

Abstract
Nonribosomal peptides are highly sought after for their therapeutic applications. As with other natural products, dereplication of known compounds and focused discovery of new agents within this class are central concerns of modern natural product-based drug discovery. Development of a chemoinformatic library-based and informatic search strategy for natural products (iSNAP) has been constructed and applied to nonribosomal peptides and proved useful for true nontargeted dereplication across a spectrum of nonribosomal peptides and within natural product extracts.
Keywords: drug discovery, antibiotic, secondary metabolite, genome mining
Nonribosomal peptides (NRPs) are a group of natural products with diverse biological activities and pharmacophores (1, 2). The evolutionarily selected status of these peptides translates to intrinsic bioactivity, and ∼5% of the 205 NRP structural families are used clinically as inhibitors of enzymes, agonists and antagonists of receptors, modulators of eukaryotic signaling cascades, potentiators of epigenetic modification, and perturbants of protein–protein interactions (3). Efforts to discover new NRPs have increasingly resulted in the rediscovery of known compounds, stifling new therapeutic advances and highlighting the need for rapid and efficient methods of dereplication (4). Further, rapid advances in microbial genome sequencing have exposed a wealth of novel gene clusters that encode for NRPs (5, 6). New analytical tools are needed to dereplicate NRPs and reveal novel potential therapeutics.
Modern proteomic research has used mass spectrometry to achieve efficient and automated peptide dereplication from complex mixtures through de novo sequencing and database-derived methods (7). Because all peptides share a common amide monomer linkage, they should follow similar MS fragmentation patterns. However, two important variations necessitate the development of divergent informatics tools for NRP dereplication. First, NRPs can be assembled from a much larger range of monomers (>500) and often incorporate polyketide building blocks. Second, nonribosomal peptide architecture is varied between linear, cyclic, and mixed or “branched” combinations thereof (8).
In linear peptides, the fragmentation pattern proceeds from the termini, providing a series of diagnostic ladder ions or “direct sequence ions” (DSs) with relatively few internal cleavages or rearrangements that generate “nondirect sequences” (NDSs). In contrast, cyclic NRPs are prone to multiple ring-opening events, with each linear form producing unique ladder ions and enrichments of other NDSs (9, 10). The resulting output is a mix of DSs and NDSs, and has proven to be a considerable challenge for de novo sequencing methods (11). Recently, Dorrestein and Pevzner and coworkers presented a de novo sequencing approach for purely cyclic nonribosomal peptides and demonstrated the utility in “comparative dereplication.” In their approach, a comparative dereplication (similarity ranking) was illustrated using 18 pure known cyclic nonribosomal peptides, with 4 of these being correctly classically dereplicated in a manual fashion (12–14). Similarly, Dorrestein and colleagues connected chemotypes with microbial genomic data by an iterative de novo sequencing approach in peptidogenomics (15).
Use of nonribosomal peptide databases and scoring of fragment matches may provide an alternative strategy to de novo approaches and result in classical dereplication of nonribosomal peptides. A structural matching design would not require differentiation of NDSs from DSs, and may work for the varied architectural forms, backbone modifications, altered connectivities, and nonpeptidic building blocks found in NRPs and hybrids thereof (NRP-polyketide, NRP-terpene). Unfortunately, no spectral library of mass-to-charge ratios of known NRPs exists and no scoring matrices are established.
In this work, we present a platform for informatic searching of natural products (iSNAP) to detect NRPs using a database-searching algorithm in an automated data-dependent mode that is nontargeted and affords a nanogram-sensitive, efficient, and high-throughput means of classical dereplication of NRPs in natural product extracts.
Results
Development of an Informatic Platform and Chemoinformatic Database for Natural Product Discovery.
Numerous challenges are confronted in constructing NRP natural product databases for automated dereplication. First, there is no compiled spectral database with information on all of the known NRPs or a ready supply of compounds to create one. Further, there are no mathematical tools available to computationally compare unknown analytes to known nonribosomal peptides and no infrastructure existing to create hypothetical MS/MS spectra of known compounds in a rapid fashion.
Nonribosomal peptides [represented in simplified molecular input line specification (SMILES) format] were taken from the NORINE database (3), PubChem, the Journal of Antibiotics, and other resources (SI Appendix, section I.F). SMILES is a linear string code that contains all of the structural information of a given small molecule (16). The assembled in-house NRP database contains 1,107 NRP structures and, for the initial part of our informatic search approach for natural products (iSNAP), we created a script that would identify all amide bonds and generate hypothetical spectral fragments (hSFs) based on amide cleavage. These hSFs are calculated estimations as to how a protonated peptide may fragment or be generated from collision-induced dissociation (CID) within the gas phase of an MS/MS experiment (17). The iSNAP algorithm labels all amide cleavage sites within a compound’s SMILES code. The hSFs are generated by enumerating the cleavage at two amide sites at a time. These fragments arise from the cleavage of N-terminal (b and a ions) and C-terminal (y ions) cleavage, and the iSNAP program takes these and adds mass offsets of +H and +H+1 to account for protonation and the first isotope ion, respectively. In this way, the initial 1,107 NRP structures resulted in a hypothetical spectral library of 100,747 hSFs. Of these, 27,036 fragments resulted from amide cleavage, with each having a corresponding fragment bearing values indicative of the sequestration ionization charges (hydrogen and hydrogen plus one species) (81,108 mass-to-charge values) and neutral losses species (water, ammonia, and carbon monoxide) generating 19,639 offset mass-to-charge values.
The collective of these hSFs comprises all of the mass-to-charge ratio ions that may be observed in real MS/MS spectra of known NRPs. As such, a direct comparison of the hypothetical versus experimental spectra for a given NRP should yield a significant number of shared high-intensity peaks.
Comparative Analysis of Hypothetical Mass-to-Charge Ratios and Tandem Mass Spectra for the Detection of Nonribosomal Peptides.
We sought to determine how computational fragmentation of NRPs (described above) would compare with actual NRP fragmentation (Fig. 1 A and B). For this, we compared the spectral fragments derived from bacitracin A, an antimicrobial NRP composed of both linear and cyclic portions, with the hSFs generated by iSNAP. An authentic standard of bacitracin A was subjected to electrospray ionization (ESI)-MS/MS analysis by direct infusion with the double-charged ion (+711.4 m/z) selected and subjected to CID. iSNAP analysis of bacitracin A generated 102 hSFs and a total of 301 mass-to-charge values from these by +H and +H+1 mass offsets (Dataset S1), in addition to neutral loss species (H2O, NH3, and CO). Of these, 89 mass-to-charge values could be detected by the iSNAP matching algorithm from the doubly charged MS/MS spectrum (Fig. 1B).
Fig. 1.

Chemoinformatic analysis of bacitracin A. (A) Structure of bacitracin A. (B) Raw matching score overview. Hypothetical spectral library fragments composed of mass-to-charge ratios are compared with peaks from real MS/MS spectra. Peak fragments in green represent matched mass-to-charge ratios within the tandem MS spectra. Matched peaks are then processed through the in-house nonribosomal peptide database and statistically scored to determine a candidate’s match significance for dereplication.
Creation of a Scoring Scheme.
Having generated an NRP hypothetical spectral library (Fig. 2A), we next focused on deriving a scoring mechanism to compare experimentally generated spectra with the hypothetical spectral library. Three scores are computed for these two purposes: raw score,  score, and
score, and  score. The raw score is an overall spectral match between the MS/MS spectrum of an analyte and the hypothetical spectrum of a known NRP. The raw score alone, however, does not remove bias toward larger-sized NRPs and spectra with large numbers of fragment peaks. In this way, the raw score is not a comparable measure across different spectra, and therefore we derived probability scores denoted
score. The raw score is an overall spectral match between the MS/MS spectrum of an analyte and the hypothetical spectrum of a known NRP. The raw score alone, however, does not remove bias toward larger-sized NRPs and spectra with large numbers of fragment peaks. In this way, the raw score is not a comparable measure across different spectra, and therefore we derived probability scores denoted  and
and 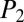 that use raw scoring but derive match significance differently. In general, as NRPs increase in mass, the number of hSFs also increases due to the presence of potentially more amide bonds and cyclic/cyclic-branching connectivities. With added offsets and neutral losses, the total number of hSFs can rapidly accumulate, and thus the chances of falsely matching fragment ions rise, creating an artificial bias.
that use raw scoring but derive match significance differently. In general, as NRPs increase in mass, the number of hSFs also increases due to the presence of potentially more amide bonds and cyclic/cyclic-branching connectivities. With added offsets and neutral losses, the total number of hSFs can rapidly accumulate, and thus the chances of falsely matching fragment ions rise, creating an artificial bias.
Fig. 2.

iSNAP scoring scheme. (A) Histogram representing the hypothetical spectral library of 1,107 compounds. (B) Dereplicating bacitracin A in Fig. 1B using doubly protonated (+711.82 m/z) MS/MS spectra. Multiple MS/MS scans are generated from an ∼1-min direct infusion of bacitracin A; each blue point indicates a match between an MS/MS spectrum and bacitracin A. The red points show the score distribution of the other matches between the MS/MS spectra and the rest of the 1,106 database NRPs. The score distribution plots indicate the capability of the  and
and  scores in distinguishing true and false matches. (C)
scores in distinguishing true and false matches. (C)  score calculation of bacitracin A. The raw score distribution is generated by scoring the MS2 spectrum against database compounds within the 0 to [M] + 100–Da mass range. The raw matching score of the bacitracin A candidate is 59.1, where the P value on the distribution is 1.74e-006. The
score calculation of bacitracin A. The raw score distribution is generated by scoring the MS2 spectrum against database compounds within the 0 to [M] + 100–Da mass range. The raw matching score of the bacitracin A candidate is 59.1, where the P value on the distribution is 1.74e-006. The  score is calculated as −10log10(P value) = 57.6. The fitted gamma distribution is shown as the red curve. (D)
score is calculated as −10log10(P value) = 57.6. The fitted gamma distribution is shown as the red curve. (D)  score calculation of bacitracin A. The raw score distribution is generated by scoring each decoy spectrum against bacitracin A. The original spectrum has a raw score of 59.1, which is greater than that of the decoy spectra. The P value on the distribution is 5.87e-008, with a
score calculation of bacitracin A. The raw score distribution is generated by scoring each decoy spectrum against bacitracin A. The original spectrum has a raw score of 59.1, which is greater than that of the decoy spectra. The P value on the distribution is 5.87e-008, with a  score calculated as −10log10(P value) = 72.31.
score calculated as −10log10(P value) = 72.31.
Raw Score Calculation.
In calculating the raw, or spectral-matching, score, the iSNAP algorithm first conducts a noise-filtering process to remove low-intensity peaks from the input MS/MS spectra. In this process, iSNAP calculates the relative peak intensity for all of the ion peaks by comparing them with the highest peak within the spectrum and filters out peaks of less than 0.5%. This prefiltering is applied to reduce the likelihood of randomly matched peaks, and such preprocessing is embedded within most proteomic ribosomal peptide algorithms (18–20). The iSNAP program collects the remaining peaks and matches only those with the hypothetical spectral library. In the event that an input MS/MS spectrum is from a multiply charged ion, the algorithm correlates and adjusts the protonated hypothetical spectrum to account for differences in charge states. When the parent ion of the MS/MS spectrum bears a charge k, the m/z values of hypothetical fragments with charges up to k are combined to form the charge-k hypothetical spectrum. By using a mass error tolerance of 0.1 Da, the algorithm finds all spectrum peaks that have matches and computes the raw score as
 |
The fraction 1/0.5% (factor 200) in the formula is used to ensure that a match to a peak of significant intensity (≥0.5% relative intensity) will not contribute negatively to the overall score. Within the iSNAP algorithm, a mass error tolerance of 0.1 Da is set to accommodate errors arising from use of low-resolution mass spectral files. Values set to “low” will limit matched fragments, and higher ones increase matches, possibly increasing random assignments.
For each MS/MS spectrum, the raw score is calculated against the database compounds within a mass range of 0 to [M] + 100 Da, where [M] represents parent mass. Having a relaxed mass range ensures sufficient raw scores are calculated for statistical distribution, and the upper limit of [M] + 100 Da avoids a potential bias for large molecules that may score higher due to more fragment-matching possibilities. The +100 Da value is chosen empirically by experimenting with +0, 50, 100, 200, and 500 Da (SI Appendix, section I.G). Only database compounds within the mass range of [M] ± 1 Da are considered candidates of known NRPs and are ultimately subjected to  and
and  calculations.
calculations.
 Score.
Score.
A  score is introduced as a normalized version of the raw score to add statistical significance. Empirically, when an MS/MS spectrum is scored against all database compounds within the 0 to [M] + 100–Da mass range, the statistical distribution of the raw scores closely fits a gamma distribution (Fig. 2C). In Fig. 2C, the fitted gamma distribution is shown as a red curve. The parameters required for a gamma distribution are estimated with the maximum-likelihood method. For each compound, the P value is the exceedance frequency at the compound’s raw score, which is the area under the curve and to the right of the raw score. The P value represents the probability of a random structure scoring higher with the MS/MS spectrum than the correct structure. A low P value indicates the match is unlikely to be random and therefore is likely a correct one. The
score is introduced as a normalized version of the raw score to add statistical significance. Empirically, when an MS/MS spectrum is scored against all database compounds within the 0 to [M] + 100–Da mass range, the statistical distribution of the raw scores closely fits a gamma distribution (Fig. 2C). In Fig. 2C, the fitted gamma distribution is shown as a red curve. The parameters required for a gamma distribution are estimated with the maximum-likelihood method. For each compound, the P value is the exceedance frequency at the compound’s raw score, which is the area under the curve and to the right of the raw score. The P value represents the probability of a random structure scoring higher with the MS/MS spectrum than the correct structure. A low P value indicates the match is unlikely to be random and therefore is likely a correct one. The  score is calculated as −10log10(P value).
score is calculated as −10log10(P value).
 Score.
Score.
Whereas the  score measures the significance of the candidate structure compared with other NRP structures in the database, a
score measures the significance of the candidate structure compared with other NRP structures in the database, a 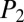 score is used to measure the significance of the MS/MS spectrum compared with artificially generated “decoy” spectra. If the MS/MS spectrum
score is used to measure the significance of the MS/MS spectrum compared with artificially generated “decoy” spectra. If the MS/MS spectrum 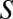 is from an NRP structure, then the structure should be scored significantly higher using
is from an NRP structure, then the structure should be scored significantly higher using  than using the artificially generated decoy spectra. Suppose the spectrum
than using the artificially generated decoy spectra. Suppose the spectrum 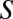 has a mass range from
has a mass range from 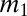 to
to  . To generate a decoy spectrum, the m/z value of each peak in
. To generate a decoy spectrum, the m/z value of each peak in  is shifted by an integer
is shifted by an integer . More specifically, an m/z value
. More specifically, an m/z value  is changed to
is changed to  if
if  , and to
, and to  if
if  . Thus, by trying every integer
. Thus, by trying every integer  between
between  and
and  , many decoy spectra can be obtained. The shifting method was inspired by the calculation of the cross-correlation score in the SEQUEST algorithm, which was the first computer algorithm for matching ribosomal peptides in a database with MS/MS spectral data (21). A gamma distribution is then estimated from the raw scores between the decoy spectra and the candidate structure. The P value is the exceedance frequency at the original MS/MS spectrum’s raw score (Fig. 2D). The
, many decoy spectra can be obtained. The shifting method was inspired by the calculation of the cross-correlation score in the SEQUEST algorithm, which was the first computer algorithm for matching ribosomal peptides in a database with MS/MS spectral data (21). A gamma distribution is then estimated from the raw scores between the decoy spectra and the candidate structure. The P value is the exceedance frequency at the original MS/MS spectrum’s raw score (Fig. 2D). The  score is calculated as −10log10(P value).
score is calculated as −10log10(P value).
Hypothetical Spectral Library Matching Studies with Known Nonribosomal Peptides.
iSNAP is designed to analyze individual spectra and reveal the significance of a match between MS/MS spectra and candidate NRP compounds (those within a mass range of [M] ± 1 Da). For each MS/MS spectrum with established candidates, a  score and a
score and a 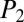 score are generated for each candidate. A training experiment using six pure NRPs (bacitracin A, cyclosporin A, gramicidin A, polymyxin B, surfactin, and seglitide) were used to reveal a threshold needed for true-positive identification from
score are generated for each candidate. A training experiment using six pure NRPs (bacitracin A, cyclosporin A, gramicidin A, polymyxin B, surfactin, and seglitide) were used to reveal a threshold needed for true-positive identification from  and
and 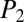 scores. We rationalized the selection of the six NRPs for the training experiment based on structural complexity, backbone modification (e.g., N-methylated amides, amides replaced by esters, and polyketide extended amino acid building blocks), and variance in chemical architecture (linear, cyclic, and branched). The expectation from this test set is that a true candidate match will have distinctively higher
scores. We rationalized the selection of the six NRPs for the training experiment based on structural complexity, backbone modification (e.g., N-methylated amides, amides replaced by esters, and polyketide extended amino acid building blocks), and variance in chemical architecture (linear, cyclic, and branched). The expectation from this test set is that a true candidate match will have distinctively higher  and
and  scores (additional details are in SI Appendix, section I.B).
scores (additional details are in SI Appendix, section I.B).
An initial test with the branched cyclic NRP bacitracin A was conducted to reveal whether the designed scoring strategy would result in the true candidate having distinctively higher  and
and  scores than those of other database structures. The resulting spectrum from an infusion experiment consisted of 56 bacitracin A MS/MS scans and, using the scoring scheme, without mass filtering ([M] ± 1 Da), produced bacitracin A as the top-ranking hit and distinguishably higher than the other 1,106 database NRPs (see multiscan score distribution plot of
scores than those of other database structures. The resulting spectrum from an infusion experiment consisted of 56 bacitracin A MS/MS scans and, using the scoring scheme, without mass filtering ([M] ± 1 Da), produced bacitracin A as the top-ranking hit and distinguishably higher than the other 1,106 database NRPs (see multiscan score distribution plot of  vs.
vs.  scores; Fig. 2B).
scores; Fig. 2B).
Applying the scoring scheme and [M] ± 1 Da filter, pure standards of the five additional test compounds cyclosporin A, gramicidin A, polymyxin B, surfactin, and seglitide underwent manual MS/MS and automated data-dependent acquisitions (DDAs). In the case of seglitide, a purely cyclic peptide, a doubly protonated [M+2H]2+ species within scan 10 underwent a single stage of tandem MS and scored ( = 57.5, and
= 57.5, and  = 48.2) with 17 out of 30 b ions and 27 matched mass-to-charge values. Another cyclic peptide, polymyxin B, whose complexity derives from repetitive blocks (six a,g-diaminobutyric acid residues), had the second-highest number of total matched peaks at 59 with 33 b ions matched, yielding
= 48.2) with 17 out of 30 b ions and 27 matched mass-to-charge values. Another cyclic peptide, polymyxin B, whose complexity derives from repetitive blocks (six a,g-diaminobutyric acid residues), had the second-highest number of total matched peaks at 59 with 33 b ions matched, yielding  = 35.1 and
= 35.1 and 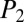 = 35.0. Matched peaks composed of repeat amino acid units were of relatively low intensity for four of six monomers. The fragmentation pattern derived from macrocyclic ring opening, acyl chain loss, and a diaminobutyric acid monomer (+963.6, +863.5, and +241 m/z) is consistent as the major pathway of fragmentation (22). In the case of cyclosporin A, iSNAP dereplicated the structure despite the N-methylated peptide backbone. N-methylation limits peptide cleavage, as the amide bond is unable to be protonated through intramolecular proton transfer, and thus additional stability is gained by increasing the basicity of its neighboring carbonyl group, favoring a C-terminal fragmentation pathway and the generation of y ions (23). The highest-scoring MS/MS scan came from acquisition 28, and a total of 27 hSFs was matched to the real MS/MS spectra. Of these, 25 were b ions, a quarter of all possible b-ion fragments, with score values of
= 35.0. Matched peaks composed of repeat amino acid units were of relatively low intensity for four of six monomers. The fragmentation pattern derived from macrocyclic ring opening, acyl chain loss, and a diaminobutyric acid monomer (+963.6, +863.5, and +241 m/z) is consistent as the major pathway of fragmentation (22). In the case of cyclosporin A, iSNAP dereplicated the structure despite the N-methylated peptide backbone. N-methylation limits peptide cleavage, as the amide bond is unable to be protonated through intramolecular proton transfer, and thus additional stability is gained by increasing the basicity of its neighboring carbonyl group, favoring a C-terminal fragmentation pathway and the generation of y ions (23). The highest-scoring MS/MS scan came from acquisition 28, and a total of 27 hSFs was matched to the real MS/MS spectra. Of these, 25 were b ions, a quarter of all possible b-ion fragments, with score values of  = 35.1 and
= 35.1 and  = 41.8. In the case of the linear polypeptide gramicidin A, only 5 of 85 b ions were generated in the MS experiment and identified (within scan 19) and, overall, 13 matched mass-to-charge values were sufficient for dereplication with scores above threshold cutoffs,
= 41.8. In the case of the linear polypeptide gramicidin A, only 5 of 85 b ions were generated in the MS experiment and identified (within scan 19) and, overall, 13 matched mass-to-charge values were sufficient for dereplication with scores above threshold cutoffs,  = 34.6 and
= 34.6 and 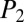 = 40.7. In the case of another cyclic-branching peptide, surfactin, 29 low-intensity (<10%) peaks were matched in scan 18, of which 22 were b ions (
= 40.7. In the case of another cyclic-branching peptide, surfactin, 29 low-intensity (<10%) peaks were matched in scan 18, of which 22 were b ions (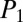 = 28.5 and
= 28.5 and  = 31.2).
= 31.2).
Establishing iSNAP Cutoffs for True- and False-Positive Rate Identification.
Early-stage dereplication of natural product extracts is a key goal of modern natural product screening programs, and we probed whether iSNAP enables nontargeted dereplication of known compounds in complex mixtures using low-resolution tandem mass spectrometry. Optimized MS/MS and LC-MS/MS settings for optimal 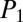 and
and 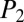 scoring and nontargeted dereplication were realized by testing mass resolution (u/s), activation energy (q), isolation width (m/z), and DDA settings (SI Appendix, section I.E and Dataset S2).
scoring and nontargeted dereplication were realized by testing mass resolution (u/s), activation energy (q), isolation width (m/z), and DDA settings (SI Appendix, section I.E and Dataset S2).
DDA acquisitions were performed under the auto-MS/MS setting with the available tuning option active, smart parameter setting. A scan range of 100–2,000 m/z was selected with precursors over 300 m/z targeted for MS/MS using the active exclusion option set to eight spectra over a release time of 0.25 min. The active exclusion feature enables the targeting of lower-abundance ions by deselecting and not fragmenting more-abundant ions. Ten precursor ions were selected for MS/MS using the enhanced resolution mode and baseline intensity threshold of 6 × 105, with an isolation width of 4 m/z.  and
and  threshold cutoffs were determined through a combination of two MS/MS experiments. In the first experiment, MS/MS spectra were generated from NRP working standards (direct infusion), and the iSNAP scores (
threshold cutoffs were determined through a combination of two MS/MS experiments. In the first experiment, MS/MS spectra were generated from NRP working standards (direct infusion), and the iSNAP scores ( and
and  ) were used as positive controls in the threshold training (Fig. 3A). In the second experiment, LC-MS/MS data derived from the scanning of 11 common fermentation media (no NRPs added) were used to investigate false matching (SI Appendix, section I.D). As no NRP compounds exist within those matrices, matches to NRPs within the iSNAP database must be considered as falsely matched and these low
) were used as positive controls in the threshold training (Fig. 3A). In the second experiment, LC-MS/MS data derived from the scanning of 11 common fermentation media (no NRPs added) were used to investigate false matching (SI Appendix, section I.D). As no NRP compounds exist within those matrices, matches to NRPs within the iSNAP database must be considered as falsely matched and these low  and
and  scores are used as negative controls (SI Appendix, Figs. S3 and S4). By combining the true or correct NRP database matches (NRP working standards) with the negative-control false matches in a
scores are used as negative controls (SI Appendix, Figs. S3 and S4). By combining the true or correct NRP database matches (NRP working standards) with the negative-control false matches in a 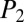 vs.
vs.  scatterplot,
scatterplot,  and
and  threshold cutoffs were empirically derived (Fig. 3A). Candidates with
threshold cutoffs were empirically derived (Fig. 3A). Candidates with  and
and  scores above 27 and 24, respectively, are considered dereplicated or positively identified. Using the estimated thresholds, 335 of 367 MS/MS scans were identified as true candidates, with a true-positive rate of 91.3%, whereas 24 of 6,744 registered as false positives, with a false-positive rate of 0.0036%, from the 11 fermentation media. In an effort to further reduce false-positive hits, additional filtering was applied to candidate matches with
scores above 27 and 24, respectively, are considered dereplicated or positively identified. Using the estimated thresholds, 335 of 367 MS/MS scans were identified as true candidates, with a true-positive rate of 91.3%, whereas 24 of 6,744 registered as false positives, with a false-positive rate of 0.0036%, from the 11 fermentation media. In an effort to further reduce false-positive hits, additional filtering was applied to candidate matches with  and
and  scores above the empirical threshold. Candidates with fewer than 4 matched peaks were determined to contribute to false matches, whereas candidate matches with fewer than 10 matched peaks, of which more than 75% were derived from low intensities (<2%), were also excluded.
scores above the empirical threshold. Candidates with fewer than 4 matched peaks were determined to contribute to false matches, whereas candidate matches with fewer than 10 matched peaks, of which more than 75% were derived from low intensities (<2%), were also excluded.
Fig. 3.

iSNAP threshold determination and complex mixture analysis. (A) MS/MS spectra from the six NRPs standards (STDs; in blue) obtained by direct infusion experiments, overlaid with over 6,500 MS/MS spectra from LC-MS/MS analysis of 11 microbial fermentation media; n = 3 (in red). The fermentation media represent the blank control. Empirical threshold cutoffs are estimated, 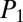 = 27 and
= 27 and  = 24. (B) NRP standards are spiked and extracted from the 11 media and subjected to LC-MS/MS analysis and iSNAP dereplication. (C) iSNAP results from B, with the highest-scoring MS/MS spectra from across the 11 media panel reported.
= 24. (B) NRP standards are spiked and extracted from the 11 media and subjected to LC-MS/MS analysis and iSNAP dereplication. (C) iSNAP results from B, with the highest-scoring MS/MS spectra from across the 11 media panel reported.
The output of the iSNAP analysis is a complete report for each MS/MS scan (SI Appendix, section I.A), showing the scan number, retention time, precursor m/z, charge state, precursor mass, outputted candidate name, mass, SMILES code, number-matched fragments, raw score,  score, and
score, and  score (SI Appendix, Figs. S1 and S2).
score (SI Appendix, Figs. S1 and S2).
Probing iSNAP Fidelity in Data-Dependent Acquisition Within Different Fermentation Conditions and Groupings of Nonribosomal Peptides.
To reveal the suitability and fidelity of the iSNAP algorithm for screening extracts, a series of liquid media varying in their spectrum of use (differing natural product producers) and nutrient and peptide composition was subjected to LC-MS/MS and iSNAP analysis to reveal their contributions to potential false positives. This panel of 11 different microbial fermentation media used for fermentation of NRP producers (myxobacteria, streptomycetes, and other actinobacteria, pseudomonads, bacilli, and filamentous fungi) used included YPD (yeast protein, milk protein), YMPG (yeast, malt, peptone, glucose), GYM (yeast, malt), TSB (soy protein), LB (peptone peptides and yeast protein), nutrient (beef and meat peptides from meat infusion solids), pharmamedia (cotton seed protein), grass seed vegetable protein (grass seed extract proteins), fish meal (fish meal protein), R2A (proteose peptone, casamino acids, yeast proteins), and CY (casitone, yeast). In each of these cases, we designed the experiment based on a typical volume of fermentation media used in screening (50-mL cultures) and a final amount of 50 ng of a given NRP analyzed by mass spectrometry. A panel of NRPs was spiked into each medium (final 50 μg/mL), and the mixture was extracted with organic solvent and subjected to LC-MS/MS analysis using DDA settings (SI Appendix, Fig. 3B). In these instances, we also determined the true- and false-positive rates for the study. For this, we sought to determine the number of MS/MS spectra acquired for each spiked medium and the number of MS/MS spectra matched to the iSNAP database, MS/MS spectra from spiked NRPs, and false matches (SI Appendix, Table S1).
Automated LC-MS/MS analysis of the 11 NRP-spiked fermentation media revealed, as expected, a variance in the numbers of product ions, with 485 being the average. In the case of R2A-spiked media, a total of 192 MS/MS spectra was matched to product-ion spectra and their m/z offsets, which were derived from the six NRP candidates; of these, 126 scans were above the  and
and  cutoffs. The false-positive rate for R2A is calculated as the total number of MS/MS spectra (minus NRP candidates) divided by the total number of candidates with false-positive hits. The false-positive rate was determined to be 0.83% for R2A, with only one false-positive hit (SI Appendix, Fig. S5). The media YMPG and grass seed had zero false positives detected, whereas the remaining media panel had between one and four false-positive hits.
cutoffs. The false-positive rate for R2A is calculated as the total number of MS/MS spectra (minus NRP candidates) divided by the total number of candidates with false-positive hits. The false-positive rate was determined to be 0.83% for R2A, with only one false-positive hit (SI Appendix, Fig. S5). The media YMPG and grass seed had zero false positives detected, whereas the remaining media panel had between one and four false-positive hits.
In each instance where an NRP’s product-ion spectrum is generated from the spiked media extracts, iSNAP made a positive identification (Fig. 3C). However, in certain cases, some of the fermentation media had no product ions generated for polymyxin B (i.e., YPD, YMPG, TSB, and grass seed) and seglitide (i.e., YPD, TSB, LB, and CY). Poor extraction efficiency, compound instability, or ion suppression in these matrices is the likely origin. Importantly, these studies reveal that iSNAP conducts true dereplication in a nontargeted fashion for a series of structurally diverse NRPs from various complex matrices with average iSNAP processing times of under a minute for each LC-MS/MS data file. The 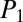 and
and 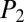 scores of the most representative candidates for each of the six NRP spike-in compounds and media candidates are plotted in Fig. 3B, with the LC-MS/MS results from the DDA analysis in Fig. 3C, highlighting the top scores across the media panels (SI Appendix, Tables S2 and S3). As multiple MS/MS scans can be generated for each NRP compound, at least one scan must have an NRP candidate scored above the
scores of the most representative candidates for each of the six NRP spike-in compounds and media candidates are plotted in Fig. 3B, with the LC-MS/MS results from the DDA analysis in Fig. 3C, highlighting the top scores across the media panels (SI Appendix, Tables S2 and S3). As multiple MS/MS scans can be generated for each NRP compound, at least one scan must have an NRP candidate scored above the and
and  thresholds for a dereplication to be made.
thresholds for a dereplication to be made.
In the NRP spiking studies, four low-scoring false positives were identified, with  and
and  scores of 27–34 and 25–34, respectively. The four false-positive hits were attributed to three compounds: esperin, empedopeptin, and tyrocidine C (SI Appendix, Fig. S6). Analysis of the detailed iSNAP report revealed that surfactin’s MS/MS spectrum was incorrectly matched to that of esperin (as revealed by retention time and fragment analysis). However, the false matching of surfactin to esperin can be rationalized, as they are structurally similar cyclic depsipeptides, with C13—C15 acyl chains and common monomer building blocks (l-Glu, d-Leu, and l-Asp), and esperin being within a [M] ± 1–Da mass range of surfactin. In comparing the
scores of 27–34 and 25–34, respectively. The four false-positive hits were attributed to three compounds: esperin, empedopeptin, and tyrocidine C (SI Appendix, Fig. S6). Analysis of the detailed iSNAP report revealed that surfactin’s MS/MS spectrum was incorrectly matched to that of esperin (as revealed by retention time and fragment analysis). However, the false matching of surfactin to esperin can be rationalized, as they are structurally similar cyclic depsipeptides, with C13—C15 acyl chains and common monomer building blocks (l-Glu, d-Leu, and l-Asp), and esperin being within a [M] ± 1–Da mass range of surfactin. In comparing the  and
and  scores, esperin’s are lower than that of surfactin. Analysis of surfactin’s iSNAP results and matching hits has also revealed that MS/MS spectral data may be useful in revealing analogs. In the case of empedopeptin and tyrocidine C, they were matched to analytes arising from two fermentation media (LB and CY).
scores, esperin’s are lower than that of surfactin. Analysis of surfactin’s iSNAP results and matching hits has also revealed that MS/MS spectral data may be useful in revealing analogs. In the case of empedopeptin and tyrocidine C, they were matched to analytes arising from two fermentation media (LB and CY).
Dereplicating Complex NRPs by Data-Dependent Acquisition: Kutzneride.
Kutznerides are among the most complex NRPs, composed entirely of nonproteinogenic amino acids, including several halogenated and oxidized groups (24). We sought to test whether iSNAP could dereplicate these from extracts in a nontargeted fashion using DDA and whether halogenated analogs could be detected (SI Appendix, section I.C). Supernatants from Kutzneria sp. 744 grown in complex Merlin Norkans medium were extracted with HP20 resin and subjected to solvent partitioning, with organic fractions subjected to LC-MS/MS analysis. Untargeted automated analysis by iSNAP dereplicated kutzneride 1 with matched fragment peaks (+837.3, 836.3, 743.2, and 609.2 m/z). The matched fragment ions can be correlated to cleavage at the lactone ring opening (−17, −18) and subsequent amide cleavages (−111 and −245 m/z) between the 6,7-dichloro-3a-hydroxy-1,2,3,3a,8,-8a hexahydropyrrolo[2,3-b]indole-2-carboxylic acid and the 3-hydroxyglutamine residue (+609.2 m/z). Positive identification of kutzneride 1 was achieved using iSNAP, with  and
and 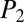 scores of 31.3 and 33.4, respectively.
scores of 31.3 and 33.4, respectively.
Frequently, in modern natural product discovery, simple variants of known NRP families are revealed in screening efforts. As such, it would therefore be useful to dereplicate “probable” variants of knowns (e.g., methylated, hydroxylated, or halogenated). We used the kutzneride producer to probe whether hypothetical variants of the known NRP could be detected using iSNAP. To promote the formation of a new kutzneride, we grew the producing strain in a medium containing bromide salts, replacing the original chloride ones. We anticipated that brominated kutznerides would be biosynthesized, as halogenases are known to accept either halide. As expected, the LC-MS/MS chromatogram of the resulting extract indicated the presence of the dibromo-kutzneride analog with a molecular weight of +942.1 [M+H]+ and the absence of kutzneride 1 (SI Appendix, Fig. S7). Analyzing this kutzneride fraction with iSNAP did not generate hits (despite a wide candidate window of [M] ± 150 Da), and did not reveal false positives by scoring with the original kutzneride 1. Adding the dibromo-kutzneride SMILES code to the database and rerunning the previous spectra revealed that four high-intensity fragment peaks were identified from the MS/MS spectra (+942.2, +925.2, +924.2, and +830.2 m/z), an analogous fragmentation sequence as seen for kutzneride 1, with  and
and  score values of 75.9 and 29.3, respectively (Dataset S2). These experiments highlight the utility of the iSNAP upload feature and how iSNAP can be used to reveal variants of known complex nonribosomal peptides.
score values of 75.9 and 29.3, respectively (Dataset S2). These experiments highlight the utility of the iSNAP upload feature and how iSNAP can be used to reveal variants of known complex nonribosomal peptides.
Probing the Utility of iSNAP to Interrogate Complex Extracts and Dereplicate Known Compounds.
Natural product screening campaigns often use bioactivity-guided fractionation to isolate active compounds. To explore how iSNAP may assist in dereplication within a bioactivity-guided fractionation campaign, we applied it to a screening of natural products for antistaphylococcal agents. One of the natural product extracts derived from an environmental bacillus produced a large zone of inhibition using agar-disk diffusion assays. The extract was subjected to LC-MS/MS and coordinate time-dependent fractionation into a 96-well plate. Bioactivity assays were conducted with the resulting 96-well plate with bioluminescent Staphylococcus aureus strain Xen29, and the LC/MS file was uploaded onto iSNAP (Fig. 4A).
Fig. 4.

Dereplicating bioactives from Bacillus sp. (A) IVIS bioluminescence imaging of crude fermentation extracts of Bacillus sp. against S. aureus (Xen29 strain), following HPLC fractionation. (B) LC-MS/MS chromatogram of Bacillus sp. extract. Total-ion chromatograms (TICs) for MS and MS(n) are shown; bioactive wells are highlighted. (C) iSNAP dereplication results identifying a series of tyrocidines from the inputted LC-MS/MS data file in .mzXML format. Rt, retention time. (D) Extracted-ion chromatogram (EIC) of the five dereplicated tyrocidines.
In the analysis of a crude pellet extract, a total of 1,964 MS/MS scans was acquired over a 75-min LC-MS/MS run and, of these, 45 had  and
and  scores above the threshold cutoffs and 41 were for members of the tyrocidine family (25). Collectively, these 41 tyrocidine matches correlated with wells D1–6, D8, and E1, which all lacked S. aureus growth (SI Appendix, Figs. S8–S10). iSNAP scoring revealed high
scores above the threshold cutoffs and 41 were for members of the tyrocidine family (25). Collectively, these 41 tyrocidine matches correlated with wells D1–6, D8, and E1, which all lacked S. aureus growth (SI Appendix, Figs. S8–S10). iSNAP scoring revealed high  and
and  scores for tyrocidine A (
scores for tyrocidine A ( = 85,
= 85,  = 43.3), B (
= 43.3), B (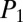 = 85.8,
= 85.8, 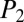 = 61.6), C (
= 61.6), C (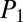 = 84.3,
= 84.3, 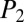 = 44.3), D (
= 44.3), D (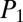 = 68.1,
= 68.1,  = 41.5), and E (
= 41.5), and E ( = 72.9,
= 72.9,  = 55.0) from their double-protonated precursor masses of +636.2, +655.8, +675.3, +686.8, and +628.2 m/z, respectively (Fig. 4 B–D). High-resolution mass determination of the dereplicated candidates using LTQ-Orbitrap HRS-FTMS measurements revealed the candidates were within ∼0.6–4 ppm of the tyrocidines (SI Appendix, Table S4). Further comparison of the MS/MS fragmentation pattern of authentic tyrocidines with the candidates laddering b ions, acylium ions (SI Appendix, Figs. S11 and S12), provided confirmatory evidence (26). The positive identification of each tyrocidine analog, and distinguishing between them, with increased
= 55.0) from their double-protonated precursor masses of +636.2, +655.8, +675.3, +686.8, and +628.2 m/z, respectively (Fig. 4 B–D). High-resolution mass determination of the dereplicated candidates using LTQ-Orbitrap HRS-FTMS measurements revealed the candidates were within ∼0.6–4 ppm of the tyrocidines (SI Appendix, Table S4). Further comparison of the MS/MS fragmentation pattern of authentic tyrocidines with the candidates laddering b ions, acylium ions (SI Appendix, Figs. S11 and S12), provided confirmatory evidence (26). The positive identification of each tyrocidine analog, and distinguishing between them, with increased 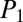 and
and  scores highlights the selectivity of iSNAP and the detection of low-abundance analogs (i.e., tyrocidine E: relative abundance is 2%).
scores highlights the selectivity of iSNAP and the detection of low-abundance analogs (i.e., tyrocidine E: relative abundance is 2%).
The remaining four MS/MS spectral matches were identified as belonging to three compounds (SI Appendix, Fig. S13): capreomycin IB ( = 28,
= 28,  = 39.4), emerimicin III (
= 39.4), emerimicin III ( = 28.6,
= 28.6,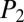 = 27.9), and nepadutant (
= 27.9), and nepadutant ( = 29.7,
= 29.7, 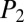 = 57.9). Of note, however, upon further investigation, capreomycin and nepadutant had only four matched fragments, with only one high-intensity peak contributing significantly to the scoring scheme. Given these findings, we suggest that MS/MS spectra with low matched peaks should be further examined for positive dereplication (SI Appendix, Figs. S14 and S15).
= 57.9). Of note, however, upon further investigation, capreomycin and nepadutant had only four matched fragments, with only one high-intensity peak contributing significantly to the scoring scheme. Given these findings, we suggest that MS/MS spectra with low matched peaks should be further examined for positive dereplication (SI Appendix, Figs. S14 and S15).
Discussion
Nonribosomal peptides comprise a highly privileged section of chemical space, which is diverse due to varied use of over 500 building blocks and molecular architectures (cyclic, linear, branched) and modifications and fusions with other chemical classes (i.e., polyketides). Critical to new nonribosomal peptide natural product discovery is efficient dereplication within complex extracts in a nondirected fashion. iSNAP is a strategy to achieve this, and we have shown that it is applicable to a spectrum of nonribosomal peptide types: linear, cyclic, and branched (linear and cyclic portions) and those with highly modified subunits (e.g., halogenation), mixed backbone linkages (e.g., lactones, N-methylated amides), and polyketide extensions. False-positive scores were evaluated in a number of matrices and shown to be relatively insignificant in all of the media tested. Through this design, we have created a platform that is robust enough to tackle a battery of differing medium compositions and dereplicated the correct NRP at low-nanogram levels from complex matrices in an untargeted fashion using a relatively low resolution mass spectrometer. Whereas the current version of iSNAP dereplicates, an enhanced ability may be realized by isotopic labeling. The design of iSNAP and its flexible use of informatic databases of natural product SMILES codes may provide a mechanism to couple needs of dereplication with the discovery potential of novel substances revealed by microbial genomic sequencing.
Methods
Details relating to the materials used, bacterial strains, culture conditions, isolation and purification of kutznerides and tyrocidines, fermentation medium-screening conditions and NRP compound spiking, mass spectrometry, MS/MS and LC-MS/MS experiments, access to the data files, and a user guide for iSNAP can be found in SI Appendix, section I. The iSNAP online research tool is available at www-novo.cs.uwaterloo.ca:8180/isnap.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Suzanne Osborne and Dr. Brian Coombes for IVIS imaging as well the Centre for Microbial Chemical Biology at McMaster University. This work was funded by NSERC discovery Grants RGPIN 371576-2009 (to N.A.M.) and RGPIN 238748-2011 (to B.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The mass spectrometry data for nonribosomal peptides reported in this paper has been deposited on iSNAP website, www-novo.cs.uwaterloo.ca:8180/isnap/data/iSNAP_PNAS_data.rar.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1206376109/-/DCSupplemental.
References
- 1.Schwarzer D, Finking R, Marahiel MA. Nonribosomal peptides: From genes to products. Nat Prod Rep. 2003;20(3):275–287. doi: 10.1039/b111145k. [DOI] [PubMed] [Google Scholar]
- 2.Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: Logic, machinery, and mechanisms. Chem Rev. 2006;106(8):3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 3.Caboche S, et al. NORINE: A database of nonribosomal peptides. Nucleic Acids Res. 2008;36(Database issue):D326–D331. doi: 10.1093/nar/gkm792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li JW, Vederas JC. Drug discovery and natural products: End of an era or an endless frontier? Science. 2009;325(5937):161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 5.McAlpine JB. Advances in the understanding and use of the genomic base of microbial secondary metabolite biosynthesis for the discovery of new natural products. J Nat Prod. 2009;72(3):566–572. doi: 10.1021/np800742z. [DOI] [PubMed] [Google Scholar]
- 6.Corre C, Challis GL. New natural product biosynthetic chemistry discovered by genome mining. Nat Prod Rep. 2009;26(8):977–986. doi: 10.1039/b713024b. [DOI] [PubMed] [Google Scholar]
- 7.Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 8.Finking R, Marahiel MA. Biosynthesis of nonribosomal peptides. Annu Rev Microbiol. 2004;58:453–488. doi: 10.1146/annurev.micro.58.030603.123615. [DOI] [PubMed] [Google Scholar]
- 9.Harrison AG, Young AB, Bleiholder C, Suhai S, Paizs B. Scrambling of sequence information in collision-induced dissociation of peptides. J Am Chem Soc. 2006;128(32):10364–10365. doi: 10.1021/ja062440h. [DOI] [PubMed] [Google Scholar]
- 10.Eckart K. Mass spectrometry of cyclic peptides. Mass Spectrom Rev. 1994;13(1):23–55. [Google Scholar]
- 11.Bleiholder C, et al. Sequence-scrambling fragmentation pathways of protonated peptides. J Am Chem Soc. 2008;130(52):17774–17789. doi: 10.1021/ja805074d. [DOI] [PubMed] [Google Scholar]
- 12.Liu WT, et al. Interpretation of tandem mass spectra obtained from cyclic nonribosomal peptides. Anal Chem. 2009;81(11):4200–4209. doi: 10.1021/ac900114t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohimani H, et al. Sequencing cyclic peptides by multistage mass spectrometry. Proteomics. 2011;11(18):3642–3650. doi: 10.1002/pmic.201000697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ng J, et al. Dereplication and de novo sequencing of nonribosomal peptides. Nat Methods. 2009;6(8):596–599. doi: 10.1038/nmeth.1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kersten RD, et al. A mass spectrometry-guided genome mining approach for natural product peptidogenomics. Nat Chem Biol. 2011;7(11):794–802. doi: 10.1038/nchembio.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci. 1988;28:31–36. [Google Scholar]
- 17.Paizs B, Suhai S. Fragmentation pathways of protonated peptides. Mass Spectrom Rev. 2004;24(4):508–548. doi: 10.1002/mas.20024. [DOI] [PubMed] [Google Scholar]
- 18.Chamrad DC, et al. Evaluation of algorithms for protein identification from sequence databases using mass spectrometry data. Proteomics. 2004;4(3):619–628. doi: 10.1002/pmic.200300612. [DOI] [PubMed] [Google Scholar]
- 19.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5(11):976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 20.Zhang J, et al. PEAKS DB: De novo sequencing assisted database search for sensitive and accurate peptide identification. Mol Cell Proteomics. 2012;11(4) doi: 10.1074/mcp.M111.010587. M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Razumovskaya J, et al. A computational method for assessing peptide- identification reliability in tandem mass spectrometry analysis with SEQUEST. Proteomics. 2004;4(4):961–969. doi: 10.1002/pmic.200300656. [DOI] [PubMed] [Google Scholar]
- 22.Govaerts C, et al. Mass spectrometric fragmentation of cyclic peptides belonging to the polymyxin and colistin antibiotics studied by ion trap and quadrupole/orthogonal-acceleration time-of-flight technology. Rapid Commun Mass Spectrom. 2002;16(9):823–833. doi: 10.1002/rcm.644. [DOI] [PubMed] [Google Scholar]
- 23.Vaisar T, Urban J. Gas-phase fragmentation of protonated mono-N-methylated peptides. Analogy with solution-phase acid-catalyzed hydrolysis. J Mass Spectrom. 1998;33(6):505–524. [Google Scholar]
- 24.Broberg A, Menkis A, Vasiliauskas R. Kutznerides 1–4, depsipeptides from the actinomycete Kutzneria sp. 744 inhabiting mycorrhizal roots of Picea abies seedlings. J Nat Prod. 2006;69(1):97–102. doi: 10.1021/np050378g. [DOI] [PubMed] [Google Scholar]
- 25.Barber M, et al. An investigation of the tyrothricin complex by tandem mass spectrometry. Int J Mass Spectrom Ion Process. 1992;122:143–151. [Google Scholar]
- 26.Tang X, Thibault P, Boyd R. Characterisation of the tyrocidine and gramicidin fraction of the tyrothricin complex from Bacillus brevis using liquid chromatography and mass spectrometry. Int J Mass Spectrom Ion Process. 1992;122:153–179. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.