

Abstract
The protein kinase v-akt murine thymoma viral oncogene homolog (AKT), a key regulator of cell survival and proliferation, is frequently hyperactivated in human cancers. Intramolecular pleckstrin homology (PH) domain–kinase domain (KD) interactions are important in maintaining AKT in an inactive state. AKT activation proceeds after a conformational change that dislodges the PH from the KD. To understand these autoinhibitory interactions, we generated mutations at the PH–KD interface and found that most of them lead to constitutive activation of AKT. Such mutations are likely another mechanism by which activation may occur in human cancers and other diseases. In support of this likelihood, we found somatic mutations in AKT1 at the PH–KD interface that have not been previously described in human cancers. Furthermore, we show that the AKT1 somatic mutants are constitutively active, leading to oncogenic signaling. Additionally, our studies show that the AKT1 mutants are not effectively inhibited by allosteric AKT inhibitors, consistent with the requirement for an intact PH–KD interface for allosteric inhibition. These results have important implications for therapeutic intervention in patients with AKT mutations at the PH–KD interface.
Keywords: interdomain, AKT-targeting, PI3K-pathway, next generation sequencing, BaF3
The mammalian v-akt murine thymoma viral oncogene homolog (AKT) family consists of three members, AKT1/protein kinase B (PKB)a, AKT2/PKBβ, and AKT3/PKBγ (Fig. S1). They belong to the protein kinase A, kinase G, and kinase C (AGC) superfamily of serine/threonine kinases and are involved in regulating key cellular processes, including cell proliferation, survival, growth, metabolism, and angiogenesis (1). The AKTs share a common domain architecture consisting of an N-terminal pleckstrin homology (PH) domain, a kinase domain (KD), and a C-terminal regulatory region that contains a hydrophobic motif (2, 3).
AKT functions downstream of class IA PI3K (4). After growth factor stimulation, activated PI3Ks catalyze the conversion of phosphatidylinositol 4,5 bisphosphate to phosphatidylinositol 3,4,5 trisphosphate that directs translocation of AKT to the plasma membrane (5). At the membrane, AKT1 undergoes phosphorylation on two regulatory sites: T308 within the catalytic domain and S473 in the hydrophobic motif through phosphoinositide-dependent kinase 1 (PDK1) and PDK2 mammalian target of Rapamycin (mTORC2), respectively. This phosphorylation leads to its activation and downstream signaling (6).
AKT is frequently activated in cancers, mostly through mutations or amplifications of upstream genes like PIK3CA (7). AKT activation can also result from inactivation or loss of the lipid phosphatase, PTEN (8). Genomic alterations that directly affect AKT also lead to its activation. Whereas AKT1 amplification is rare, AKT2 is frequently amplified in a variety of cancers (9). Recently, a somatic mutation in the PH domain of AKT1 was identified in a subset of human carcinomas (10). This mutation results in the substitution of glutamic acid at codon 17 of AKT1 with lysine (E17K) and alters the lipid-binding specificity of AKT, leading to pathological membrane association and constitutive signaling (10, 11). Other than human cancers, germ-line and somatic E17K mutations in AKT have been identified in Proteus syndrome, human hypoglycemia, and hemimegalencephaly (12–14).
Recent molecular modeling and structure-based studies suggest that, under basal conditions, interactions between the PH and KD maintain AKT in a closed conformation (PH-in) (15–17). In this state, PDK1 is unable to access and phosphorylate T308. In response to upstream signaling, AKT shifts from an autoinhibited PH-in confirmation to an open PH-out state, leading to its phosphorylation and activation. Molecular dynamic studies and crystal structure of AKT suggest that the interactions between the PH and KD are important for maintaining the kinase in an inactive state.
In this study, we have performed a systematic analysis to understand the effects of perturbing PH–KD interactions on activation of AKT. We show that disrupting interdomain contacts by mutating residues at the PH–KD interface leads to AKT activation. Given this finding, we sequenced a large number of human tumors to see if mutations at the PH–KD contact sites occur in cancers. Interestingly, we found human tumors that carry mutations in AKT at these sites, indicating that disruption of PH–KD interactions is a mechanism for AKT activation in cancers. Furthermore, we show that these tumor-specific mutations are oncogenic and that they alter sensitivity to allosteric AKT inhibitors.
Results
Perturbing PH–KD Contacts Lead to AKT Activation.
To assess the activation status of AKT, we measured its ability to promote growth factor-independent survival of IL-3–dependent BaF3 cells. The BaF3 pro-B cells can be rendered growth factor-independent by enforced expression of oncogenes (18). We generated BaF3 cells expressing WT AKT1, Myristoylated (Myr), or the E17K AKT1 mutant and found that activated AKT by itself was unable to promote factor independence (Fig. 1A). However, coexpression of Myr or E17K AKT1 and an activated form of the MAP2 kinase mitogen-activated protein kinase (MAPK)/extracellular-signal-regulated kinase (ERK) kinase (MEK1) (Mek1 ΔN3, S218E, S222D) (19) promoted factor-independent growth of BaF3 cells (Fig. 1A). Although WT AKT1 along with active MEK1 (MEK1 N3) showed some activity in this assay, it was markedly less compared with mutant AKT1. This result is consistent with previous reports that show cooperation between AKT and MEK in oncogenic transformation (20, 21).
Fig. 1.

PH–KD contact site mutations lead to AKT activation. (A) IL-3–independent proliferation of BaF3 cells expressing empty vector (EV), WT, Myr, or E17K AKT1 alone or combined with MEK1 N3. (B) An open book representation of PH (purple) and KD (orange) of AKT1 in complex with an allosteric inhibitor (gray sticks; Protein Data Bank ID code 3O96). Sites discussed in this work are labeled and colored pink if the mutation promotes survival in the BaF3 assay (Dataset S1). Sites for which no electron density is observed in the crystal structure are in parentheses, and the gatekeeper residue M227 is colored green for reference. KD residues that contact substrate peptide are colored blue (determined from the structure 3OCB). (C) Schematic depicting the screen used to assess the effect of AKT1 PH–KD interface mutations. (D) PH–KD interface mutations promote IL-3–independent proliferation of BaF3 cells. (E) Immunoblot analysis of BaF3 cells coexpressing the indicated AKT1 mutants and MEK1 N3. (F and G) Plots depicting correlation between IL-3–independent survival of BaF3 cell expressing the mutants indicated in E and D and phospho-AKT levels observed in E.
We used the BaF3 assay to investigate the consequence of disrupting PH–KD interactions. Using the recently published full-length structure of AKT1 (17), we identified residues at the PH–KD interface. Mutations at these sites were designed to compromise the PH–KD interaction (Materials and Methods). We generated a library of 35 such AKT1 mutants (Fig. 1B and Dataset S1), including negative (WT AKT1) and positive (Myr and E17K AKT1) controls for activity. We used this library to derive a pool of BaF3 cells that stably coexpressed the mutants along with MEK1 N3. After allowing growth in the absence of IL-3 for 3–4 d, the proportion of various mutants in the pool was determined relative to the input at 0 h using next-generation sequencing (Fig. 1C). Each mutation was scored based on a normalized ratio of observed frequency at a given time point compared with the input frequency, and these ratios were then normalized to the ratios for WT AKT1. As expected, AKT1 E17K was more than 50 times enriched over WT. Similarly, mutants, such as T81Y and D323A, were also strongly enriched (>15-fold over WT), indicating that these mutations lead to AKT activation. Other mutants (R23A, N53A, F55Y, L78T, Q79E, W80A, E191A, T195I, V270A, V271A, L321A, D325A, and R328A) showed moderate enrichment (two- to sixfold over WT) in the assay and are likely activating (Fig. 1D and Dataset S1).
To further understand the effect of PH–KD interface mutants, we generated BaF3 cell lines expressing some of the AKT1 mutants that promoted survival and some that did not promote survival in our initial screen. We then assessed the T308 and S473 phosphorylation status (pT308 and pS473) of AKT in these lines. Consistent with the survival activity observed in the screen, we found that mutants, such as F55Y, T81Y, T195I, L321A, and D325A, showed elevated levels of pT308 and pS473 (Fig. 1E), confirming the positive correlation between phosphorylated AKT (pAKT) status and cell survival (T308: R2 = 0.71, P value = 8.735e-05; S473: R2 = 0.66, P value = 0.0002158) (Fig. 1 F and G). However, R23A, although scored positive for survival in our initial screen, failed to show increased pAKT, indicating that it potentially was a false positive. Furthermore, AKT mutants Y18S, I19E, Q59E, R200A, L202F, and L362R, which did not support IL-3–independent survival, also did not show an increase in pAKT levels (Fig. 1E). These results indicate that disrupting the PH–KD contacts leads to constitutive phosphorylation of AKT. In agreement with this result, a recent study showed that deletion of AKT1 PH domain led to its constitutive S473 phopshorylation (22).
Identification of AKT Somatic Mutations in Cancer.
Given that perturbation of the PH–KD interface led to AKT activation, we wanted to assess if such mutations occur in human primary tumors. To identify potential AKT mutations, we sequenced all of the coding exons of AKT1, AKT2, and AKT3 in a total of 394 human primary tumor samples consisting of 65 colorectal, 51 breast, 48 nonsmall-cell lung (NSCLC) adenocarcinoma (adeno), 43 NSCLC (squamous), 43 renal carcinoma, 37 melanoma, 33 gastric, 32 ovarian, 15 esophageal, 11 hepatocellular, 10 small-cell lung cancer, and 6 others (5 lung large cell and 1 lung cancer other) (Dataset S2). We found protein-altering, somatic AKT1 mutations in 2 of 51 (4%) breast and 1 of 65 (1.5%) colon samples. We also found AKT2 somatic mutations in 2 of 43 (∼5%) NSCLC (adeno) samples (Fig. 2A and Dataset S3). Previous studies have reported protein-altering mutations in AKT1, primarily in codon 17 (E17K) in ∼5% of breast cancers (10, 23). Mutations at other residues in AKT family members, although quite rare, have been reported (Fig. 2A and Dataset S4) (24–27).
Fig. 2.

Somatic AKT mutations in human cancer. (A) Somatic mutations in AKT family members. Horizontal black bars indicate residues conserved across AKT1, AKT2, and AKT3. (B) Localization of somatic mutations mapped onto AKT1 crystal structure. The central portion depicts open book views of PH (purple) and KD (orange), whereas the outer portions indicate the outside of the PH–KD structure. Mutations occurring in AKT1 (pink), AKT2 (magenta), and AKT3 (dark blue) are labeled. Modeled loops missing from the deposited structure are in gray. Somatic mutations in these loops have labels in parentheses. For reference, the gatekeeper residue (M227) is shown in green, and the allosteric inhibitor is shown as gray tubes. (C) IL-3–independent proliferation of BaF3 cells stably expressing the indicated AKT1 mutants combined with MEK1 N3. (D) Immunoblot analysis of the same BaF3 cells expressing WT or mutant AKT1 tested in C.
Other than the E17K mutation, we have previously reported an AKT1 mutation at codon 52, L52R (Fig. 2A and Datasets S3 and S4) (28). L52 is at the PH–KD interface and makes hydrophobic contacts with V270, V271, Y326, and the methylene portion of R328 in the KD (Fig. 1B). Although the L52A mutant did not support IL-3–independent survival of BaF3 cells (Fig. 1D), the L52R mutation is likely to weaken PH–KD interaction, because it is predicated to replace favorable hydrophobic interactions with an unfavorable interaction with R328 (Fig. 2B). The synthetic mutant D323A was active in our screen (Fig. 1D), suggesting that D323H will also be constitutively active, because a histidine is even more disruptive to the interdomain contacts than alanine. The R96 residue located in the main PH domain helix is far removed from the KD interface and hence, unlikely to promote activation through disruption of PH–KD interactions (Fig. 2B). Electron density for residues K189 to E198 is not observed in the full-length AKT1 crystal structure (Fig. 2B), and hence, a structure-based assessment of its effect on AKT1 activity could not be made.
To understand the biological significance of the somatic mutations identified in this study and a few published somatic AKT1 mutations, we tested their ability to support IL-3–independent growth of BaF3 cells coexpressing MEK1 N3. We found that L52R, Q79K, and D323H, like Myr and E17K AKT1, supported growth factor-independent survival of BaF3 cells (Fig. 2C), and this support was dependent on their kinase activity (Fig. S2). Consistently, mutants that promoted survival also showed elevated levels of pAKT (Fig. 2D).
AKT1 Somatic Mutants Signal Constitutively and Lead to Transformation.
To further assess the functional relevance of the AKT1 somatic mutations, we tested their effect on signaling in NIH 3T3 cells expressing FLAG-tagged WT, Myr, E17K, L52R, K189N, or D323H AKT1. Immunoblot analysis showed that, similar to Myr AKT1, all mutants (except K189N) showed elevated pAKT levels compared with WT (Fig. 3A). Consistent with this finding, we observed increased phosphorylation of the AKT substrates FOXO and S6 ribosomal protein (Fig. 3A). Also, we observed elevation of phospho PRAS40 levels, although the activation was delayed in E17K and D323H mutant-expressing NIH 3T3 cells (Fig. 3A and Fig. S3 A and B).
Fig. 3.

AKT1 somatic mutations are activating. (A) NIH 3T3 cells stably expressing AKT1 somatic mutations show elevated pT308 and pS473. (B) Representative image showing anchorage-independent growth of NIH 3T3 cells expressing the indicated AKT1 constructs. (C) 3D morphogenesis assay with MCF10A cells expressing indicated Myr or mutant AKT1 (Upper) and kinase dead (K179M) Myr or mutant AKT1 (Lower). (D) Immunoblot analysis of MCF10A cells expressing the indicated constructs cultured on matrigel (3D assay). (E) AKT1 PH–KD mutants show impaired interaction in a mammalian two-hybrid assay. (F) Still images from live-cell imaging of serum-starved NIH 3T3 cells expressing GFP-PH domain (WT, E17K, or L52R) before and after PDGF stimulation. Arrows indicate membrane localization.
To further characterize the AKT1 mutants, we tested their transforming ability in an anchorage-independent growth assay. We found that NIH 3T3 cells expressing Myr or mutant AKT1 formed ∼20-fold more colonies compared with controls (Fig. 3B). To further characterize these mutants, we used a 3D morphogenesis assay using MCF10A breast epithelial cells. MCF10A cells, when cultured on a 3D matrix, form polarized acini with a hollow lumen (29). Expression of oncogenes (including Myr AKT1) in this system is known to disrupt acinar architecture (30). We found that, like Myr AKT1, expression of E17K, L52R, K189N, D323H, or Q79K led to formation of disorganized multiacinar structures (Fig. 3C), and it was dependent on the kinase activity of these mutants (Fig. 3C). In contrast, the mutants F35L and R96W, consistent with their inability to promote survival (Fig. 2C), did not affect morphogenesis (Fig. 3C). Interestingly, although the K189N mutant did not support BaF3 survival or increase downstream signaling (Figs. 2 C and D and 3A and Fig. S3C), it promoted anchorage-independent growth and acinar disruption (Fig. 3 B and C). Consistent with this finding, MCF10A cells expressing the K189N mutant showed elevated pAKT and increased pS6RP levels compared with WT when cultured on a 3D matrix (Fig. 3D).
AKT1 PH–KD Mutants Weaken Interdomain Interactions and Show Impaired Plasma Membrane Translocation.
Although the cancer-specific AKT1 mutations (L52R and D323H) occur at positions predicted to disrupt PH–KD interactions, the amino acid substitutions observed were different from the synthetic mutants generated and analyzed earlier (Fig. 1 B, D, and E). To directly test whether these somatic mutations weaken interdomain interactions, we performed a mammalian two-hybrid assay using AKT PH and KD constructs fused to the VP16 activation domain and Gal4 DNA-binding domain, respectively. The strength of the interaction was measured using a luciferase reporter, where the luciferase activity is proportional to the strength of the interaction. We found that the L52R PH/WT-KD, Q79K-PH/WT-KD, and WT-PH/D323H-KD combinations showed significant reduction in the interaction signal compared with WT-PH/WT-KD, confirming that these mutants are deficient in the PH–KD interaction (Fig. 3E). In contrast, F35L-PH/WT-KD, R96W-PH/WT-KD, and WT-PH/K189N-KD combinations in the two-hybrid assay showed interaction that was comparable with the WT-PH/WT-KD pair, indicating that these mutations do not affect PH–KD interactions. Interestingly, the E17K-PH/WT-KD combination also showed a reduced interaction compared with the WT-PH/WT-KD combination, suggesting that the E17K mutation may also weaken PH–KD interaction, although this effect was modest (Fig. 3E).
To further understand the mechanism of activation of AKT mutants, we tested their cellular localization. Previous studies indicate that mutations in lipid-binding PH domains that result in dysregulated membrane recruitment can lead to carcinogenesis (31). To assess the effects of the PH domain mutant L52R on subcellular localization and membrane translocation, we generated GFP-tagged L52R PH domain along with GFP-tagged WT and E17K PH domains as controls. In the absence of stimulation, whereas WT AKT1 was distributed throughout the cytoplasm and nucleus, AKT1 E17K was constitutively localized to the plasma membrane as previously reported (10). In contrast, the L52R PH domain was distributed throughout the cell, behaving like the WT PH domain. However, on growth factor stimulation, unlike WT, the mutant L52R PH domain did not translocate to the membrane (Fig. 3F and Movies S1, S2, and S3). This result suggests that, unlike E17K, which is activated in response to altered lipid affinity and localization, the L52R mutant is most likely activated in the cytoplasm because of absence of autoinhibitory interactions. Consistent with this finding, a recent study has shown that activation of AKT can occur in the cytoplasm independent of membrane localization (32).
Disruption of AKT2 and AKT3 PH–KD Interactions Leads to Their Activation.
Given the common domain architecture of the AKT family members, we tested whether disrupting PH–KD interactions in AKT2 and AKT3 can lead to their activation. To test, we generated AKT2 mutants L52R and D324H and AKT3 mutants L51R and D320H (equivalent of AKT1 L52R and D323H), all of which are predicted to disrupt PH–KD interactions (Fig. S4). Because AKT3 E17K mutations in melanoma (33) and AKT2 E17K mutations in human hypoglycemina (12) have been reported, we generated the E17K AKT2 and AKT3 mutants along with additional AKT2 and AKT3 somatic mutations that have been identified in human cancers (Fig. 2A). Additionally, we generated Myr AKT2 and Myr AKT3 as positive controls. We stably expressed the AKT2 and AKT3 mutants in NIH 3T3 cells and assessed pAKT. AKT2 E17K, L52R, and D324H, and AKT3 E17K, L51R, and D320H all showed elevated pT308 and pS473 compared with WT AKT2 or AKT3 (Fig. S5A). Consistent with the activation status, these mutants (combined with MEK1 N3) support IL-3–independent survival of BaF3 cells (Fig. S5B). Interestingly, the cancer-specific AKT2 R371H mutant, although it showed elevated pAKT, was not capable of promoting growth factor-independent survival of BaF3. The remaining mutants (AKT2 V90L and R101L and AKT3 Q124L and G171R) did not increase pAKT and were unable to support growth factor-independent survival of BaF3 cells (Fig. S5). Inspections of the homology models generated for full-length AKT2 and AKT3 indicate that these mutations occur in surface-exposed loops and are not proximal to the PH–KD interface (Fig. 2B). Note that, although AKT2 V90L and AKT3 Q124L are in loops not defined in the AKT1 electron density, the termini of these loops are not proximal to the PH–KD interface. Thus, structural analysis does not offer insights into the role of these mutants in cancers.
AKT1 Somatic Mutants Promote Oncogenesis in Vivo.
Previous studies have shown that BaF3 cells stably expressing oncogenes promote a leukemia-like disease when implanted in mice, leading to reduced overall survival (34, 35). Because the AKT1 mutants cooperate with active MEK1 to promote factor-independent growth of BaF3 cells (Figs. 1A and 2C), we used this model system to test their tumorigenic potential in vivo. Mice implanted with BaF3 cells coexpressing MEK1 N3 and Myr or mutant AKT1 (E17K, L52R or D323H) showed a median survival of 19–20.5 d. In contrast, mice implanted with cells coexpressing MEK1 N3 and AKT1 WT had a significantly longer median survival of 29 d (Fig. 4). This result is consistent with the fact that AKT1 WT in the context of active MEK1 was able to support IL-3-independent survival of BaF3 cells, although the effect was modest compared with AKT mutants (Fig. 1A and 2C). As expected, mice expressing MEK1 N3 alone (control) were alive at the end of the 55-d study period. Necropsies were performed at 19 d posttransplantation on a cohort of three mice per treatment group to follow disease progression. Consistent with the reduced overall survival, mice implanted with mutant AKT1 had a significant proportion of GFP-tagged BaF3 cells in the bone marrow and spleens (Fig. S6A) and had massively enlarged liver and spleen compared with controls (Fig. S6 B–D). Histological examination of H&E-stained liver, spleen, and bone marrow sections from these mice showed evidence of infiltration with leukemic blasts (Fig. S6E). These results confirm the transforming potential of the AKT1 mutants in vivo.
Fig. 4.

AKT1 mutants promote oncogenesis in vivo. Kaplan–Meier survival curve depicting reduction in overall survival of mice implanted with BaF3 cells expressing AKT1 mutants. (n = 10 for arms; log-rank test P < 0.0001).
AKT1 PH–KD Interaction-Deficient Mutants Are Less Sensitive to Allosteric Inhibitors.
Several ATP-competitive and allosteric small-molecule inhibitors of AKT are in development and/or clinical trials (36, 37). Previous studies have shown that allosteric AKT inhibitors require an intact PH–KD interface for their activity (17, 38–40). Given that some AKT1 somatic mutants have impaired PH–KD contacts, we predicted that allosteric inhibitors are likely to be less efficacious in inhibiting their activity. We tested this prediction by assaying the activity of two ATP-competitive inhibitors [GNE-692 (41) and GSK690693 (42)] and two allosteric inhibitors [Inhibitor VIII (43) and GNE-929 (Fig. S7)] on recombinant full-length WT and mutant AKT1 enzymes. We also tested the effect of the inhibitors on the proliferation of NIH 3T3 cells expressing WT or mutant AKT1.
We found that, in biochemical activity assays, the ATP-competitive inhibitors GNE-692 and GSK690693 were effective in blocking activity of WT AKT1 (GNE-692 IC50 = 24.3 nM) as well as the mutant enzymes (E17K, L52R, and D323H; GNE-692 IC50 = 3.7–15.8 nM) (Fig. S8A, Fig. S8E, and Dataset S5A). Similarly, the ATP-competitive inhibitors were equally effective against both WT and mutant AKT1 in the cell-based proliferation assay (Fig. S8B and Dataset S5B). In contrast, the allosteric inhibitors, Inhibitor VIII and GNE-929, were less effective against recombinant full-length mutant enzymes (Inhibitor VIII: IC50 = 268.4 nM for L52R; IC50 > 1 μM for D323H) compared with WT AKT1 (Inhibitor VIII: IC50 = 119.3 nM) (Fig. S8C, Fig. S8F, and Dataset S5A). Consistent with this finding, in cell-based assay, we found Inhibitor VIII to be at least 50% less effective at blocking proliferation of cells expressing mutant AKT1 compared with WT AKT (Fig. S8D and Dataset S5B).
To confirm that the reduced sensitivity of the mutants to allosteric inhibitors was caused by impaired PH–KD interactions, we performed an in vitro biochemical reconstitution assay using purified recombinant PH and KD. In this system, allosteric Inhibitor VIII, when assayed against AKT1 KD alone, was unable to block its activity (Fig. 5 A and B). Reconstituting the enzyme by adding back WT PH domain restored the ability of Inhibitor VIII to block enzyme activity, albeit with a threefold higher IC50 (IC50 = 238.8 nM), compared with the full-length WT enzyme (IC50 = 80.8 nM) (Fig. 5 A and B and Dataset S5C). In contrast, reconstitution with mutant PH domain (L52R or E17K) further impaired the ability of Inhibitor VIII in blocking AKT1 (L52R IC50 = 713.5 nM and E17K IC50 > 1 μM) (Fig. 5A and Dataset S5C). Similarly, Inhibitor VIII showed no activity when the WT PH domain was reconstituted with a mutant D323H KD (Fig. 5B). The lack of E17K inhibition by allosteric inhibitors suggests that, in addition to increased affinity for phosphatidylinositol 4,5 bisphosphate (10, 11), this mutation may also affect the PH–KD interaction, leading to its activation. These data confirm the importance of an intact PH–KD interface for AKT1 allosteric inhibitors.
Fig. 5.
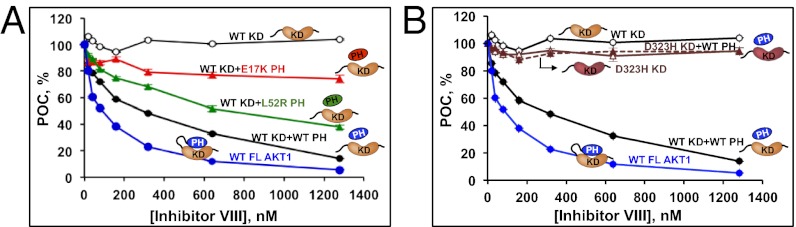
Effective AKT inhibition by allosteric inhibitors requires intact PH–KD interaction. (A) Effect of Inhibitor VIII on the kinase activity of recombinant AKT1 WT KD reconstituted with WT or mutant PH domains or (B) AKT1 D323H KD reconstituted with WT PH domain. POC, percent of control.
Discussion
Recent structural studies indicate that inhibitory interdomain interactions play a crucial role in regulating AKT activation (15, 17). Using a mutational screen, we show here that activation of AKT can result from mutations in residues involved in PH–KD contacts. Furthermore, we report the identification of mutations in human cancers, some of which involve residues at the PH–KD interface.
In addition to the previously identified mutation E17K, we show that the AKT1 PH domain mutant L52R and the KD mutant D323H identified in clinical samples mediate cellular transformation and are oncogenic in vivo. Inspection of the structure of full-length AKT1 reveals that E17, L52, and D323 are at the PH–KD interface and that substitutions at these positions are predicted to perturb PH–KD binding. Consistent with this finding, both L52R and D323H weaken PH–KD binding in two-hybrid assays. Previously, the mechanism of activation of E17K has been attributed to an altered lipid-binding specificity (10, 11). Our results indicate perturbation of interdomain interactions to be an additional mechanism underlying E17K activation.
Taken together, our findings suggest that the oncogenicity of the AKT1 PH–KD interface mutations identified here is caused by their constitutive activation resulting from destabilization of interdomain contacts. The activity of many multidomain proteins is regulated through autoinhibitory intramolecular interactions (44). It is likely that mutational destabilization of such interactions is a general mechanism leading to their inappropriate activation in human disease. In agreement with this likelihood, activating mutations in interdomain contact residues have been described in nonreceptor tyrosine kinases, like JAK2 (in polycythemia vera) and BCR-ABL, as well as phosphatases, such as SHP-2 in Noonan’s syndrome and juvenile myelo-monocytic leukemia (45–48).
Inhibitors targeting the PI3K-AKT pathway members, including AKT, are currently in various stages of development (36, 37). Previous studies have shown that AKT allosteric inhibitors require an intact PH–KD interface, because such inhibitors preferentially bind the closed PH-in conformation (16, 17, 38). Consistent with this result, we found that mutations in AKT that favor an open (PH-out) conformation show reduced sensitivity to allosteric AKT inhibitors, although they retain sensitivity to ATP-competitive inhibitors. This finding indicates that the AKT mutational status has important implications for the choice of inhibitor in the clinic. AKT mutations, although they may function as drivers in naive tumors, can also arise in tumors in response to agents that target upstream components of the AKT pathway. Future studies will be required to assess such mutations and fully understand the predictive and prognostic significance of these mutations in the clinic.
Materials and Methods
Coding exons of AKT1, AKT2 and AKT3 were amplified and sequenced as described before (Dataset S6) (35). Retroviral constructs expressing WT and mutant AKT were used to generate stable BaF3, MCF10A, and NIH 3T3 cell lines for studying signaling, transformation and modeling oncogenesis in vivo. Baculovirus expressed proteins were used in biochemical assays.
Additional details on the reagents and assays can be found in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank George Kan, Kanan Pujara, Deepali Bhatt, and Jeffrey Eastham-Anderson for their support during the course of this project.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1204384109/-/DCSupplemental.
References
- 1.Vasudevan KM, Garraway LA. AKT signaling in physiology and disease. Curr Top Microbiol Immunol. 2010;347:105–133. doi: 10.1007/82_2010_66. [DOI] [PubMed] [Google Scholar]
- 2.Brazil DP, Hemmings BA. Ten years of protein kinase B signalling: A hard Akt to follow. Trends Biochem Sci. 2001;26(11):657–664. doi: 10.1016/s0968-0004(01)01958-2. [DOI] [PubMed] [Google Scholar]
- 3.Hanada M, Feng J, Hemmings BA. Structure, regulation and function of PKB/AKT—a major therapeutic target. Biochim Biophys Acta. 2004;1697(1–2):3–16. doi: 10.1016/j.bbapap.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 4.Fayard E, Xue G, Parcellier A, Bozulic L, Hemmings BA. Protein kinase B (PKB/Akt), a key mediator of the PI3K signaling pathway. Curr Top Microbiol Immunol. 2010;346:31–56. doi: 10.1007/82_2010_58. [DOI] [PubMed] [Google Scholar]
- 5.Bellacosa A, et al. Akt activation by growth factors is a multiple-step process: The role of the PH domain. Oncogene. 1998;17(3):313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- 6.Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett. 2003;546(1):108–112. doi: 10.1016/s0014-5793(03)00562-3. [DOI] [PubMed] [Google Scholar]
- 7.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14(5):381–395. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 8.Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22(14):2954–2963. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 9.Chalhoub N, Baker SJ. PTEN and the PI3-kinase pathway in cancer. Annu Rev Pathol. 2009;4:127–150. doi: 10.1146/annurev.pathol.4.110807.092311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carpten JD, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448(7152):439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 11.Landgraf KE, Pilling C, Falke JJ. Molecular mechanism of an oncogenic mutation that alters membrane targeting: Glu17Lys modifies the PIP lipid specificity of the AKT1 PH domain. Biochemistry. 2008;47(47):12260–12269. doi: 10.1021/bi801683k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hussain K, et al. An activating mutation of AKT2 and human hypoglycemia. Science. 2011;334(6055):474. doi: 10.1126/science.1210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindhurst MJ, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365(7):611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JH, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44(8):941–945. doi: 10.1038/ng.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calleja V, et al. Intramolecular and intermolecular interactions of protein kinase B define its activation in vivo. PLoS Biol. 2007;5(4):e95. doi: 10.1371/journal.pbio.0050095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Calleja V, Laguerre M, Parker PJ, Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: Structural mechanism for allosteric inhibition. PLoS Biol. 2009;7(1):e17. doi: 10.1371/journal.pbio.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu WI, et al. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One. 2010;5(9):e12913. doi: 10.1371/journal.pone.0012913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warmuth M, Kim S, Gu XJ, Xia G, Adrián F. Ba/F3 cells and their use in kinase drug discovery. Curr Opin Oncol. 2007;19(1):55–60. doi: 10.1097/CCO.0b013e328011a25f. [DOI] [PubMed] [Google Scholar]
- 19.Mansour SJ, et al. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265(5174):966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 20.Boehm JS, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129(6):1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 21.Robinson JP, et al. Activated MEK cooperates with Ink4a/Arf loss or Akt activation to induce gliomas in vivo. Oncogene. 2011;30(11):1341–1350. doi: 10.1038/onc.2010.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Warfel NA, Niederst M, Newton AC. Disruption of the interface between the pleckstrin homology (PH) and kinase domains of Akt protein is sufficient for hydrophobic motif site phosphorylation in the absence of mTORC2. J Biol Chem. 2011;286(45):39122–39129. doi: 10.1074/jbc.M111.278747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bamford S, et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br J Cancer. 2004;91(2):355–358. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parsons DW, et al. Colorectal cancer: Mutations in a signalling pathway. Nature. 2005;436(7052):792. doi: 10.1038/436792a. [DOI] [PubMed] [Google Scholar]
- 25.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446(7132):153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anonymous Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455(7216):1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kan Z, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466(7308):869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 29.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30(3):256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 30.Debnath J, Walker SJ, Brugge JS. Akt activation disrupts mammary acinar architecture and enhances proliferation in an mTOR-dependent manner. J Cell Biol. 2003;163(2):315–326. doi: 10.1083/jcb.200304159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Falke JJ. Membrane recruitment as a cancer mechanism: A case study of Akt PH domain. Cellscience. 2007;4(2):25–30. [PMC free article] [PubMed] [Google Scholar]
- 32.Ding Z, et al. Physical association of PDK1 with AKT1 is sufficient for pathway activation independent of membrane localization and phosphatidylinositol 3 kinase. PLoS One. 2010;5(3):e9910. doi: 10.1371/journal.pone.0009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davies MA, et al. A novel AKT3 mutation in melanoma tumours and cell lines. Br J Cancer. 2008;99(8):1265–1268. doi: 10.1038/sj.bjc.6604637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horn S, et al. Mutations in the catalytic subunit of class IA PI3K confer leukemogenic potential to hematopoietic cells. Oncogene. 2008;27(29):4096–4106. doi: 10.1038/onc.2008.40. [DOI] [PubMed] [Google Scholar]
- 35.Jaiswal BS, et al. Somatic mutations in p85alpha promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009;16(6):463–474. doi: 10.1016/j.ccr.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pal SK, Reckamp K, Yu H, Figlin RA. Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs. 2010;19(11):1355–1366. doi: 10.1517/13543784.2010.520701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mattmann ME, Stoops SL, Lindsley CW. Inhibition of Akt with small molecules and biologics: Historical perspective and current status of the patent landscape. Expert Opin Ther Pat. 2011;21(9):1309–1338. doi: 10.1517/13543776.2011.587959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnett SF, et al. Identification and characterization of pleckstrin-homology-domain-dependent and isoenzyme-specific Akt inhibitors. Biochem J. 2005;385(Pt 2):399–408. doi: 10.1042/BJ20041140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lindsley CW, Barnett SF, Layton ME, Bilodeau MT. The PI3K/Akt pathway: Recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Curr Cancer Drug Targets. 2008;8(1):7–18. doi: 10.2174/156800908783497096. [DOI] [PubMed] [Google Scholar]
- 40.Calleja V, Laguerre M, Larijani B. 3-D structure and dynamics of protein kinase B-new mechanism for the allosteric regulation of an AGC kinase. J Chem Biol. 2009;2(1):11–25. doi: 10.1007/s12154-009-0016-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bencsik JR, et al. Discovery of dihydrothieno- and dihydrofuropyrimidines as potent pan Akt inhibitors. Bioorg Med Chem Lett. 2010;20(23):7037–7041. doi: 10.1016/j.bmcl.2010.09.112. [DOI] [PubMed] [Google Scholar]
- 42.Rhodes N, et al. Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res. 2008;68(7):2366–2374. doi: 10.1158/0008-5472.CAN-07-5783. [DOI] [PubMed] [Google Scholar]
- 43.Lindsley CW, et al. Allosteric Akt (PKB) inhibitors: Discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett. 2005;15(3):761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 44.Pufall MA, Graves BJ. Autoinhibitory domains: Modular effectors of cellular regulation. Annu Rev Cell Dev Biol. 2002;18:421–462. doi: 10.1146/annurev.cellbio.18.031502.133614. [DOI] [PubMed] [Google Scholar]
- 45.Tartaglia M, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29(4):465–468. doi: 10.1038/ng772. [DOI] [PubMed] [Google Scholar]
- 46.Tartaglia M, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003;34(2):148–150. doi: 10.1038/ng1156. [DOI] [PubMed] [Google Scholar]
- 47.Smith KM, Yacobi R, Van Etten RA. Autoinhibition of Bcr-Abl through its SH3 domain. Mol Cell. 2003;12(1):27–37. doi: 10.1016/S1097-2765(03)00274-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levine RL, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell. 2005;7(4):387–397. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.