

Abstract
Painful peripheral neuropathy often occurs without apparent underlying cause. Gain-of-function variants of sodium channel Nav1.7 have recently been found in ∼30% of cases of idiopathic painful small-fiber neuropathy. Here, we describe mutations in Nav1.8, another sodium channel that is specifically expressed in dorsal root ganglion (DRG) neurons and peripheral nerve axons, in patients with painful neuropathy. Seven Nav1.8 mutations were identified in 9 subjects within a series of 104 patients with painful predominantly small-fiber neuropathy. Three mutations met criteria for potential pathogenicity based on predictive algorithms and were assessed by voltage and current clamp. Functional profiling showed that two of these three Nav1.8 mutations enhance the channel’s response to depolarization and produce hyperexcitability in DRG neurons. These observations suggest that mutations of Nav1.8 contribute to painful peripheral neuropathy.
Keywords: dorsal root ganglia, patch clamp
Painful peripheral neuropathy involving small-diameter nociceptive nerve fibers represents a significant public health challenge (1); in about one-half of cases, no cause can be identified (1). Faber et al. (2) recently described gain-of-function variants (single amino acid substitutions) in sodium channel Nav1.7, which is abundantly expressed in spinal sensory [dorsal root ganglion (DRG)] neurons and their axons (3) in nearly 30% of patients with biopsy-confirmed, painful small-fiber neuropathy (SFN) and no other apparent cause. These variants increase the excitability of DRG neurons, providing an explanation for pain in these patients. Molecular substrates for pain in the remaining SFN patients remain unknown. Here, we describe gain-of-function mutations in Nav1.8, another sodium channel that is specifically expressed in DRG neurons and peripheral nerve axons, in human subjects with painful neuropathy, including a father–son pair. These mutations alter gating properties of the channel in a proexcitatory manner and increase the excitability of DRG neurons. These genetic and functional observations suggest that Nav1.8 mutations contribute to pain in some peripheral neuropathies.
Results
Genomic analysis of SCN10A was performed in 104 patients with idiopathic painful predominantly SFN, all negative for mutations in SCN9A. All patients underwent clinical, nerve conduction, and skin biopsy assessment. Among them, we identified nine patients carrying seven variants in SCN10A. Here, we describe two gain-of-function mutations in SCN10A, which we identified in three patients with painful neuropathy.
Clinical Description.
Patient 1, a 67-y-old man with unremarkable medical history, complained of burning and intense paroxysmal itch in the feet that interfered with sleep for more than 10 y Neurological examination and nerve conduction studies were normal [sural sensory nerve action potential amplitude, 11 μV (normal >6); conduction velocity (CV), 43 m/s (>40); compound muscle action potential peroneal nerve, 5.4 mV (>4); CV, 46.3 m/s (>42)]. Skin biopsy showed reduced intraepidermal nerve fiber (IENF) density in the distal leg (2.0 IENF/mm; fifth percentile, 2.8 IENF/mm for age and sex; median per age span, 8.3 IENF/mm) (4), confirming the diagnosis of SFN. Thorough screening excluded any underlying systemic causes and ruled out diabetes and impaired glucose intolerance.
Patient 2, the son of case 1, developed intense paroxysmal burning pain (scored 8 on a visual analog scale) in the feet and legs at age 39 y, with severe allodynia and hyperalgesia. Diagnosis of predominantly SFN was confirmed by skin biopsy showing complete epidermal denervation (0 IENF/mm; fifth percentile, 4.4 IENF/mm for age and sex; median per age span, 9.6 IENF/mm) (4) (Fig. 1). The patient had >20-y type I diabetes with well-controlled glucose and HBA1c levels. In addition to debilitating SFN, signs of large-fiber involvement were also found at examination. Sural nerve action potential amplitude (2 μV; normal, >10 μV), CV (39 m/s; normal, >42 m/s), and peroneal compound motor action potential amplitude (2.1 mV; normal, >4 mV) were reduced.
Fig. 1.
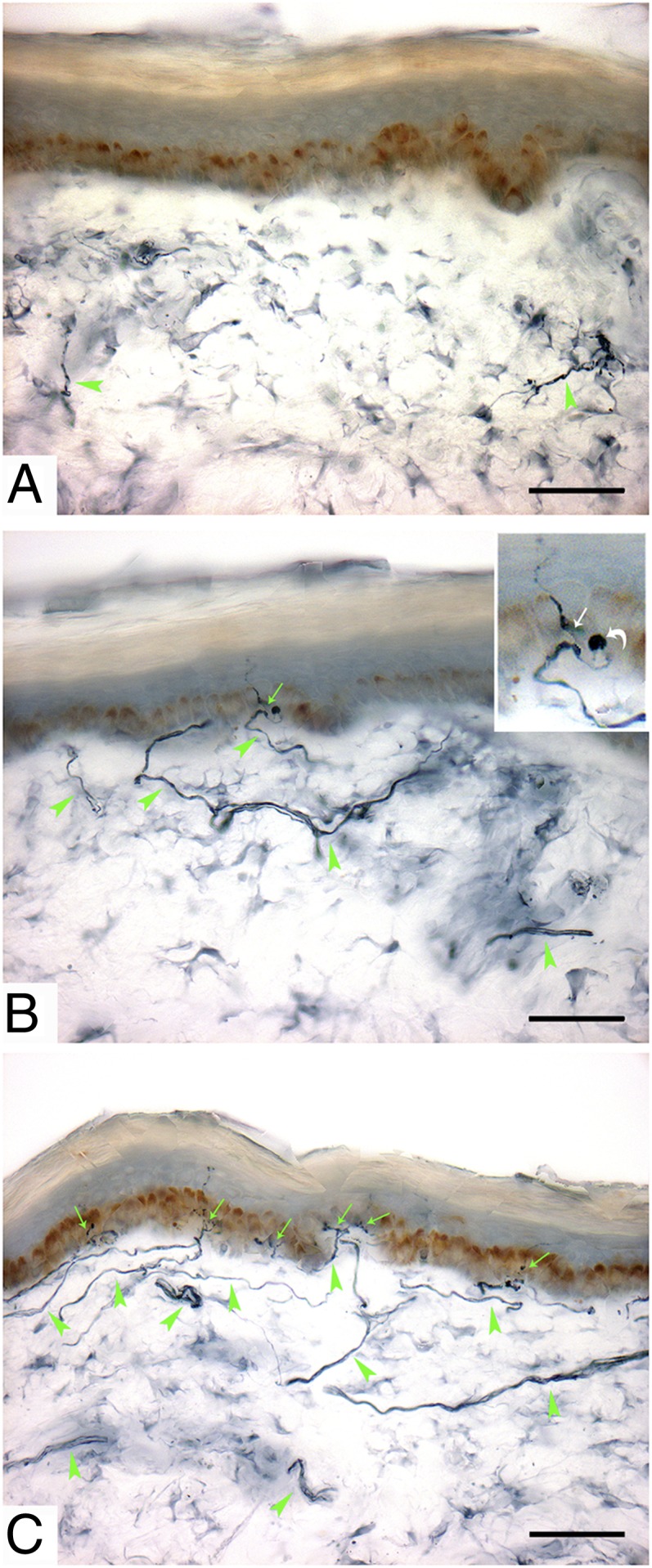
Loss of IENFs. Immunohistochemical studies (polyclonal anti-protein gene product 9.5 antibody) in sections of skin biopsy at distal leg in a father–son pair (A and B), both harboring L554P mutation in Nav1.8, and in healthy subject (C). Arrows indicate IENF; arrowheads indicate dermal nerve bundles. Patient 2 (A) showed complete depletion of IENFs and severe reduction in density of dermal nerve bundles, which appeared fragmented due to axonal degeneration. Patient 1 (B) showed almost complete loss of IENFs. Inset shows IENF (white arrow) branching after penetrating the epidermis, with large swelling (bent arrow) representing typical axonal predegeneration state. A and B are typical of SFN. For comparison, C shows diffuse innervation of epidermis and dermis in a healthy subject. (Scale bar, 50 μm.)
Patient 3 is a 69-y-old woman with stabbing pain in her feet, lower legs, and hands, and intolerance to sheets over her feet for 4 y. Warmth gave some relief. Her soles occasionally showed red discoloration. Family history was unremarkable. Nerve conduction studies showed normal sural distal motor latency and CV (distal motor latency, 2.20 ms; amplitude, 5.6 μV; CV, 55 m/s). Quantitative sensory testing showed abnormal thresholds for warmth and cold sensation in both feet (5, 6). Skin biopsy showed IENF density of 5.3 per mm, which is normal compared with normative values (fifth percentile cutoff, 3.2 per mm; median per age span, 8.7 IENF/mm) (4).
DNA Analysis.
Nine patients, including a father–son pair, carried seven missense variants in SCN10A gene encoding for Nav1.8. Substitutions in SCN10A were selected for voltage- and current-clamp assessment, after expression in DRG neurons based on the following criteria: (i) substitution either previously unreported or reported at allelic frequency of <1% in public databases (The 1000 Genomes Project, dbSNP, Exome database) and in a control population; (ii) familial substitutions, when present, segregate in affected individuals; (iii) substitutions that alter conserved residues in transmembrane segments, or in previously identified functional motifs that regulate Nav1.8 current density or gating.
Of the seven variants identified [c.1661T > C (p.Leu554Pro) (n = 2); c.2816C > T (p.Pro939Leu); c.2819A > T (p.Gln940Leu); c.3166G > A (p.Asp1056Asn); c.3910G > A (p.Ala1304Thr); c.4568G > A (p.Cys1523Tyr) (n = 2); c.4984G > A (p.Gly1662Ser)], three fulfilled selection criteria for functional testing: c.1661T > C (p.Leu554Pro), c.3910G > A (p.Ala1304Thr), and c.4568G > A (p.Cys1523Tyr). The first two mutations demonstrated gain-of-function in SCN10A and are described in this paper.
A previously unreported missense mutation (c.1661T > C; p.Leu554Pro) in exon 11 of SCN10A gene, found in patients 1 and 2 (both heterozygous), was not found in 325 healthy blood donors (650 chromosomes) from the same geographical region as the index patient. This substitution has been reported in dbSNP (rs138404783) with a frequency of <0.001 (once in 4,552 chromosomes). The mutation substitutes leucine 554 by proline within a proline-rich motif, which is highly conserved in loop 1 (L1) among Nav1.8 mammalian orthologs.
Mutation c.3910G > A (p.Ala1304Thr) found in patient 3, was not found in 600 controls (100 from ethically matched Dutch; 500 from the twin research KCL) and has not been reported in the literature, nor in dbSNP, The 1000 Genomes Project, and Exome Variant Server. The variant substitutes alanine 1304 by threonine, likely resulting in alteration of polarity. Alanine 1304 is highly conserved in multiple species and in other SCN proteins, and is located in transmembrane domain DIII/S5.
Electrophysiology.
Nav1.8 is resistant to block by TTX (TTX-R) and displays depolarized voltage dependence of activation and fast inactivation. Nav1.8 is the major contributor to the action potential upstroke during repetitive firing in DRG neurons (7, 8).
L554P.
To determine whether the L554P and A1304T mutations alter biophysical properties of Nav1.8, constructs encoding WT, L554P and A1304T channels were transiently transfected into homozygous Nav1.8-cre DRG neurons (lacking endogenous Nav1.8) in sister cultures prepared in parallel. In 1 μM TTX, large voltage-dependent, slowly inactivating inward currents were recorded from DRG neurons transfected with WT or mutant channels (Fig. 2A), indicating expression of functional channels at current densities that were not significantly different (WT: 261 ± 35 pA/pF, n = 38; L554P: 257 ± 33 pA/pF, n = 40; P > 0.05).
Fig. 2.

L554P mutation alters hNav1.8 channel properties. (A) Representative hNav1.8 WT (black) and L554P (red) currents recorded from DRG neurons. (B) Activation and steady-state fast inactivation were not significantly different. (C) L554P displays enhanced recovery from fast inactivation at −50 mV, close to resting membrane potential of DRG neurons, compared with WT channel (P < 0.05). Data are presented as means ± SEM. (D) Averaged ramp currents (normalized to maximal transient peak current) produced by WT Nav1.8 (black; n = 25) and L554P (red; n = 23) channels. SEM is indicated by error bars every 2 mV. Mean normalized amplitude of peak ramp currents are significantly larger for L554P (WT: 22.3 ± 1.6%, n = 25; L554P: 28.9 ± 2.2%, n = 23; P < 0.05).
WT and L554P channels produced large persistent currents (WT: 10.7 ± 0.7% of peak transient current, n = 19; L554P: 12.4 ± 0.5%, n = 14; P > 0.05), probably due to slow inactivation. The half-voltage for activation (V1/2,act) and fast inactivation (V1/2,fast) of WT channels (Table 1) were consistent with the previously reported depolarized voltage dependence of Nav1.8 (9–11). L554P had no significant effect on activation or steady-state fast inactivation (Fig. 2B, Table 1), or slow inactivation (Table 1). L554P increased recovery from fast inactivation (repriming) at −50 mV (Fig. 2C), close to resting potential.
Table 1.
Biophysical properties of L554P channels in mouse DRG neurons
| Activation |
Steady-state fast inactivation |
Slow inactivation |
||||||||||
| NaV1.8 | Reversal potential, mV | V1/2,act | k | n | V1/2,fast | k | A% | n | V1/2,slow | k | A% | n |
| WT | 59.1 ± 0.7 | −9.8 ± 1.4 | −7.47 ± 0.32 | 27 | −35.3 ± 1.2 | 7.07 ± 0.28 | 6.69 ± 0.56 | 27 | −49.6 ± 1.1 | 9.64 ± 0.47 | 4.25 ± 0.84 | 10 |
| L554P | 60.2 ± 0.6 | −11.6 ± 1.6 | −6.95 ± 0.37 | 25 | −33.6 ± 1.0 | 6.61 ± 0.30 | 6.80 ± 0.96 | 24 | −49.7 ± 1.4 | 9.62 ± 0.40 | 3.06 ± 0.67 | 12 |
The response to slow ramp stimuli (−80 to +40 mV over 600 ms) was one-third larger at peak for L554P compared with WT channels (Fig. 2D) (WT: 22.3 ± 1.6%, n = 25; L554P: 28.9 ± 2.2%, n = 23; P < 0.05). The potential of peak ramp current (VRamp) was −19.1 ± 1.3 mV (n = 25) for WT and −19.3 ± 1.4 mV (n = 23) for L554P. The ramp response of Nav1.8 is ∼20 times larger than that of Nav1.7, and the enhanced ramp response of L554P is predicted (12, 13) to increase excitability in DRG neurons.
To assess the effect of the L554P mutation on excitability, we expressed WT and L554P channels in small DRG neurons (<30 μm diameter) and performed current-clamp recordings (Fig. 3). Input resistance was not significantly different in DRG neurons expressing WT (669 ± 84 MΩ; n = 33) or L554P mutant channels (696 ± 90 MΩ; n = 29). Resting potential was not significantly different in DRG neurons expressing WT (−50.1 ± 1.3 mV; n = 33) or L554P mutant channels (−49.6 ± 1.4 mV; n = 29).
Fig. 3.

L554P mutation increases the excitability of small DRG neurons. (A) Response of cells expressing WT (Upper) and L554P (Lower) channels, respectively, to 500-ms depolarizing current steps at 1×, 2×, and 3× (left, center, and right traces, respectively) current threshold for action potential generation. (B) Comparison of responses (number of impulses evoked by 500-ms stimulus) in DRG neurons expressing WT (n = 33) and L554P channels (n = 29) across a range of step current injections from 25 to 500 pA (*P < 0.05). (C) Resting membrane potential (RMP) of neurons expressing WT (−50.1 ± 1.3 mV; n = 33) and L554P (−49.6 ± 1.4 mV; n = 29) was not significantly different. (D) Current threshold was significantly reduced after expression of L554P (80 ± 14 pA; n = 29), compared with WT (141 ± 20 pA; n = 33; P < 0.05). (E) Representative recording showing spontaneous firing (30 s) of DRG neuron expressing L554P mutant channels. Trace was recorded for 30 s without current injection. (Inset) Comparison of portions of WT and L554P neurons that produce spontaneous firing.
Current threshold was significantly reduced after expression of L554P (80 ± 14 pA; n = 29), compared with WT channels (141 ± 20 pA; n = 33; P < 0.05) (Fig. 3D). Voltage threshold (the voltage at which action potential take-off occurs) of DRG neurons expressing L554P (−26.3 ± 1.0 mV; n = 29) or WT channels (−24.7 ± 0.9 mV; n = 33) was not significantly different. There was no significant difference in action potential amplitude (WT: 105.5 ± 1.9 mV, n = 33; L554P: 104.5 ± 2.0 mV, n = 29), or half-width (WT: 7.82 ± 0.63 ms, n = 33; L554P: 9.07 ± 1.14 ms, n = 29).
To evaluate the effect of L554P mutation on repetitive firing, we injected neurons with 500-ms current stimuli from 25 to 500 pA in 25-pA increments. Fig. 3A shows the responses of representative DRG neurons that expressed WT and L554P channels respectively, to 500-ms steps at 1×, 2×, and 3× current threshold for the respective neuron. DRG neurons expressing L554P fired significantly more action potentials over a range of stimulation levels (Fig. 3B).
L554P mutation also produced an increase in the proportion of spontaneously firing DRG neurons (Fig. 3E). Only ∼11% (4 of 37 cells) of DRG neurons expressing WT channels displayed spontaneous firing. In contrast, ∼34% of DRG neurons expressing L554P channels displayed spontaneous firing (15 of 44 cells), significantly more than for WT (P < 0.05). Fig. 3E shows an example of sustained spontaneous firing from a representative DRG neuron expressing L554P mutant channels.
A1304T.
Current densities (WT: 776 ± 84 pA/pF, n = 18; A1304T: 809 ± 88 pA/pF, n = 19) and persistent currents (WT: 11.1 ± 1.0%, n = 11; A1304T: 9.3 ± 0.6%, n = 13; P > 0.05) in neurons expressing WT and A1304T channels were not significantly different. The half-voltage for activation (V1/2,act) for A1304T was hyperpolarized by nearly 6 mV compared with WT (A1304T: −23.0 ± 1.7 mV, n = 13; WT: −17.2 ± 2.2 mV, n = 11) (Fig. 4B, Table 2). A1304T had no significant effect on steady-state fast inactivation, or slow inactivation (Table 2).
Fig. 4.

A1304T mutation alters hNav1.8 channel properties. (A) Representative hNav1.8 WT (black) and A1304T (red) currents recorded from DRG neurons. (B) Activation for A1304T channels was hyperpolarized by ∼6 mV compared with WT, whereas steady-state fast inactivation of WT and A1304T were not significantly different. (C) Averaged ramp currents (normalized to maximal transient peak current) produced by WT Nav1.8 (black; n = 11) and A1304T (red; n = 12) channels. SEM is indicated by error bars. Voltage dependence of ramp currents for A1304T was hyperpolarized by 6 mV compared with WT. (D) Mean normalized voltage of peak ramp current for A1304T channels (−28.5 ± 1.1 mV; n = 12; *P < 0.05) was significantly different from that for WT channels (−22.5 ± 1.9 mV; n = 11). Data are presented as means ± SEM. *P < 0.05.
Table 2.
Biophysical properties of A1304T channels in mouse DRG
| Activation |
Steady-state fast inactivation |
Slow inactivation |
||||||||||
| NaV1.8 | Reversal potential, mV | V1/2,act | k | n | V1/2,fast | k | A% | n | V1/2,slow | k | A% | n |
| WT | 62.6 ± 2.0 | −17.2 ± 2.2 | −5.7 ± 0.6 | 11 | −37.2 ± 2.2 | 5.7 ± 0.3 | 5.1 ± 0.7 | 13 | −52.1 ± 1.5 | 8.1 ± 0.6 | 7.2 ± 1.9 | 13 |
| A1304T | 58.0 ± 2.1 | −23.0 ± 1.7* | −4.1 ± 0.4* | 13 | −37.1 ± 1.0 | 5.3 ± 0.2 | 4.5 ± 0.8 | 14 | −52.8 ± 1.4 | 7.4 ± 0.4 | 3.3 ± 0.9 | 12 |
*P < 0.05.
The amplitude of the response to slow ramp stimuli was not significantly different for A1304T compared with WT channels (WT: 27.0 ± 3.5%, n = 11; A1304T: 26.6 ± 2.6%, n = 12; P > 0.05). However, the potential of peak ramp current (VRamp) was hyperpolarized by 6 mV for A1304T (WT: −22.5 ± 1.9 mV, n = 11; A1304T: −28.5 ± 1.1 mV, n = 12) (Fig. 4 C and D).
Current-clamp recording showed that resting potential was significantly depolarized by 5 mV (P < 0.01) in DRG neurons expressing A1304T (−46.3 ± 1.3 mV; n = 35) compared with WT (−51.3 ± 1.2 mV; n = 34) (Fig. 5C). Input resistance was not significantly different in DRG neurons expressing WT (697 ± 74 MΩ; n = 34) or A1304T mutant channels (543 ± 55 MΩ; n = 35).
Fig. 5.

A1304T increases the excitability of small DRG neurons. (A) Response of cells expressing WT (Upper) and A1304T (Lower) channels, respectively, to 500-ms depolarizing current steps that are 1×, 2×, and 3× (left, center, and right traces, respectively) of current threshold for action potential generation. (B) Comparison of responses (number of impulses evoked by a 500-ms stimulus) in DRG neurons expressing WT (n = 34) and A1304T channels (n = 35) across a range of step current injections from 25 to 500 pA; *P < 0.05. (C) Resting membrane potential for A1304T channels was depolarized by 5 mV compared with WT (WT: −51.3 ± 1.2 mV, n = 34; A1304T: −46.3 ± 1.3 mV, n = 35; **P < 0.01). (D) Current threshold to 200-ms stimuli was reduced in DRG neurons expressing A1304T (161 ± 21 pA; n = 35) compared with WT channels (270 ± 32 pA; n = 34; **P < 0.01).
Current threshold was significantly reduced after expression of A1304T (161 ± 21 pA; n = 35), compared with WT channels (270 ± 32 pA; n = 34; P < 0.01) (Fig. 5D). Voltage threshold of DRG neurons expressing A1304T (−29.2 ± 1.5 mV; n = 35) or WT channels (−29.7 ± 1.2 mV; n = 34) was not significantly different. There was no significant difference in action potential amplitude (WT: 103.0 ± 3.9 mV, n = 34; A1304T: 105.5 ± 2.6 mV, n = 35), or half-width (WT: 5.2 ± 0.4 ms, n = 34; A1304T: 5.6 ± 0.6 ms, n = 35).
DRG neurons expressing A1304T fired significantly more action potentials than neurons expressing WT channels over a range of graded stimulation levels (Fig. 5 A and B).
Discussion
Three sodium channels, Nav1.7, Nav1.8, and Nav1.9, are preferentially expressed in peripheral neurons. Nav1.7 has been genetically linked to human pain (14, 15), whereas a role for Nav1.8 and Nav1.9 in pain transmission is supported by animal studies (14). A crucial role for Nav1.8 in pain signaling has been supported by several lines of evidence. First, Nav1.8 carries most of the sodium current underlying the depolarizing phase of action potential in DRG neurons (7, 8). Second, Nav1.8 is crucial for full manifestation of functional effects of Nav1.7 mutations (16). Third, knockout of Nav1.8 or ablation of Nav1.8-positive neurons impair thermal hyperalgesia following inflammation (10, 17) and cold-induced pain (18). However, a direct link of Nav1.8 and human pain has not been previously reported. Here, we report mutations in SCN10A, the gene that encodes Nav1.8, from patients with painful peripheral neuropathies.
We found gain-of-function changes in Nav1.8 in two of three mutations studied by voltage and current clamp, based on predictive algorithms that focused assessment on conserved residues in transmembrane segments, or in functional sequence motifs that had a high likelihood of regulating Nav1.8 gating or current density. A third mutation found in two patients (C1523Y) did not display gain-of-function changes by voltage clamp. Depending on whether the additional four mutations identified in 4 patients from our cohort of 104 patients produced gain-of-function changes, at least 3 patients and at most 7 patients from this cohort of 104 with predominantly SFN carry gain-of-function mutations of Nav1.8.
In adults, Nav1.8 is specifically expressed within peripheral sensory (DRG) neurons (9, 11). Nav1.8 recovers rapidly from inactivation (19) and contributes the majority of the inward current responsible for the action potential upstroke during repetitive firing of DRG neurons (7, 8). Nav1.8 mutations produced gain-of-function changes at the channel level (enhanced ramp responses and repriming for L554P; enhanced activation for A1304T), and increased excitability of small DRG neurons, which include nociceptors. Interestingly, the A1304T mutation hyperpolarized ramp response, by 6 mV, and depolarized resting potential to a similar degree, by 5 mV.
The mechanisms by which these mutations alter channel gating are not understood. It is possible that L554P enhances ramp currents or repriming as a result of a substitution of a structurally rigid residue, proline, instead of the more flexible residue, leucine, in a L1 proline-rich motif. The A1304T mutation substitutes a highly conserved residue within the DIII S5 membrane-spanning segment with a larger polar residue. Although S5 segments have not been directly implicated in activation, mutations in DIII/S5 of Nav1.5 (20), Nav1.1 (21, 22), and Nav1.7 (23, 24) have been linked to channelopathies, with the latter hyperpolarizing activation in a manner similar to A1304T.
The patients carrying these Nav1.8 mutations displayed distal neuropathic pain. The L554P mutation was carried by two family members with painful neuropathy. The pattern of segregation, together with our functional profiling, suggests a link to painful neuropathy. The A1304T mutation was observed in a single case. Nav1.7 and Nav1.8 function in tandem, with Nav1.7 amplifying small depolarizations to bring the cell to threshold, and Nav1.8 producing most of the inward current underlying the action potential upstroke during repetitive firing (7, 8, 12, 16). However, the Nav1.8 mutations we describe were identified in patients negative for mutations in SCN9A, the gene encoding Nav1.7. Definitive linkage of these Nav1.8 mutations to peripheral neuropathy would be further supported by the characterization of larger families, or development of an in vivo animal model. Nonetheless, our functional characterizations strongly suggest that these Nav1.8 mutations contribute to the pathophysiology of painful neuropathies.
After expression of the mutant channels within DRG neurons, where Nav1.8 is normally expressed (25, 26), we found at the channel level, by voltage clamp, enhancement of the response to depolarization, a proexcitatory effect. By current clamp, we found that the mutations reduce current threshold and increase firing frequency in response to suprathreshold stimuli (L554P, A1304T), depolarize resting potential (A1304), and induce spontaneous firing of small DRG neurons (L554P), which include nociceptors. The first three changes would be expected to lower threshold for, or increase intensity of, evoked pain, whereas the latter change would be expected to contribute to spontaneous pain. Whether these mutations contribute to axonal degeneration has not been established. Nav1.8 is abundantly expressed in nociceptive DRG neurons and their peripheral axons, together with sodium channels Nav1.6, Nav1.7, and Nav1.9 and the NCX2 Na/Ca exchanger (3, 27). Fine-caliber distal axons are especially sensitive to small changes in sodium channel activity due to high input resistance, short electrotonic and diffusional length constant, and high surface-to-volume ratio (28, 29). Persistent sodium current in myelinated CNS and PNS axons can trigger injurious, Ca2+-importing reverse Na/Ca exchange (30, 31). I228M Nav1.7 mutant channels, found in a patient with painful neuropathy, reduce the length of DRG neurites in vitro, whereas blockade of sodium channels, and of reverse Na/Ca exchange, are protective of axons expressing mutant, but not wild-type channels, suggesting that activity of mutant channels and reverse Na/Ca exchange contribute to axonal injury in this model (32). Whether mutant Nav1.8 channels contribute to axonal injury remains to be established.
In summary, our results demonstrate mutations in Nav1.8, a sodium channel specifically expressed in DRG neurons and peripheral nerve axons, in human subjects with painful neuropathy. Gain-of-function changes caused by these mutations increase the excitability of DRG neurons. These observations suggest that Nav1.8 mutations contribute to pain in some peripheral neuropathies.
Materials and Methods
Clinical Description.
Human studies were approved by Institutional Review Boards of “Carlo Besta” Neurological Institute and Maastricht University Medical Center. All aspects of the study were explained, and written informed consent was obtained before study initiation. The two institutes combined to enroll patients with predominantly idiopathic painful predominantly SFN [n = 104 (n = 64 from Maastricht; n = 40 from Milan)], all negative for mutations in SCN9A. All patients were profiled clinically and intraepidermal nerve fiber density (IENFD) assessed (4, 33) in a standardized fashion. Laboratory screening was performed to evaluate for diabetes mellitus, impaired glucose tolerance, hyperlipidemia, liver, kidney, or thyroid dysfunction, malignancy, monoclonal gammopathy, connective tissue disorders, sarcoidosis, amyloidosis, Fabry’s disease (α-galactosidase, in females combined with GLA-gene sequencing), celiac disease, HIV, alcohol abuse, hemochromatosis, antiphospholipid syndrome, and B6 intoxication.
Detailed methods are available in SI Materials and Methods.
IENF Quantification.
Skin biopsies from the distal leg (10 cm above the lateral malleolus), obtained with a 3-mm punch after topical anesthesia with lidocaine, were fixed [2% (wt/vol) paraformaldehyde–lysine–sodium periodate, 4 °C overnight] and cryoprotected (4, 34). Immunostaining was performed on serial sections using polyclonal anti-protein gene product 9.5 antibodies (Ultraclone), and IENF density (IENF per millimeter) determined and compared with sex- and age-adjusted normative values (4) using bright-field microscopy applying established counting rules (34). Intraepidermal nerves that cross or originate at the dermal–epidermal junction are counted, and secondary branches and fragments are not counted. At least three sections were analyzed. IENFD is reported as the mean IENF of these three sections per millimeter (4, 33).
SCN10A Exon Screening.
Genomic DNA was extracted from peripheral blood samples (35). Mutation screening in all 27 exons that constitute SCN10A was based on National Center for Biotechnology Information’s reference sequences (genomic, NG_031891.1; coding, CCDS33736.1). Exons were amplified using 200 ng of genomic DNA template by PCR with primers complementary to flanking intronic sequences as detailed in SI Materials and Methods. DNA control panels [Milan: 325 controls; Maastricht: 600 controls (100 from ethnically matched Dutch; 500 from the twin research KCL)] were screened for all mutations.
Primary Sensory Neuron Isolation and Transfection.
The WT construct (pcDNA5-SCN10A) that encodes human Nav1.8 protein was purchased from Genionics. The L554P or A1304T mutation was introduced into the construct using QuikChange II XL site-directed mutagenesis (Stratagene).
Animal studies were approved by Yale University and Veterans Administration West Haven Hospital Animal Use Committees. For voltage-clamp recording, DRG neurons were isolated from homozygous Nav1.8-cre mice (4–8 wk of age) that lack endogenous Nav1.8 and transfected by electroporation (36). Filtered DRG cell suspension was divided into two tubes, sequentially transfected with WT or mutant Nav1.8 constructs (2 μg of hNav1.8 WT or mutant construct plus 0.2 μg of EGFP) using Nucleofector IIS (Lonza) and Amaxa SCN Nucleofector (VSPI-1003) (36). Transfected neurons were seeded onto poly-d-lysine/laminin-coated coverslips (BD) and incubated at 37 °C in a 95% air/5% (vol/vol) CO2 incubator. For current-clamp recordings, DRG from 4- to 8-wk-old Sprague Dawley rats were harvested, dissociated (37), and transfected as described above. Transfected cell suspension were diluted in medium supplemented with NGF (50 ng/mL) and glial cell line-derived neurotrophic factor (50 ng/mL), plated on 12-mm circular coverslips coated with laminin and poly-ornithine, and incubated at 37 °C in 95% air/5% (vol/vol) CO2 incubator.
Electrophysiology.
Voltage-clamp recordings were obtained at 22 ± 1 °C, 36–54 h after transfection from small DRG neurons (surface area, 250–490 μm2) with robust green fluorescence (38) and were used to assess activation, steady-state inactivation, recovery from inactivation, slow inactivation, and the responses to slow ramp (0.2 mV/ms) depolarizations. Current-clamp recordings were obtained 40–48 h after transfection. Resting potential, current threshold, voltage threshold, and response to graded suprathreshold depolarizations were assessed as previously described (2, 37, 38) and in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Dr. J. A. Black for helpful advice; L. Tyrrell, L. Macala, P. Zhao, P. Shah, and L. K. M. Meekels for technical assistance; S. Scollen (Neusentis) for valuable help regarding exome data; T. D. Spector for TwinsUK exome data funded by Pfizer, Inc. (www.twinsuk.ac.uk); and B. Schulman for superb administrative support. This work was supported in part by grants from the Rehabilitation Research Service and Medical Research Service, Department of Veterans Affairs, and The Erythromelalgia Association (S.G.W. and S.D.D.-H.), and the “Profileringsfonds” University Hospital Maastricht (C.G.F. and I.S.J.M.). The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America with Yale University.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1216080109/-/DCSupplemental.
References
- 1.Hoeijmakers JG, Faber CG, Lauria G, Merkies IS, Waxman SG. Small-fibre neuropathies—advances in diagnosis, pathophysiology and management. Nat Rev Neurol. 2012;8(7):369–379. doi: 10.1038/nrneurol.2012.97. [DOI] [PubMed] [Google Scholar]
- 2.Faber CG, et al. Gain of function Naν1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol. 2012;71(1):26–39. doi: 10.1002/ana.22485. [DOI] [PubMed] [Google Scholar]
- 3.Persson AK, et al. Sodium-calcium exchanger and multiple sodium channel isoforms in intra-epidermal nerve terminals. Mol Pain. 2010;6:84. doi: 10.1186/1744-8069-6-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lauria G, et al. Intraepidermal nerve fiber density at the distal leg: A worldwide normative reference study. J Peripher Nerv Syst. 2010;15(3):202–207. doi: 10.1111/j.1529-8027.2010.00271.x. [DOI] [PubMed] [Google Scholar]
- 5.Reulen JP, Lansbergen MD, Verstraete E, Spaans F. Comparison of thermal threshold tests to assess small nerve fiber function: Limits vs. levels. Clin Neurophysiol. 2003;114(3):556–563. doi: 10.1016/s1388-2457(02)00405-4. [DOI] [PubMed] [Google Scholar]
- 6.Yarnitsky D, Sprecher E. Thermal testing: Normative data and repeatability for various test algorithms. J Neurol Sci. 1994;125(1):39–45. doi: 10.1016/0022-510x(94)90239-9. [DOI] [PubMed] [Google Scholar]
- 7.Renganathan M, Cummins TR, Waxman SG. Contribution of Na(v)1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol. 2001;86(2):629–640. doi: 10.1152/jn.2001.86.2.629. [DOI] [PubMed] [Google Scholar]
- 8.Blair NT, Bean BP. Roles of tetrodotoxin (TTX)-sensitive Na+ current, TTX-resistant Na+ current, and Ca2+ current in the action potentials of nociceptive sensory neurons. J Neurosci. 2002;22(23):10277–10290. doi: 10.1523/JNEUROSCI.22-23-10277.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akopian AN, Sivilotti L, Wood JN. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379(6562):257–262. doi: 10.1038/379257a0. [DOI] [PubMed] [Google Scholar]
- 10.Akopian AN, et al. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2(6):541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- 11.Sangameswaran L, et al. Structure and function of a novel voltage-gated, tetrodotoxin-resistant sodium channel specific to sensory neurons. J Biol Chem. 1996;271(11):5953–5956. doi: 10.1074/jbc.271.11.5953. [DOI] [PubMed] [Google Scholar]
- 12.Cummins TR, Howe JR, Waxman SG. Slow closed-state inactivation: A novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J Neurosci. 1998;18(23):9607–9619. doi: 10.1523/JNEUROSCI.18-23-09607.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waxman SG. Neurobiology: A channel sets the gain on pain. Nature. 2006;444(7121):831–832. doi: 10.1038/444831a. [DOI] [PubMed] [Google Scholar]
- 14.Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33:325–347. doi: 10.1146/annurev-neuro-060909-153234. [DOI] [PubMed] [Google Scholar]
- 15.Hoeijmakers J, Merkies I, Gerrits M, Waxman S, Faber C. Genetic aspects of sodium channelopathy in small fiber neuropathy. Clin Genet. 2012;82(4):351–358. doi: 10.1111/j.1399-0004.2012.01937.x. [DOI] [PubMed] [Google Scholar]
- 16.Rush AM, et al. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci USA. 2006;103(21):8245–8250. doi: 10.1073/pnas.0602813103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abrahamsen B, et al. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321(5889):702–705. doi: 10.1126/science.1156916. [DOI] [PubMed] [Google Scholar]
- 18.Zimmermann K, et al. Sensory neuron sodium channel Nav1.8 is essential for pain at low temperatures. Nature. 2007;447(7146):855–858. doi: 10.1038/nature05880. [DOI] [PubMed] [Google Scholar]
- 19.Cummins TR, Waxman SG. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci. 1997;17(10):3503–3514. doi: 10.1523/JNEUROSCI.17-10-03503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keller DI, et al. A novel SCN5A mutation, F1344S, identified in a patient with Brugada syndrome and fever-induced ventricular fibrillation. Cardiovasc Res. 2006;70(3):521–529. doi: 10.1016/j.cardiores.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 21.Fukuma G, et al. Mutations of neuronal voltage-gated Na+ channel alpha 1 subunit gene SCN1A in core severe myoclonic epilepsy in infancy (SMEI) and in borderline SMEI (SMEB) Epilepsia. 2004;45(2):140–148. doi: 10.1111/j.0013-9580.2004.15103.x. [DOI] [PubMed] [Google Scholar]
- 22.Wallace RH, et al. Neuronal sodium-channel alpha1-subunit mutations in generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2001;68(4):859–865. doi: 10.1086/319516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Estacion M, et al. NaV1.7 gain-of-function mutations as a continuum: A1632E displays physiological changes associated with erythromelalgia and paroxysmal extreme pain disorder mutations and produces symptoms of both disorders. J Neurosci. 2008;28(43):11079–11088. doi: 10.1523/JNEUROSCI.3443-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fischer TZ, et al. A novel Nav1.7 mutation producing carbamazepine-responsive erythromelalgia. Ann Neurol. 2009;65(6):733–741. doi: 10.1002/ana.21678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Djouhri L, et al. The TTX-resistant sodium channel Nav1.8 (SNS/PN3): Expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J Physiol. 2003;550(Pt 3):739–752. doi: 10.1113/jphysiol.2003.042127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shields SD, et al. Na(v)1.8 expression is not restricted to nociceptors in mouse peripheral nervous system. Pain. 2012;153(10):2017–2030. doi: 10.1016/j.pain.2012.04.022. [DOI] [PubMed] [Google Scholar]
- 27.Zhao P, et al. Voltage-gated sodium channel expression in rat and human epidermal keratinocytes: Evidence for a role in pain. Pain. 2008;139(1):90–105. doi: 10.1016/j.pain.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 28.Waxman SG, Black JA, Kocsis JD, Ritchie JM. Low density of sodium channels supports action potential conduction in axons of neonatal rat optic nerve. Proc Natl Acad Sci USA. 1989;86(4):1406–1410. doi: 10.1073/pnas.86.4.1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donnelly DF. Orthodromic spike generation from electrical stimuli in the rat carotid body: Implications for the afferent spike generation process. J Physiol. 2007;580(Pt 1):275–284. doi: 10.1113/jphysiol.2006.119263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: Role of Na+ channels and Na+-Ca2+ exchanger. J Neurosci. 1992;12(2):430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lehning EJ, Doshi R, Isaksson N, Stys PK, LoPachin RM., Jr Mechanisms of injury-induced calcium entry into peripheral nerve myelinated axons: Role of reverse sodium-calcium exchange. J Neurochem. 1996;66(2):493–500. doi: 10.1046/j.1471-4159.1996.66020493.x. [DOI] [PubMed] [Google Scholar]
- 32.Persson A-K, et al. Neuropathy-associated Nav1.7 variant I228M impairs integrity of DRG neuron axons. Ann Neurol. 2012 doi: 10.1002/ana.23725. [DOI] [PubMed] [Google Scholar]
- 33.Bakkers M, et al. Intraepidermal nerve fiber density and its application in sarcoidosis. Neurology. 2009;73(14):1142–1148. doi: 10.1212/WNL.0b013e3181bacf05. [DOI] [PubMed] [Google Scholar]
- 34.Lauria G, et al. 2010. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on the use of skin biopsy in the diagnosis of small fiber neuropathy. Eur J Neurol 17(7):903–912, e44–49.
- 35.Strauss WM. Preparation of genomic DNA from mammalian tissue. Curr Protoc Mol Biol. 2001 doi: 10.1002/0471142727.mb0202s42. Chapter 2:Unit2.2. [DOI] [PubMed] [Google Scholar]
- 36.Dib-Hajj SD, et al. Transfection of rat or mouse neurons by biolistics or electroporation. Nat Protoc. 2009;4(8):1118–1126. doi: 10.1038/nprot.2009.90. [DOI] [PubMed] [Google Scholar]
- 37.Han C, et al. Nav1.7-related small fiber neuropathy: Impaired slow-inactivation and DRG neuron hyperexcitability. Neurology. 2012;78(21):1635–1643. doi: 10.1212/WNL.0b013e3182574f12. [DOI] [PubMed] [Google Scholar]
- 38.Cummins TR, Rush AM, Estacion M, Dib-Hajj SD, Waxman SG. Voltage-clamp and current-clamp recordings from mammalian DRG neurons. Nat Protoc. 2009;4(8):1103–1112. doi: 10.1038/nprot.2009.91. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.