (s−1 mm−1) of VP-I formation (k1app) by H2O2, and VP-I (k2app) and VP-II reduction (k3app) by HQ. Reactions at 25 °C in 0.1 m tartrate, pH 3.5 (pH 3 for VP-I formation), using 1 μm VP, final concentration, were conducted as described in the text.
| k1app | k2app | k3appAb | k3appB | |
|---|---|---|---|---|
| VP | 3,030 ± 30 | 3,340 ± 30 | 29 ± 0 | 400 ± 0 |
| W164S | 3,840 ± 60 | 3,680 ± 110 | 10 ± 0 | –b |
| E140G | 3,650 ± 60 | 8,480 ± 40 | 200 ± 0 | 800 ± 0 |
| P141G | 3,240 ± 60 | 618 ± 9 | 120 ± 6 | 3,000 ± 100 |
| K176G | 3,480 ± 110 | 4,720 ± 70 | 67 ± 1 | 500 ± 0 |
| E140G/K176G | 2,610 ± 60 | 14,400 ± 200 | 282 ± 4 | 1,500 ± 100 |
| E140G/P141G/K176G | 3,390 ± 140 | 5,470 ± 140 | 2,500 ± 0 | NDc |
| E140G/W164S/K176G | 3,060 ± 30 | 9,890 ± 250 | 225 ± 6 | –b |
a k3appA refers to the reduction of VP-IIA, whereas k3appB refers to the reduction of VP-IIB.
b Not determined because of a lack of activity.
c Only one k3app value could be determined for this variant, which was assigned to VP-IIA reduction (k3appA). Means and 95% confidence limits.
VP-I Reduction
The kinetic traces for one-electron reduction of VP-I by HQ (Equation 2) exhibited single-exponential character from which the pseudo first-order rate constant (k2obs) was calculated. Plots of k2obs versus HQ concentration were linear, corresponding to nonsaturation kinetics (Fig. 5A) and apparent second-order rate constants (k2app) were determined as the slope of a second-order plot (Table 5). The W164S mutation did not cause any significant change in the k2app value compared with native VP, whereas the P141G variant showed a 5-fold decrease for this constant. The rest of mutations enlarging the heme access channel improved the k2app values to different extents, the E140G, E140G/W164S/K176G, and E140G/K176G variants causing the highest increases (2.5-, 3-, and 4.3-fold, respectively). These results confirm the important role of the heme access channel in VP-I reduction by HQ, its enlargement allowing better access, interaction and/or oxidation of this, and by extension other simple phenols, reacting directly with the heme cofactor. Moreover, the differences between the E140G/K176G and E140G/W164S/K176G variants (the latter lacking the catalytic Trp-164) suggest that both the high and low efficiency oxidation sites are involved in VP-I reduction by HQ, despite reduction by W164S had suggested that only the low efficiency site was involved.
FIGURE 5.
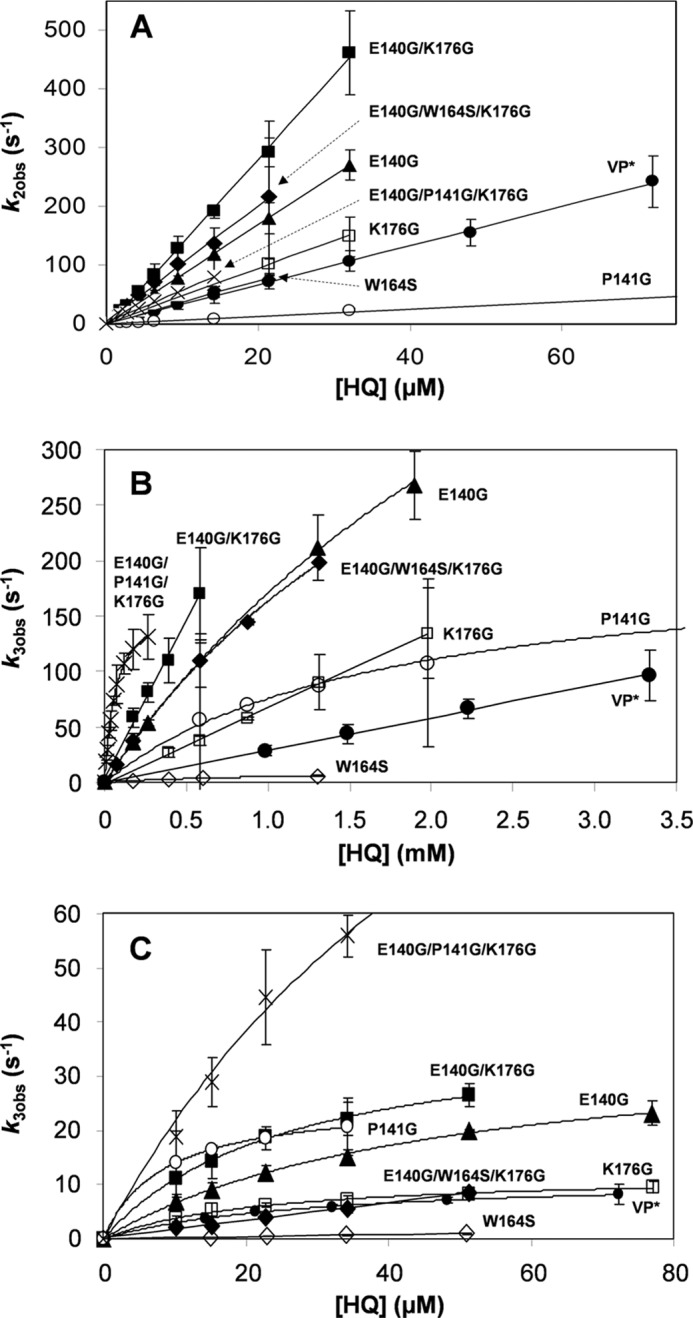
Kinetics of VP-I (A), VP-IIA (B) and VP-IIB (C) reduction by HQ by native VP and seven mutated variants. Native VP (●) and W164S (224), E140G (▴), K176G (□), P141G (○), E140G/K176G (■), E140G/P141G/K176G (×), and E140G/W164S/K176G (♦) variants. Reactions were at 25 °C in 0.1 m tartrate (pH 3.5). Means and 95% confidence limits are shown.
VP-II Reduction
Pseudo first-order rate constants for one-electron reduction of VP-II by HQ (k3obs) were also estimated. Plots of k3obs versus HQ concentration for native VP and the mutated variants exhibited a biphasic behavior, those of the W164S and E140G/W164S/K176G variants (lacking the catalytic Trp-164) being the only exception. With increasing substrate concentration, the reduction rates were saturated or nonsaturated at the millimolar range (Fig. 5B) and always saturated in the micromolar range (Fig. 5C). The rates at the millimolar range were assigned to VP-IIA reduction, whereas those at the micromolar range were attributed to VP-IIB reduction at low and high efficiency oxidation sites, respectively (see Fig. 1). The transient kinetic constants (k3app, k3, and KD3) of both sites are shown in Tables 5 and 6.
TABLE 6.
Comparison of transient-state kinetic constants for reduction of VP-II by HQ of those variants exhibiting saturation kinetics, first-order rate constants (k3, s−1) and equilibrium dissociation constants (KD3, μm) are indicated
Reactions at 25 °C in 0.1 m tartrate (pH 3.5) using 1 μm VP, final concentration, conducted as described in the text.
| k3Aa | KD3A | k3B | KD3B | |
|---|---|---|---|---|
| VP | NDb | NDb | 11.4 ± 0.1 | 28.8 ± 0.8 |
| W164S | 11 ± 1 | 1020 ± 100 | –c | –c |
| E140G | 787 ± 32 | 3600 ± 200 | 47.0 ± 3.3 | 37.5 ± 1.4 |
| P141G | 206 ± 8 | 1730 ± 150 | 25.6 ± 0.2 | 8.4 ± 0.2 |
| K176G | NDb | NDb | 12.1 ± 0.4 | 22.2 ± 1.9 |
| E140G/K176G | NDb | NDb | 39.9 ± 1.3 | 26.4 ± 1.9 |
| E140G/P141G/K176G | 166 ± 3 | 67 ± 3 | NDd | NDd |
| E140G/W164S/K176G | 589 ± 41 | 2610 ± 240 | –c | –c |
a K3A and KD3A refer to the reduction of VP-IIA, whereas K3B and KD3B refer to the reduction of VP-IIB.
b Not determined because saturation was not reached during the reaction.
c Not determined because of a lack of activity.
d Only one set of k3 and KD3 constants could be determined for this variant which was assigned to VP-IIA reduction (k3A and KD3A). Means and 95% confidence limits.
VP-II reduction by those variants exhibiting saturation kinetics can be explained by Equations 4–6, k3 and KD3 being their first-order rate constant and equilibrium dissociation constant, respectively.
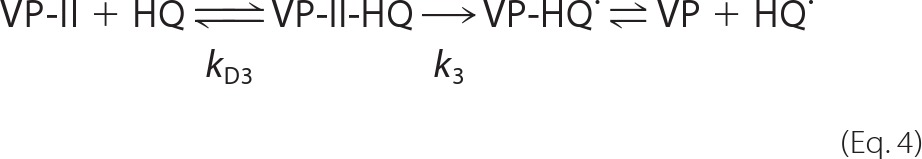 |
The apparent second-order rate constant for VP-II reduction, k3app (k3/KD3), was calculated by nonlinear least-squares fitting to Equation 5 adapted as follows: k3obs = (k3/KD3)[S]/(1 + [S]/KD3).
Unlike the saturation kinetics described above, the kinetics of VP-II reduction associated to the low efficiency oxidation sites of native VP and the K176G and E140G/K176G variants were not saturated (Fig. 5B), so only apparent second-order rate constants (k3appA) were determined.
In agreement with the steady-state results, only one set of the transient-state kinetic constants, corresponding to the low efficiency site, could be calculated for the W164S and E140G/W164S/K176G variants (Tables 5 and 6) where the Trp-164 had been removed.
The apparent second-order rate constant of the low efficiency oxidation site (k3appA) increased in all the variants with an enlarged heme access channel, the E140G/P141G/K176G variant exhibiting the highest increments (86-fold with respect to native VP) (Table 5). The improvement of this variant was due to a significant decrease in the KD3A value (66.5 μm) compared with the other variants exhibiting saturation kinetics (with KD3A values in the 1–3.6 mm range) (Table 6). These variants showed significant improvements of k3A and to a lesser extent of KD3A, when compared with the W164S variant (Table 6; native VP was not used as reference because it presents nonsaturation kinetics). Direct comparison of the k3A/KD3A values between the W164S (11 s−1 and 1020 μm, respectively) and E140G/W164S/K176G (588 s−1 and 2610 μm, respectively) variants, differing only in the size of the heme access channel, definitively demonstrated that a larger heme access channel improves VP-II reduction at the low efficiency oxidation site.
The k3appB also experienced an increase with the enlargement of the heme channel, although the k3appA increments were always higher (as shown for the K176G, E140G, and E140G/K176G variants) (Table 5). This phenomenon could be explained by interferences of VP-IIA reduction by HQ at the heme active site on VP-IIB reduction at the catalytic Trp-164. So, it was observed that the more the k3appA values improve, the more the k3appB values increase, the P141G variant being the only exception to this behavior.
Crystal Structures
Several attempts to crystallize the above mutated variants were performed in the presence and absence of reducing substrates (DMP, catechol, guaiacol, HQ, and ABTS) to investigate how the engineered mutations could affect the VP catalytic properties, as described above. Only four of the variants, in which the ability to oxidize these substrates have been improved (E140G, P141G, K176G, and E140G/K176G), could be crystallized as well as the E140G variant in complex with guaiacol. Crystals obtained in the absence of substrate were soaked with the different substrates, but additional enzyme-substrate complexes were not obtained.
No major structural rearrangements were observed when the crystal structures of the mutated variants were compared with that of native VP. The main differences correspond to the size and shape of the main channel giving access to the heme active site (Fig. 6). Differences in the orientation of the Glu-140 and Glu-83 side chains were observed (the Glu-83 side chain has two alternative positions with the same occupancy in the E140G/K176G crystal structure). However, they were not significant taking into account the variable orientation of these residues in the eight VP crystal structures previously available in PDB (Fig. 7), none of them including mutations at this molecular region.
FIGURE 6.
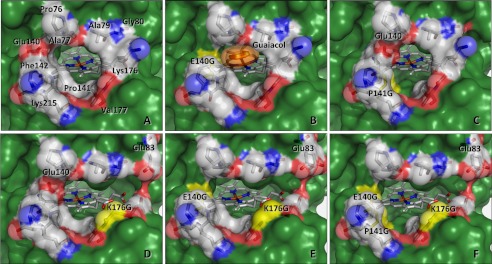
Heme access channel in the crystal structures of native VP (A) and the E140G (B), P141G (C), K176G (D), and E140G/K176G (E) variants, and the E140G/P141G/K176G model (F) mutated in silico. The different mutations are indicated and highlighted in yellow, and the solvent access surfaces are in green. The heme group is shown as sticks and amino acid at the channel opening are both as sticks and semitransparent van der Waals spheres using Corey-Pauling-Koltun (CPK) colors. The side chain of Glu-40 shows different orientations in native VP and the P141G and K176G variants, and Glu-83 presents two orientations with the same occupancy in the E140G/K176G variant. The E140G variant was co-crystallized with a guaiacol molecule (shown as orange sticks and semitransparent van der Waals spheres) that is located inside the heme channel, interacting with the enzyme as described in Fig. 8. From PDB entries: 2BOQ (A), 4FDQ (B), 4FEF (C), 4FCS (D), and 4FCN (E).
FIGURE 7.

Superimposition of residues forming the heme channel entrance in different VP crystal structures. Selected residues from wild VP from P. eryngii cultures (PDB 3FJW), native VP (PDB 2BOQ), and six variants (PDB entries 2VKA, 2W23, 3FKG, 3FM1, 3FM4, and 3FMU), none of them containing mutations at this region of the protein, are shown as Corey-Pauling-Koltun (CPK) sticks, whereas the Glu-83 and Glu-140 residues in the P141G (PDB 4FEF), K176G (PDB 4FCS), and E140G/K176G (PDB 4FCN) variants are shown as orange, magenta, and yellow sticks, respectively. The heme cofactor is shown at the bottom of the channel (CPK sticks). All the variants were expressed in E. coli.
The crystal structure of the E140G-guaiacol complex was solved at 2.3-Å resolution (Fig. 8). The substrate molecule is located at the entrance of the heme distal cavity with its aromatic ring accommodated in a hydrophobic region formed by the heme pyrrolic ring-D, and five amino acid residues (His-47, Pro-76, Ala-77, Gly-140, and Pro-141). The methoxyl and hydroxyl groups of guaiacol are orientated toward the internal cavity, hydrogen bonded to Arg-43 and Pro-139, the latter most probably through a water molecule (Fig. 8). A comparison of the E140G-guaiacol crystal structure with those of native VP and the other crystallized variants revealed that two conserved water molecules were displaced by the substrate. Both positions are occupied by the methoxyl and hydroxyl groups of guaiacol in the enzyme-substrate complex.
FIGURE 8.

Guaiacol at the heme access channel of the E140G-guaiacol complex. A, 2Fo − Fc electron density map, contoured at the 1.1 σ level, of the guaiacol molecule within the E140G-guaiacol complex. The guaiacol molecule was located near the position of Arg-43, Pro-139, and two water molecules (W145 and W172), and the heme cofactor (waters are shown as red spheres; and the rest as Corey-Pauling-Koltun (CPK) colored sticks). B, LIGPLOT (41) diagram of guaiacol interactions with the E140G variant. HBPLUS (42) was used to calculate hydrogen bonds and hydrophobic contacts (the latter are interpreted by following the spokes protruding from a ligand atom toward a protein residue, which is shown as an arc). Guaiacol bonds are depicted in purple line and those of the protein in brown line; and hydrogen bonds, and their lengths, are shown in green.
DISCUSSION
Overview
VP oxidizes Mn2+ and both high and low redox potential aromatic compounds (11). The catalytic sites responsible for oxidation of Mn2+ and high redox potential substrates have been exhaustively investigated (12, 13, 15, 18). However, not much attention has been paid to clarify how low redox potential substrates are oxidized by this enzyme.
Oxidation of low redox potential substrates, including phenols and small negatively charged dyes, by VP has been suggested to occur at the exposed heme edge (22) although not enough evidence has been provided to date. The same channel connecting the distal heme cavity with the protein surface for cofactor activation by exogenous H2O2 allows phenols and other low redox potential compounds to gain access to the heme in other members of the superfamily of microbial, fungal, and plant peroxidases (30). Comparison of the heme channel entrance of two of these peroxidases and two ligninolytic peroxidases reveals that the channel is wider in HRP and CiP than in LiP and VP (Fig. 3). The two ligninolytic peroxidases have residues with bulky side chains resulting in more difficult substrate access to the heme group due to steric hindrances. However, molecular dynamic simulations suggest that LiP heme channel exhibits some degree of plasticity (31). Even so, the channel is too narrow in LiP to allow simple aromatic substrates (including phenols) to directly interact with the heme group (32), but not so in VP that has a wider channel. Moreover, certain mobility of the side chains located at the heme channel opening in VP is suggested by the superimposition of crystal structures available at PDB, somehow emulating the snapshots of a molecular dynamics simulation (Fig. 7). These crystal structures also reveal that Glu-140 could play a role facilitating or hindering access of substrates to the heme channel. An additional aspect of the heme channel region refers to its partial electrostatic charge. VP heme channel shares characteristics of the other three peroxidases (HRP, CiP, and LiP) by including one acidic (Glu-140) and two basic (Lys-176 and Lys-215) amino acid residues. Therefore, we initially thought that the positive net charge at this VP region could favor the oxidation of anionic substrates (e.g. ABTS) in direct contact with the heme group, as discussed below.
Two Sites for Low Redox Potential Substrates
The biphasic kinetic curves obtained for different phenols and ABTS under steady-state conditions showed the presence of two independent catalytic sites for these substrates in native VP, characterized by high (Km in the micromolar range) and low (Km in the millimolar range) specificity constants. Similar curves had been described for wild-type VP isolated from fungal cultures, and kinetic constants for a low and a high efficiency oxidation sites (not yet identified at that time in the VP molecular structure) had been provided (11, 33).
Taking the above considerations together, size and local charge of the VP main heme access channel were analyzed in depth to elucidate its eventual role in oxidation of low redox potential substrates. VP-directed variants were prepared by substituting amino acid residues with bulky side chains by glycines, and by replacing basic residues to reduce or reverse the positive partial charge. Moreover, the above mutations were combined with the removal of Trp-164, which has been reported as responsible for oxidation of high redox potential substrates (12) and also ABTS (18) by P. eryngii VP.
High Efficiency Oxidation at Trp-164
After H2O2 activation, both VP transient states present a protein radical at the Trp-164 residue (15), which is exposed to the solvent at ∼25 Å from the channel edge (see supplemental Fig. S1A). According to the results presented herein, the substrate specificity of this catalytic site can be extended to simple phenols, such as DMP, HQ, catechol, and guaiacol. Suppression of high efficiency oxidation of DMP and HQ, and impaired catechol and guaiacol oxidation by the W164S variant confirm this fact. The steady- and transient-state kinetic constants for oxidation of phenols by native VP are similar to those reported for LiP oxidation of phenols (34) (namely Km/KD3B values in the micromolar range, and kcat/k3B values of 5–20 s−1). This suggests that the homologous tryptophan in P. chrysosporium LiP (Trp-171), being responsible for high redox potential substrate oxidation (21), would be also involved in oxidation of phenols by this enzyme. However, unlike VP, LiP is rapidly inactivated during oxidation of phenolic substrates (35, 36), this being the reason why phenols are not considered among its natural substrates.
Low Efficiency Oxidation at the δ-Heme Edge
Exhaustive analysis of variants mutated at the heme channel has confirmed this site as involved in the low efficiency oxidation of phenols and ABTS by VP. The affinity constants of native VP at this site were near (HQ) or in the millimolar (ABTS and other phenols) range. These values are similar to the Km and KD values reported for generic peroxidases oxidizing the same type of substrates at the δ-meso-position of the porphyrin macrocycle (the so-called δ-heme edge) (supplemental Fig. S1B) (37–39). These data suggest both similar binding of these substrates by generic peroxidases and VP, despite the aforementioned differences in their heme access channels, and minimal interferences by contaminants present at substrate concentrations in the millimolar range (substrates were not 100% pure, as described under “Experimental Procedures”). Relevant changes in the affinity and catalytic constants of the low efficiency site were observed when the VP channel was enlarged by selected amino acid substitutions by glycines. The effect became more evident when multiple substitutions were included in double (E140G/K176G) and triple (E140G/P141G/K176G) variants. The catalytic properties of the E140G/W164S/K176G variant (lacking Trp-164) left no doubts about the existence of this second site for oxidation of low redox potential substrates by VP. These results revealed a general tendency of the enzyme (depending on each variant and substrate) to improve its catalytic efficiency at the low efficiency site when an enlargement of the heme channel was produced.
The exposed δ-heme edge has been also described as the site for oxidation of a negatively charged difluoroazo dye by LiP, and this reaction has been influenced by a charge neutralization mutation in the “classic” heme edge access channel (21). In contrast, we observed that changes in the local charge of the entrance to the heme channel in VP did not have any relevant effect on ABTS oxidation. However, this could be due to the large size of the ABTS molecule that could interact with oppositely charged residues at some distance from the entrance of the heme access channel.
VP-I/VP-II Reduction at Trp-164 or δ-Heme Edge
As discussed above, the steady-state kinetics of native VP and its mutated variants allowed us to identify two independent sites, characterized by their different kinetic properties (a combination of high affinity and low catalytic constants for the high efficiency site at Trp-164; and low affinity and high catalytic constants for the low efficiency site at the heme edge). These differences were also used for estimating two sets of transient-state kinetic constants, revealing that both VP-I and VP-II are reduced by phenols reacting in the two catalytic sites. So, the VP catalytic cycle initially proposed by Ruiz-Dueñas et al. (24), and then extended by Pérez-Boada et al. (12) (Fig. 1), can be now completed as shown in Fig. 9. According to this scheme, phenols at micromolar concentrations are oxidized by the Trp-164 radical in both VP-IB and VP-IIB, as previously demonstrated for high redox potential substrates (15). By contrast, phenols at millimolar concentrations are oxidized in direct contact with the heme cofactor by VP-IA and VP-IIA. According to our studies, phenols gain access to the δ-heme edge through the main channel, whereas Mn2+, which is also oxidized directly by the heme, accesses to the internal propionate through a small secondary channel formed by the three acidic residues participating in its coordination (13).
FIGURE 9.
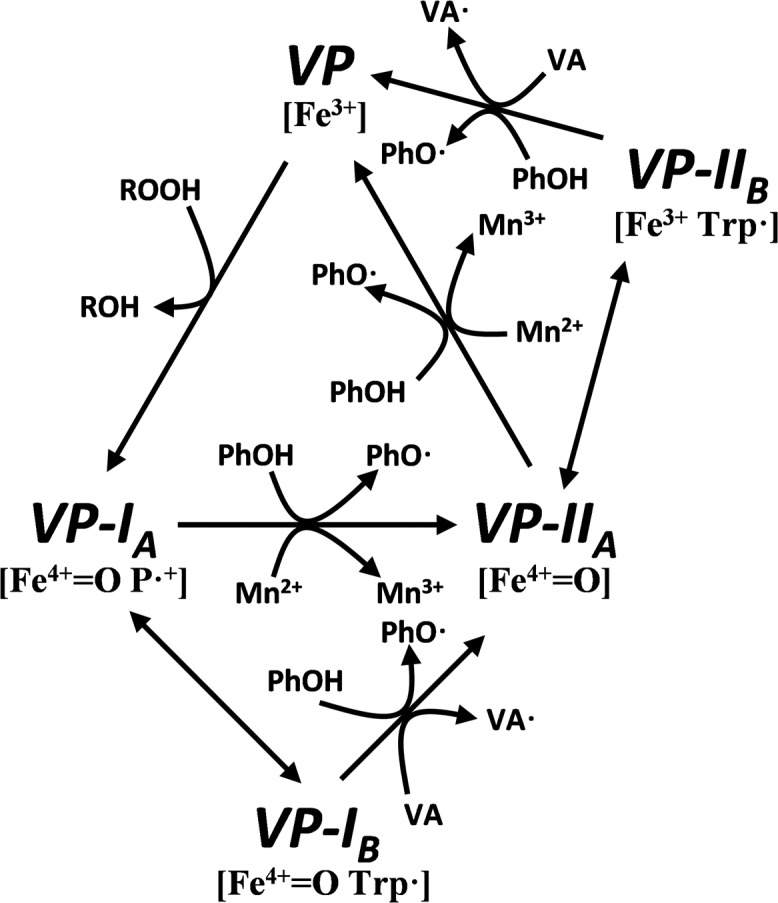
Updated VP catalytic cycle including phenols as VP-I and VP-II reducing substrates. At high concentrations (millimolar range) phenols gain access to the heme active site and are oxidized in direct contact with the δ-position of the porphyrin macrocycle by VP-IA [Fe4+ = O P•] and VP-IIA [Fe4+ = O], but when present at low concentrations (micromolar range) they are preferentially oxidized at a protein radical centered on Trp-164 exposed to the solvent by VP-IB [Fe4+ = O Trp•] and VP-IIB [Fe3+ Trp•]. The rest of the catalytic cycle is as previously described by Pérez-Boada et al. (12) (Fig. 1).
Structural Evidence for Heme Channel Oxidation Site
Optimized enlargement of the VP heme channel yielded a variant (E140G) that could be co-crystallized with a guaiacol molecule interacting with the pyrrolic ring-D of heme. This is the second in vivo phenolic substrate, ferulic acid was the first one (40), ever crystallized at the active site of a peroxidase. The weakness of the binding interactions for a stable enzyme-substrate complex is the most plausible reason for the absence of crystallographic information for a peroxidase with a phenolic substrate, in agreement with the high dissociation constants determined for some of them (38, 39). Our results with the VP-guaiacol complex seem to be consistent with this hypothesis. Only the E140G variant, showing a significant affinity improvement for guaiacol, yielded crystals with this compound located at the exposed δ-heme edge.
On the other hand, we have demonstrated the VP catalytic improvement associated to the Glu-140 removal. As shown in Fig. 7, the Glu-140 side chain occupies different positions in different VP crystal structures, in agreement with the absence of steric restrictions for free side chain mobility. This suggests that Glu-140 could temporally occlude the substrate access to the heme active site or destabilize the enzyme-substrate interaction after access of the substrate in native VP. In consequence, a Glu-140 “open” orientation seems necessary to allow the substrate to enter and optimally interact with heme and surrounding amino acid residues in native VP, as observed in the E140G-guaiacol complex.
When analyzed, the guaiacol binding mode in the E140G variant showed common features with crystal structures of microbial, fungal, and plant peroxidase (cytochrome c peroxidase, CiP, HRP, and ascorbate peroxidase) complexes with isoniazid, ferulic, and hydroxamic acids (reviewed in Ref. 19). The most similar structures to the E140G-guaiacol complex were those of HRP-ferulic acid and HRP-cyanide-ferulic acid complexes (40). These structures share the same orientation of the aromatic ring and its substituents in the ferulic acid and guaiacol complexes. Ferulic acid orientation in the HRP-cyanide-ferulic acid complex was suggested to be the same as it would be in a complex with compounds I or II (cyanide parallels the structural effect of the ferryl oxygen in these compounds) (40). In consequence, the same orientation of the guaiacol molecule observed in the crystallized E140G-guaicol complex is expected to be maintained in the two other states of VP catalytic cycle. In this context, conserved residues at the heme distal pocket are directly involved in enzyme-substrate interaction, including a proline (VP Pro-139) backbone carbonyl and the guanidinium side chain of the distal arginine (VP Arg-43), which are hydrogen bonded to the guaiacol hydroxyl and methoxyl oxygens, respectively, in the VP-phenol complex. Additionally, two distal water molecules seem to be important in the stabilization of the E140G-guaiacol and HRP-ferulic acid complexes, mediating the substrate interaction with the heme group.
Conclusions
Previous studies demonstrated how Mn2+ and high redox potential substrates are oxidized by VP, but left aside the oxidation of low redox potential phenols. Here two catalytic sites responsible for low redox potential substrate oxidation were identified. The high efficiency site corresponds to the same Trp-164 involved in high redox potential substrate oxidation. We describe for the first time in VP (and related LiP that shares the exposed catalytic tryptophan) how the substrate range of this site extends to simple phenols. In parallel, a low efficiency site, involved in oxidation of the same phenols, was localized at the entrance of the VP heme distal pocket, being absent from P. chrysosporium LiP and MnP. This catalytic site has been exhaustively characterized, and the structural determinants responsible for the enzyme-substrate interaction have been identified in the molecular structure of the E140G variant after co-crystallization with guaiacol. With the results herein presented, we conclude the description of the different catalytic sites present in VP, as a model ligninolytic peroxidase, and we provide additional clues on the possible catalytic mechanisms/sites for oxidation of similar substrates in structurally related peroxidases.
Acknowledgments
We thank Dr. K. Piontek (University of Freiburg, Germany) for making available in PDB several unpublished VP crystal structures. We are grateful to the scientists of beamline ID23-2 (ESRF, Grenoble, France) for help during data collections. We also acknowledge SOLEIL for provision of synchrotron radiation facilities (proposal ID “20100576”).
This work was supported by the RAPERO (BIO2008-01533), HIPOP (BIO2011-26694), and STRUANTIVIRU (BFU2011-24615) project grants of the Spanish Ministry of Economy and Competitiveness (MINECO) (to A. T. M., F. J. R.-D., and A. R., respectively) and by the PEROXICATS (KBBE-2010-4-265397) European project (to A. T. M.).

This article contains supplemental Fig. S1.
- MnP
- manganese peroxidase
- ABTS
- 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonate)
- CiP
- Coprinopsis cinerea peroxidase
- DMP
- 2,6-dimethoxyphenol
- HQ
- p-hydroquinone
- kapp
- apparent second-order rate constant
- kcat
- catalytic constant
- KD
- equilibrium dissociation constant
- Km
- Michaelis constant
- kobs
- pseudo first-order rate constant
- LiP
- lignin peroxidase
- PDB
- Protein Data Bank
- VP
- versatile peroxidase
- VP-I/VP-II
- VP compounds I and II.
REFERENCES
- 1. Martínez A. T., Ruiz-Dueñas F. J., Martínez M. J., Del Río J. C., Gutiérrez A. (2009) Enzymatic delignification of plant cell wall. From nature to mill. Curr. Opin. Biotechnol. 20, 348–357 [DOI] [PubMed] [Google Scholar]
- 2. Hammel K. E., Cullen D. (2008) Role of fungal peroxidases in biological ligninolysis. Curr. Opin. Plant Biol. 11, 349–355 [DOI] [PubMed] [Google Scholar]
- 3. Ruiz-Dueñas F. J., Martínez A. T. (2009) Microbial degradation of lignin. How a bulky recalcitrant polymer is efficiently recycled in nature and how we can take advantage of this. Microbial Biotechnol. 2, 164–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gold M. H., Youngs H. L., Gelpke M. D. (2000) Manganese peroxidase. Met. Ions Biol. Syst. 37, 559–586 [PubMed] [Google Scholar]
- 5. Hildén K., Martinez A. T., Hatakka A., Lundell T. (2005) The two manganese peroxidases Pr-MnP2 and Pr-MnP3 of Phlebia radiata, a lignin-degrading basidiomycete, are phylogenetically and structurally divergent. Fungal Genet. Biol. 42, 403–419 [DOI] [PubMed] [Google Scholar]
- 6. Ruiz-Dueñas F. J., Fernández E., Martínez M. J., Martínez A. T. (2011) Pleurotus ostreatus heme peroxidases. An in silico analysis from the genome sequence to the enzyme molecular structure. C. R. Biol. 334, 795–805 [DOI] [PubMed] [Google Scholar]
- 7. Floudas D., Binder M., Riley R., Barry K., Blanchette R. A., Henrissat B., Martínez A. T., Otillar R., Spatafora J. W., Yadav J. S., Aerts A., Benoit I., Boyd A., Carlson A., Copeland A., Coutinho P. M., de Vries R. P., Ferreira P., Findley K., Foster B., Gaskell J., Glotzer D., Górecki P., Heitman J., Hesse C., Hori C., Igarashi K., Jurgens J. A., Kallen N., Kersten P., Kohler A., Kües U., Kumar T. K., Kuo A., LaButti K., Larrondo L. F., Lindquist E., Ling A., Lombard V., Lucas S., Lundell T., Martin R., McLaughlin D. J., Morgenstern I., Morin E., Murat C., Nagy L. G., Nolan M., Ohm R. A., Patyshakuliyeva A., Rokas A., Ruiz-Dueñas F. J., Sabat G., Salamov A., Samejima M., Schmutz J., Slot J. C., St. John F., Stenlid J., Sun H., Sun S., Syed K., Tsang A., Wiebenga A., Young D., Pisabarro A., Eastwood D. C., Martin F., Cullen D., Grigoriev I. V., Hibbett D. S. (2012) The Paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336, 1715–1719 [DOI] [PubMed] [Google Scholar]
- 8. Fernandez-Fueyo E., Ruiz-Dueñas F. J., Ferreira P., Floudas D., Hibbett D. S., Canessa P., Larrondo L. F., James T. Y., Seelenfreund D., Lobos S., Polanco R., Tello M., Honda Y., Watanabe T., Watanabe T., Ryu J. S., San R. J., Kubicek C. P., Schmoll M., Gaskell J., Hammel K. E., St John F. J., Vanden Wymelenberg A., Sabat G., Splinter BonDurant S., Syed K., Yadav J. S., Doddapaneni H., Subramanian V., Lavín J. L., Oguiza J. A., Perez G., Pisabarro A. G., Ramirez L., Santoyo F., Master E., Coutinho P. M., Henrissat B., Lombard V., Magnuson J. K., Kües U., Hori C., Igarashi K., Samejima M., Held B. W., Barry K. W., LaButti K. M., Lapidus A., Lindquist E. A., Lucas S. M., Riley R., Salamov A., Hoffmeister D., Schwenk D., Hadar Y., Yarden O., de Vries R. P., Wiebenga A., Stenlid J., Eastwood D. C., Grigoriev I. V., Berka R., Blanchette R. A., Kersten P., Martínez A. T., Vicuña R., Cullen D. (2012) Comparative genomics of Ceriporiopisis subvermispora and Phanerochaete chrysosporium provide insight into selective ligninolysis. Proc. Natl. Acad. Sci. U.S.A. 109, 5458–5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mester T., Ambert-Balay K., Ciofi-Baffoni S., Banci L., Jones A. D., Tien M. (2001) Oxidation of a tetrameric nonphenolic lignin model compound by lignin peroxidase. J. Biol. Chem. 276, 22985–22990 [DOI] [PubMed] [Google Scholar]
- 10. Hammel K. E., Kalyanaraman B., Kirk T. K. (1986) Oxidation of polycyclic aromatic hydrocarbons and dibenzo[p]-dioxins by Phanerochaete chrysosporium ligninase. J. Biol. Chem. 261, 16948–16952 [PubMed] [Google Scholar]
- 11. Heinfling A., Ruiz-Dueñas F. J., Martínez M. J., Bergbauer M., Szewzyk U., Martínez A. T. (1998) A study on reducing substrates of manganese-oxidizing peroxidases from Pleurotus eryngii and Bjerkandera adusta. FEBS Lett. 428, 141–146 [DOI] [PubMed] [Google Scholar]
- 12. Pérez-Boada M., Ruiz-Dueñas F. J., Pogni R., Basosi R., Choinowski T., Martínez M. J., Piontek K., Martínez A. T. (2005) Versatile peroxidase oxidation of high redox potential aromatic compounds. Site-directed mutagenesis, spectroscopic and crystallographic investigations of three long-range electron transfer pathways. J. Mol. Biol. 354, 385–402 [DOI] [PubMed] [Google Scholar]
- 13. Ruiz-Dueñas F. J., Morales M., Pérez-Boada M., Choinowski T., Martínez M. J., Piontek K., Martínez A. T. (2007) Manganese oxidation site in Pleurotus eryngii versatile peroxidase. A site-directed mutagenesis, kinetic, and crystallographic study. Biochemistry 46, 66–77 [DOI] [PubMed] [Google Scholar]
- 14. Pogni R., Baratto M. C., Teutloff C., Giansanti S., Ruiz-Dueñas F. J., Choinowski T., Piontek K., Martínez A. T., Lendzian F., Basosi R. (2006) A tryptophan neutral radical in the oxidized state of versatile peroxidase from Pleurotus eryngii. A combined multi-frequency EPR and DFT study. J. Biol. Chem. 281, 9517–9526 [DOI] [PubMed] [Google Scholar]
- 15. Ruiz-Dueñas F. J., Pogni R., Morales M., Giansanti S., Mate M. J., Romero A., Martínez M. J., Basosi R., Martínez A. T. (2009) Protein radicals in fungal versatile peroxidase. Catalytic tryptophan radical in both Compound I and Compound II and studies on W164Y, W164H and W164S variants. J. Biol. Chem. 284, 7986–7994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ruiz-Dueñas F. J., Morales M., García E., Miki Y., Martínez M. J., Martínez A. T. (2009) Substrate oxidation sites in versatile peroxidase and other basidiomycete peroxidases. J. Exp. Bot. 60, 441–452 [DOI] [PubMed] [Google Scholar]
- 17. Blodig W., Smith A. T., Doyle W. A., Piontek K. (2001) Crystal structures of pristine and oxidatively processed lignin peroxidase expressed in Escherichia coli and of the W171F variant that eliminates the redox active tryptophan 171. Implications for the reaction mechanism. J. Mol. Biol. 305, 851–861 [DOI] [PubMed] [Google Scholar]
- 18. Ruiz-Dueñas F. J., Morales M., Mate M. J., Romero A., Martínez M. J., Smith A. T., Martínez A. T. (2008) Site-directed mutagenesis of the catalytic tryptophan environment in Pleurotus eryngii versatile peroxidase. Biochemistry 47, 1685–1695 [DOI] [PubMed] [Google Scholar]
- 19. Gumiero A., Murphy E. J., Metcalfe C. L., Moody P. C., Raven E. L. (2010) An analysis of substrate binding interactions in the heme peroxidase enzymes. A structural perspective. Arch. Biochem. Biophys. 500, 13–20 [DOI] [PubMed] [Google Scholar]
- 20. Smith A. T., Veitch N. C. (1998) Substrate binding and catalysis in heme peroxidases. Curr. Opin. Chem. Biol. 2, 269–278 [DOI] [PubMed] [Google Scholar]
- 21. Doyle W. A., Blodig W., Veitch N. C., Piontek K., Smith A. T. (1998) Two substrate interaction sites in lignin peroxidase revealed by site-directed mutagenesis. Biochemistry 37, 15097–15105 [DOI] [PubMed] [Google Scholar]
- 22. Ruiz-Dueñas F. J., Morales M., Rencoret J., Gutiérrez A., del Río J. C., Martínez M. J., Martínez A. T. (2008) Improved peroxidases. Patent (Spain) P200801292, 6 May 2008
- 23. Pérez-Boada M., Doyle W. A., Ruiz-Dueñas F. J., Martínez M. J., Martínez A. T., Smith A. T. (2002) Expression of Pleurotus eryngii versatile peroxidase in Escherichia coli and optimization of in vitro folding. Enzyme Microb. Technol. 30, 518–524 [Google Scholar]
- 24. Ruiz-Dueñas F. J., Martínez M. J., Martínez A. T. (1999) Molecular characterization of a novel peroxidase isolated from the ligninolytic fungus Pleurotus eryngii. Mol. Microbiol. 31, 223–235 [DOI] [PubMed] [Google Scholar]
- 25. Sambrook J., Russell D. W. (2001) Molecular Cloning, 3rd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 26. Collaborative Computational Project, Number 4 (1994) The CCP4 suite. Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 27. Murshudov G. N., Vagin A. A., Dodson D. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 28. Emsley P., Cowtan K. (2004) COOT. Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 29. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dunford H. B. (1999) Heme Peroxidases, Wiley-VCH, New York [Google Scholar]
- 31. Francesca Gerini M., Roccatano D., Baciocchi E., Di Nola A. (2003) Molecular dynamics simulations of lignin peroxidase in solution. Biophys. J. 84, 3883–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martínez A. T. (2002) Molecular biology and structure-function of lignin-degrading heme peroxidases. Enzyme Microb. Technol. 30, 425–444 [Google Scholar]
- 33. Ruiz-Dueñas F. J., Camarero S., Pérez-Boada M., Martínez M. J., Martínez A. T. (2001) A new versatile peroxidase from Pleurotus. Biochem. Soc. Trans. 29, 116–122 [DOI] [PubMed] [Google Scholar]
- 34. Koduri R. S., Tien M. (1995) Oxidation of guaiacol by lignin peroxidase. Role of veratryl alcohol. J. Biol. Chem. 270, 22254–22258 [DOI] [PubMed] [Google Scholar]
- 35. Harvey P. J., Palmer J. M. (1990) Oxidation of phenolic compounds by ligninase. J. Biotechnol. 13, 169–179 [Google Scholar]
- 36. Chung N., Aust S. D. (1995) Inactivation of lignin peroxidase by hydrogen peroxide during the oxidation of phenols. Arch. Biochem. Biophys. 316, 851–855 [DOI] [PubMed] [Google Scholar]
- 37. Rodríguez-López J. N., Gilabert M. A., Tudela J., Thorneley R. N., García-Cánovas F. (2000) Reactivity of horseradish peroxidase compound II toward substrates. Kinetic evidence for a two-step mechanism. Biochemistry 39, 13201–13209 [DOI] [PubMed] [Google Scholar]
- 38. Leigh J. S., Maltempo M. M., Ohlsson P. I., Paul K. G. (1975) Optical, NMR and EPR properties of horseradish-peroxidase and its donor complexes. FEBS Lett. 51, 304–308 [DOI] [PubMed] [Google Scholar]
- 39. Patel P. K., Mondal M. S., Modi S., Behere D. V. (1997) Kinetic studies on the oxidation of phenols by the horseradish peroxidase compound II. Biochim. Biophys. Acta 1339, 79–87 [DOI] [PubMed] [Google Scholar]
- 40. Henriksen A., Smith A. T., Gajhede M. (1999) The structures of the horseradish peroxidase C-ferulic acid complex and the ternary complex with cyanide suggest how peroxidases oxidize small phenolic substrates. J. Biol. Chem. 274, 35005–35011 [DOI] [PubMed] [Google Scholar]
- 41. Wallace A. C., Laskowski R. A., Thornton J. M. (1995) LIGPLOT. A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8, 127–134 [DOI] [PubMed] [Google Scholar]
- 42. McDonald I. K., Thornton J. M. (1994) Satisfying hydrogen bonding potential in proteins. J. Mol. Biol. 238, 777–793 [DOI] [PubMed] [Google Scholar]