

Abstract
The expansion of a polyglutamine (polyQ) tract in the N-terminal region of ataxin-7 (atxn7) is the causative event in spinocerebellar ataxia type 7 (SCA7), an autosomal dominant neurodegenerative disorder mainly characterized by progressive, selective loss of rod-cone photoreceptors and cerebellar Purkinje and granule cells. The molecular and cellular processes underlying this restricted neuronal vulnerability, which contrasts with the broad expression pattern of atxn7, remains one of the most enigmatic features of SCA7, and more generally of all polyQ disorders. To gain insight into this specific neuronal vulnerability and achieve a better understanding of atxn7 function, we carried out a functional analysis of this protein in the teleost fish Danio rerio. We characterized the zebrafish atxn7 gene and its transcription pattern, and by making use of morpholino-oligonucleotide-mediated gene inactivation, we analysed the phenotypes induced following mild or severe zebrafish atxn7 depletion. Severe or nearly complete zebrafish atxn7 loss-of-function markedly impaired embryonic development, leading to both early embryonic lethality and severely deformed embryos. More importantly, in relation to SCA7, moderate depletion of the protein specifically, albeit partially, prevented the differentiation of both retina photoreceptors and cerebellar Purkinje and granule cells. In addition, [1–232] human atxn7 fragment rescued these phenotypes showing strong function conservation of this protein through evolution. The specific requirement for zebrafish atxn7 in the proper differentiation of cerebellar neurons provides, to our knowledge, the first in vivo evidence of a direct functional relationship between atxn7 and the differentiation of Purkinje and granule cells, the most crucial neurons affected in SCA7 and most other polyQ-mediated SCAs. These findings further suggest that altered protein function may play a role in the pathophysiology of the disease, an important step toward the development of future therapeutic strategies.
Introduction
SCA7 is an autosomal dominant neurodegenerative disorder caused by the expansion of a translated CAG repeat in the SCA7/ataxin-7 gene, leading to expansion of a polyQ tract located in the N-terminal region of the encoded protein, ataxin-7 (atxn7) [1]. SCA7 thus belongs to the family of polyQ expansion disorders, also named polyQ diseases, a group of neurodegenerative disorders comprising spinobulbar muscular atrophy (SBMA) [2], Huntington’s disease (HD) [3], dentatorubral-pallidoluysian atrophy (DRPLA) [4] and spinocerebellar ataxia (SCA) 1, 2, 3, 6, 7, and 17 [1], [5]–[13].
All polyQ diseases are characterized by progressive, selective degeneration of distinct, albeit disease-specific, neuronal populations. Vulnerable neurons in SCA7 include Purkinje cells, a neuronal population that is affected in most polyQ-mediated SCAs, excepted SCA3 [14], and several other neuronal populations such as cerebellar granule cells, neurons of inferior olive and cranial nerve nuclei, and also rod-cone photoreceptors, a cell population that is spared in other SCA types [15]–[17]. Beside this disease-specific neuronal vulnerability, all polyQ disorders share several common features: (i) progressive neuronal dysfunction and degeneration, (ii) expression of the disease phenotype when the size of the polyCAG/polyQ expansion reaches a precise threshold, which varies according to the gene, (iii) a strong negative correlation between age at onset and size of the polyQ tract, (iv) instability of the CAG repeat during transmission, with a strong tendency to expansion, resulting in an effect called anticipation (cf. [18]–[22]). Paradoxically, apart from their polyQ tract, the disease proteins display neither structural nor functional similarities.
Atxn7 is a subunit of a multiprotein complex, the Spt-Ada-Gcn5-acetyltransferase (SAGA) complex, which is involved in histone acetylation and transcription regulation [23]–[26]. A body of work on several mouse models has demonstrated that rod-cone photoreceptor degeneration in SCA7 is at least partially a consequence of interference of polyQ-expanded atxn7 with CRX, a homeodomain protein that plays a key role for proper transactivation of photoreceptor genes [27]–[30]. By contrast, the molecular and cellular bases of the selective vulnerability of other neuronal populations, such as cerebellar Purkinje cells, remain poorly understood. In mammals, challenging the specific neuronal loss, the atxn7 gene is, like almost all the genes underlying polyQ disorders, expressed in numerous neuronal populations, including neurons, which are spared in SCA7, but also in a large set of non-neuronal tissues, [16], [31], [32].
To further address this issue, a better understanding of the normal function of atxn7 could provide important insights. However, though the group of Zoghbi generated an atxn7 KO mice line [33], the phenotype of these mice has not yet been described. Here, we show that the D. rerio atxn7 gene was broadly expressed throughout development from the one-cell stage onward, although in adults it was transcribed in several neuronal populations, including granule, but not Purkinje cells. Loss of function experiments demonstrated that severe depletion of zebrafish atxn7 impaired early development, leading to embryonic lethality combined with highly deformed embryos. Significantly, in relation to the disease, moderate depletion of the protein specifically compromised the differentiation of photoreceptors and cerebellar Purkinje and granule cells, the main crucial neuronal populations that are affected in SCA7. These findings lend new insight into the specific vulnerability of cerebellar neurons in SCA7 and also suggest that altered ataxin-7 function may play a role in the disease process.
Results
Characterization of the Zebrafish atxn7 gene
To identify the Danio rerio atxn7 gene, we performed a blast analysis of the release Zv9 of the zebrafish genome sequence for genes showing sequence similarities with human atxn7. Our results identified 4 atxn7 paralogs in zebrafish (Figure S1A), which are expressed in 24, 48 and 72 hours post-fecundation (hpf) embryos (Figure S1B and Figure 1G). However, molecular phylogeny deduced from ClustalW2 analysis showed that a single and bona fide ortholog of the human atxn7 (hatxn7) gene is found in D. rerio (Figure S1A). This gene is referred to hereafter as zebrafish atxn7 (zatxn7) (Ensembl ENSDARG00000074804). Sequencing of RT-PCR fragments encompassing the complete protein-coding region and part of the 5′ and 3′ UTRs showed that the zebrafish atxn7 mRNA comprises 12 exons and encodes an 866 amino acid protein (Figure S1C) referred to hereafter as zebrafish atxn7. At the amino acid levels, the protein displayed 51.1 and 49.8% identities and 65.9 and 64.6% similarities compared with human and mouse atxn7, respectively. RT-PCR demonstrated that zebrafish atxn7 transcripts were expressed at low levels in 1-, 4- to 8- and 16- to 64-cell embryos and at higher levels in embryos aged 10, 24, 48 and 72 hpf (Figure 1G). In dissected adult tissues, zebrafish atxn7 RNAs were found in the brain, cerebellum, spinal cord, eye and non-neuronal tissues (Figure 1G). RNA in situ hybridization revealed a uniform accumulation of transcripts in 4-cell and 3, 8 and 16 hpf embryos (Figure 1A–1D). High levels of zebrafish atxn7 transcription were detected in the brain of 24 hpf embryos (Figure 1E). In the dissected brain of 120 hpf embryos, zebrafish atxn7 mRNAs were found in various regions, including the anterior region of the telencephalon, optic tectum and cerebellum (Figure 1F). On adult brain sections, zebrafish atxn7 mRNAs accumulated in several neuronal populations (Figure 2A) including cerebellar granule cells, but not Purkinje cells (Figure 2B).
Figure 1. Transcription of the zebrafish atxn7 gene during development.

(A-F) In situ detection of zebrafish atxn7 transcripts on either whole mount embryos at the four-cell stage (A), or 3 (B), 8 (C), 16 (D), and 48 hpf (E) or dissected brain of 120 hpf embryos (F). (I) RT-PCR analysis of zebrafish atxn7 transcript accumulation in 1- (1 c), 4- to 16- (4–16 c), and 8- to 64-cell embryos (8–64 c), or 10 (10 h), 24 (24 h), 48 (48 h) and 72 to 96 hpf embryos (3–4 d) and dissected adult brain (Brain), cerebellum (Cer), spinal cord (SC), eye (Eye) and remaining tissues (Ad-brain). RT–PCR for β-actin is shown as a positive control. Abbreviations: Cer, cerebellum; MO, medulla oblongata; TeO, tectum opticum; Tel, telencephalon.
Figure 2. Transcription of the zebrafish atxn7 gene in adult brain sections.

In situ detection of zebrafish atxn7 transcripts on midsagittal section of adult brain (A). Dorsal view of the adult zebrafish brain showing the position level of the parasagittal section shown in A (A’). A magnified view of the section showed in panel A (B). Schematic representation of zebrafish cerebellum (C). Anterior is to the left. Abbreviations: CC, crista cerebellaris; CCe, corpus cerebelli; CM, corpus mamillare; Dm, medial zone of dorsal telencephalic area; Hd, dorsal zone of the periventricular hypothalamus; LCa, lobus caudalis cerebelli; LX, lobus vagus; MON, medial octavolateralis nucleus; PGZ, periventricular grey zone; PR, posterior recess of diencephalic ventricle; TPp, periventricular nucleus of the posterior tuberculum; Val, lateral division of valvula cerebelli; Vam, medial division of valvula cerebellaris; Vv, ventral nucleus of the ventral telencephalon.
Zebrafish atxn7 Plays an Essential Role for Embryo Development
To gain insight into zebrafish atxn7 function, we made use of morpholino-oligonucleotide (MO)-mediated gene knockdown to investigate the phenotypes caused by various levels of zebrafish atxn7 depletion in embryos. First, we microinjected wild-type zebrafish embryos of the AB strain (referred to below as morphants) with MOzatxn7AUG, a MO designed to inhibit translation of zebrafish atxn7 mRNA (Figure 3A). Injection of 1 pmol MOzatxn7AUG induced high percentages of embryonic lethality, with 24.8% and 60% of morphants dying before 10 and 24 hpf, respectively (n = 165) (Table S1). In addition, 78% of 1 pmol MOzatxn7AUG morphants that were still alive at 24 hpf (n = 66) displayed severe developmental defects, including impaired head or tail differentiation or both (Fig. 3E). These phenotypes were not observed in either non-injected siblings or morphants that had received 1 pmol 5 mismatch-containing MOzatxn7AUG (mmMOzatxn7AUG) (Figure 3C and 3D), suggesting an essential requirement for zebrafish atxn7 in proper embryonic development.
Figure 3. Morpholino-mediated inactivation of zebrafish atxn7 impairs embryonic development.

Schematic representation of the zebrafish atxn7 gene showing exons 1 (ex 1), 3 to 5 (ex 3–5) and 12 (ex 12) (black boxes), location of MOzatxn7AUG and MOzatxn7SPL (red lines) and position of oligonucleotides (black arrows) used for RT-PCR analysis of MOzatxn7SPL-mediated inhibition of zebrafish atxn7 intron 4 splicing (black arrows) (A). Untranslated exonic regions and intronic sequences are depicted as empty boxes and single lines, respectively. RT-PCR analysis of zebrafish atxn7 intron 4 splicing in non-injected (NI) and morphant embryos that had received 0.3, 0.6 and 1 pmol MOzatxn7SPL (B). RT-PCR for β-actin is shown as a positive control. Phenotypes of 48 hpf wild-type embryo (C), and age-matched 1 pmol mmMOzatxn7AUG (D), 0.3 pmol MOzatxn7AUG (G) and 0.3 pmol MOzatxn7SPL morphants (H). Phenotypes of 24 hpf 1 pmol MOzatxn7AUG (E) and 1 pmol MOzatxn7SPL morphants (F).
To assess the specificity of the phenotypes observed in MOzatxn7AUG morphants and also test whether maternal transcripts underlie the requirement for zebrafish atxn7 in early development, embryos of the wild-type AB strain were microinjected with a second MO targeting the donor splice site of zebrafish atxn7 intron 4 (MOzatxn7SPL) (Figure 3A). Following microinjection of 1 pmol MOzatxn7SPL, morphants displayed high levels of embryonic lethality, with 27.6% and 61% of injected embryos dying before 10 and 24 hpf, respectively (n = 141) (Table S1). Also, 76% of 1 pmol MOzatxn7SPL morphants that were still alive at 24 hpf (n = 55) showed developmental defects similar to those seen in 1 pmol MOzatxn7AUG morphants (Figure 3F). To further confirm the specificity of MOzatxn7SPL and estimate the levels of zebrafish atxn7 depletion in the corresponding morphants, we carried out a RT-PCR analysis of zebrafish atxn7 intron 4 splicing in 24 hpf embryos that had received 0.3, 0.6 or 1 pmol MOzatxn7SPL. RT-PCR experiments were performed using a pair of primers designed to amplify a cDNA fragment encompassing zebrafish atxn7 exons 3 to 5 (Figure 3A). The splice-blocking activity of MOzatxn7SPL was evidenced by the dose-dependent inhibition of zebrafish atxn7 intron 4 splicing in MOzatxn7SPL morphants (Figure 3B). We note that a nearly complete inhibition of intron 4 splicing was observed in 1 pmol MOzatxn7SPL morphants.
Mild Zebrafish atxn7 Depletion Compromises Photoreceptor Differentiation
While embryos that had received 0.3 pmol MOzatxn7AUG or 0.3 pmol MOzatxn7SPL did not display obvious developmental defects nor excessive lethality (Table S1), 15% (n = 113) and 12% (n = 125) of these embryos showed partially depigmented retina, respectively (Figure S2). To further investigate the requirement for zebrafish atxn7 in retina differentiation, eye sections of 3 days post-fecundation (dpf) 0.3 pmol MOzatxn7SPL morphants were analysed by immunocytochemistry using an anti-rhodopsin antibody. In all the retinas analysed (n = 8), whatever their pigmentation, we observed a marked disorganization of the photoreceptor layer (Figure 4C and 4D). These phenotypes were absent in both age-matched non-injected siblings (n = 6) (Figure 4A and 4B) and 1 pmol mmMOzatxnAUG morphants (n = 7) (Figure 4E and 4F). Importantly, rhodopsin immunostaining also revealed that 0.3 pmol MOzatxnSPL morphants displayed a marked reduction in the number of photoreceptors (Figure 4C and 4D), demonstrating an essential requirement for zebrafish atxn7 in the full differentiation of retina photoreceptors.
Figure 4. Partial zebrafish atxn7 depletion impairs photoreceptor differentiation.

DAPI staining (A, C, and E) and rhodopsin immunostaining (B, D, and F) of eye cryosections of 48 hpf wild type embryo (A and B) and age-matched 0.3 pmol MOzatxn7SPL (C and D) and 1 pmol mmMOzatxn7AUG morphant embryos (E and F).
Moderate Zebrafish atxn7 Depletion Specifically Impairs Purkinje and Granule Cell Differentiation
Because cerebellar neurons are particularly prone to degeneration in SCA7, we next tested whether partial depletion of zebrafish atxn7 in 0.3 pmol MOzatxn7SPL morphants induced an impaired differentiation of cerebellar neurons. First, we determined the number of Purkinje cells on consecutive serial optic sections of brains dissected from 5 dpf 0.3 pmol MOzatxn7SPL morphants by immunocytochemistry using an anti-paravalbumin-7 (Pvalb7) antibody that specifically labels these neurons [34]. In 5 dpf embryos that had received 1 pmol mmMOzatxnAUG, we detected 191+/−8 Purkinje cells (n = 7) (Figure 5A’ and 5C). By contrast, the number of Pvalb7-expressing cells was very significantly reduced to 79+/−25 (n = 8, p<0.0001) in 5 dpf embryos that had received 0.3 pmol MOzatxn7SPL (Figure 5B’ and 5D), suggesting a requirement for zebrafish atxn7 in Purkinje cell differentiation. To further investigate the physiology of Purkinje cells in 0.3 pmol MOzatxnSPL morphants, we analysed the expression of three additional proteins that have been shown to accumulate to high levels in zebrafish Purkinje cells, namely zebrin II, carbonic anhydrase 8 (Ca8) and retinoic-related orphan receptor α (RORα) [34]. In brains of 5 dpf 0.3 pmol MOzatxn7SPL morphants, we observed a marked decrease in the number of cells expressing RORα (Fig. S3B and S3B’’) compared with 5 dpf embryos that had received 1 pmol mmMOzatxnAUG (Figure S3A and S3A’’). Similarly, the number of cells expressing either zebrin II (Figure 6A and 6D) or Ca8 (not shown) was markedly reduced in 0.3 pmol MOzatxn7SPL morphants, compared with embryos that received 1 pmol mmMOzatxn7AUG. These results confirm that differentiation of Purkinje cells was severely compromised following moderate depletion of zebrafish atxn7. In addition to photoreceptor and Purkinje cell degeneration, SCA7 patients also show progressive loss of granule cells [18], [31], a defect also observed in SCA7 mouse models [35] and in vitro [36]. To test whether moderate zebrafish atxn7 depletion also affects the differentiation of granule cells, dissected brains of 0.3 pmol MOzatxn7SPL and 1 pmol mmMOzatxnAUG morphants were analysed by immunocytochemistry using an antibody directed against Vglut1, a vesicular glutamate transporter, which is expressed at high levels in zebrafish granule cells [34]. Our results demonstrate a marked decrease in the number of cells expressing Vglut1 in 5 dpf 0.3 pmol MOzatxn7SPL morphants (Figure 5B’’ and 5D’) compared with age-matched controls that received 1 pmol mmMOzatxn7AUG (Figure 5A’’ and 5C’), showing that granule cell differentiation also was partially compromised following moderate zebrafish atxn7 depletion.
Figure 5. Moderate zebrafish atxn7 depletion impairs the differentiation of cerebellar neurons.

Dorsal views of dissected brains from 5 dpf 1 pmol mmMOzatxnAUG (A, A’, A’’, A’’’, C, C’ and C’’) and 0.3 pmol MOzatxn7SPL morphants (B, B’, B’’, B’’’, D, D’ and D’’). DAPI staining (A and B), Pav7 immunostaining of Purkinje cells (A’, B’, C and D) and Vglut1 immunostaining of granule cells (A’’, B’’, C’ and D’). Anterior is to the left. Enlarged views of the brains showed in A’ (C), A’’ (C’), A’’’ (C’’), B’ (D), B’’ (D’), and B’’’ (D’’). Merge images of the photographs A’ and A’’ (A’’’), B’ and B’’ (B’’’), C and C’ (C’’), and D and D’ (D’’).
Figure 6. Partial zebrafish atxn7 depletion impairs the differentiation of cerebellar neurons.
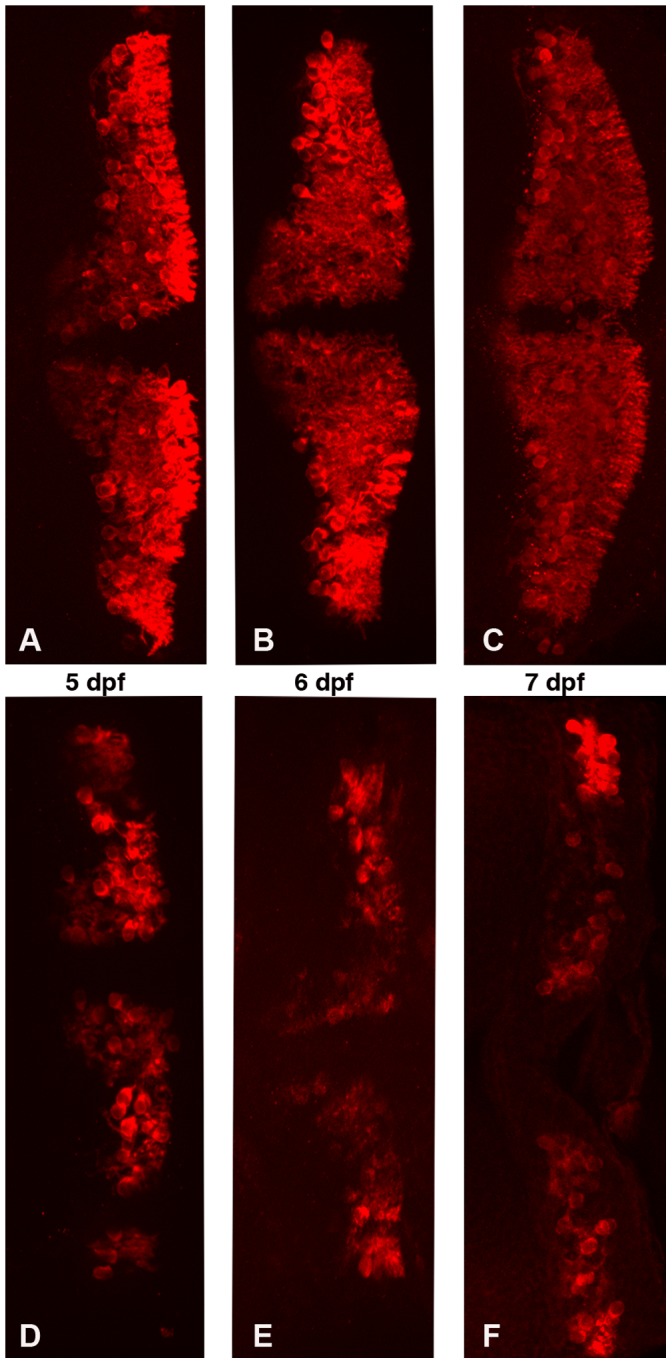
Dorsal views of dissected brains from 5 (A and D), 6 (B and E), and 7 dpf (C and F) 1 pmol mmMOzatxn7AUG (A, B, and C) and 0.3 pmol MOzatxn7SPL morphants (D, E, and F) immunostained with an anti-zebrin II antibody, which specifically labels Purkinje cells.
As the reduced number of Purkinje cells observed in 5 dpf 0.3 pmol MOzatxn7SPL morphants might be caused by delayed differentiation of these neurons, we compared the number of zebrin II-expressing cells in dissected brains of 1 pmol mmMOzatxnAUG and 0.3 pmol MOzatxn7SPL morphants at 5, 6 and 7 dpf. In embryos injected with 1 pmol mmMOzatxnAUG and aged 5, 6, and 7 dpf, we observed 183+/−11 (n = 5), 204+/−7 (n = 6), and 244+/−13 (n = 5) zebrin II-expressing cells, respectively (Figure 6A–6C). By contrast, in 0.3 pmol MOzatxn7SPL morphants, we observed a highly significant decrease in the number of Purkinje cells at 5 (90+/−18, n = 5, p<0.0001), 6 (104+/−25, n = 6, p<0.0001), and 7 dpf (143+/−23, n = 6, p<0.0001) (Figure 6D–6F), strongly suggesting that partial zebrafish atxn7 depletion partly compromised, but did not slow down Purkinje cell differentiation.
We could not rule out the possibility that the reduced number of cerebellar neurons observed in 5 dpf 0.3 pmol MOzatxn7SPL morphants was caused by apoptosis of these cells soon after their differentiation. Accordingly, to assess whether moderate depletion of zebrafish atxn7 induced increased levels of brain neuron apoptosis, we performed a TUNEL assay on dissected brains of 5 dpf 1 pmol mmMOzatxnAUG (n = 7) and 0.3 pmol MOzatxnSPL morphants (n = 6), and age-matched DNase-treated wild-type controls (n = 6). While numerous labelled cells scattered throughout the brain were detected following DNase treatment (Figure S4B), a low and roughly similar number of apoptotic cells was detected in both 1 pmol mmMOzatxnAUG (Figure S4D) and 0.3 pmol MOzatxnSPL morphants (Figure S4F), demonstrating that increased levels of apoptosis was not the cause of the reduced number of cerebellar neurons observed in 0.3 pmol MOzatxn7SPL morphants.
[1–232] Fragment of Human atxn7 can Compensate Partial Loss of Zebrafish atxn7
To further confirm that loss of both photoreceptors and cerebellar neurons observed in 0.3 pmol MOzatxn7SP L morphants (Figure 4 and Figure 5) was caused by partial depletion of zebrafish atxn7, and also to test whether human atxn7 could compensate for loss of function of the zebrafish protein, wild-type embryos were injected with a mixture comprising 0.3 pmol MOzatxn7SPL and 2 fmol of in vitro transcribed human atxn7 mRNA encoding truncated human atxn7 (ATXN7T: amino acids 1–232), which has successfully been used to model SCA7 in vivo in Drosophila [37] and is close to the shortest human atxn7 fragment found in human brain or transgenic SCA7 mice [38]. In all the embryos that received 0.3 pmol MOzatxn7SPL and 2 fmol truncated human atxn7 mRNA encoding [1–232] N-Terminal fragment of the protein (n = 78), retina pigmentation was similar to that observed in non-injected siblings or 1 pmol mmMOzatxn7AUG morphants (not shown). Also, in all the retinas analysed (n = 7), rhodopsin immunostaining of eye sections revealed that differentiation of photoreceptors was similar in 0.3 pmol MOzatxn7SPL morphants that also received 2 fmol truncated human atxn7 mRNA (Figure 7C–7C’’), compared with non-injected embryos (data not shown) or 1 pmol mmMOzatxnAUG morphants (Figure 7A–7A’’). Next, we investigated whether [1–232] fragment of human atxn7 was also able to rescue cerebellar neuron differentiation defects observed in 0.3 pmol MOzatxn7SPL morphants (Figure 5). In the dissected brains of morphants that received 0.3 pmol MOzatxn7SPL and 2 fmol truncated human atxn7 RNA (n = 6), we detected 186+/−12 (n = 6) Purkinje cells as revealed by Pvalb7 immunostaining (Figure 8C and 8C’’), a number similar to that observed in 1 pmol mmMOzatxnAUG morphants (191+/−8, n = 7, p = 0.4) (Figure 8A and 8A’’). Similarly, Vglut1 immunostaining demonstrated that the number of granule cells seen in dissected brains of 0.3 pmol MOzatxn7SPL morphants that also received 2 fmol truncated human atxn7 RNA (Fig. 8C’ and 8C’’) was similar to that observed in 1 pmol mmMOzatxn7AUG morphants (Fig. 8A’ and 8A’’). Taken together, these data demonstrate that [1–232] N-terminal fragment of human atxn7 was able to fully rescue cerebellar neuron and photoreceptor differentiation defects caused by partial depletion of the zebrafish protein.
Figure 7. [1–232] N-terminal fragment of human atxn7 can rescue photoreceptor differentiation defect in 0.3 pmol MOzatxn7SPL morphant.

Rhodopsin immunostaining (A, B, and C) and DAPI staining (A’, B’, and C’) of eye cryosections of 48 hpf 1 pmol mmMOzatxn7AUG (A, A’ and A’’) and 0.3 pmol MOzatxn7SPL morphants (B, B’ and B’’) and age matched 0.3 pmol MOzatxn7SPL morphant co-injected with 2 fmol human atxn7 mRNA (C, C’ and C’’). Merge images of the photographs A and A’ (A’’), B and B’ (B’’), and C and C’ (C’’).
Figure 8. [1–232] human atxn7 fragment can rescue differentiation defects of cerebellar neurons in 0.3 pmol MOzatxn7SPL morphant.

Dorsal views of dissected brains from 5 dpf 1 pmol mmMOzatxnAUG (A, A’ and A’’) and 0.3 pmol MOzatxn7SPL morphants (B, B’ and B’’) and age-matched 0.3 pmol MOzatxn7SPL morphant co-injected with human atxn7 mRNA (2 fmol) (C, C’ and C’’). Pav7 immunostaining of Purkinje cells (A, B and C) and Vglut1 immunostaining of granule cells (A’, B’ and C’). Anterior is to the left. Merge images of the photographs A and A’ (A’’), B and B’ (B’’), and C and C’ (C’’).
Partial Zebrafish atxn7 Depletion does not Affect Overall Brain, Spinal Cord and Muscle Development
To determine whether the differentiation of other brain neurons and/or glial cells were also compromised following mild zebrafish atxn7 depletion, we analysed the accumulation pattern of both the glial acidic fibrillary protein (GFAP), a protein accumulated at high levels in all glial cells [39], and HuC, a pan-neuronal protein, which is expressed in all brain neurons [40], in dissected brains of 5 dpf 0.3 pmol MOzatxn7SPL morphants (n = 5) and age-matched control embryos (n = 6). No differences in either the size and organization of brain regions or the accumulation pattern of the two proteins could be detected between 5 dpf wild-type controls (Figure S5A-S5C) and age-matched 0.3 pmol MOzatxn7SPL morphants (Figure S5D-S5F), suggesting that overall brain organization was not impaired following moderate zebrafish atxn7 loss of function.
We also analysed whether partial depletion of zebrafish atxn7 caused defects in spinal cord development, motor neuron differentiation or body muscle structure and/or organization. We first made use of the Tg[NBT:MAPT-GFP]zc1 transgenic line to visualize the spinal cord and motor neuron axons [41]. Following injection of 0.3 pmol MOzatxn7SPL in embryos of the Tg[NBT:MAPT-GFP]zc1 line, we observed that both spinal cord anatomy and motor neuron axon arborisation were fully similar in 0.3 pmol MOzatxn7SPL morphants (n = 6) (Figure S6B) compared with non-injected embryos (n = 5) (Figure S6A). Next, we examined trunk muscle organization in 48 hpf 0.3 pmol MOzatxn7SPL morphants (n = 6) and age-matched 1 pmol mmMOzatxn7AUG controls (n = 5) using labelling with rhodamine-coupled phalloidin, an F-actin binding molecule that allows visualization of muscle fibres. We were unable to detect any differences between the morphology of trunk muscle of 48 hpf 0.3 pmol MOzatxn7SPL morphants (Figure S7B) and that observed in 1 pmol mmMOzatxn7AUG morphants at the same stage (Figure S7A). Taken together, these data show that differentiation of body muscles, spinal cord and motor neurons were not impaired following mild depletion of zebrafish atxn7 in embryos.
Discussion
The potential prevalence of mutations that lead to both loss and gain of function in human neurological disease (as shown by the phenotypes of presenilin−/− mice) [42], [43] underscores the importance of understanding endogenous functions of causative genes through careful analysis of loss-of-function models, which may uncover critical pathways leading to pathogenesis. Here, we performed a functional analysis of zebrafish atxn7 in the vertebrate teleost fish D. rerio (zebrafish) and investigated the phenotypes caused by various levels of protein depletion. We established a specific requirement for atxn7 in proper differentiation of the three main neuronal populations that are vulnerable in SCA7, i.e. photoreceptors, and cerebellar Purkinje and granule cells. Although the loss of differentiated neurons observed in SCA7 is clearly distinct from the neuronal differentiation defect seen in MOzatxn7 morphants, the similarity in the neuronal populations affected in the two processes is highly intriguing and suggest that reduced, or altered atxn7 function, in SCA7 plays a role in the pathophysiology of the disease. Moreover, these data are in good agreement with recent in vitro results suggesting that atxn7 plays role in Purkinje cell development and differentiation (Latouche et al., unpublished data). This loss could be the result of either dominant-negative activity of expanded atxn7 as discussed below or partial loss of function caused by heterozygocity of the non-expanded allele combined with progressive trapping of the wild-type protein in neuronal intranuclear inclusions (NII) [44], [45], or both. The dominant mode of inheritance of all polyQ-expansion diseases together with the deleterious effect of isolated polyQ peptides in vitro [46] and in vivo [46]–[49], led to the suggestion that pathologically expanded polyQ tract endowed the causative proteins with either a toxic gain of function or a dominant negative activity detrimental to life-long living post-mitotic neurons. However, a body of work also suggests that protein loss of function may also play a role in the disease phenotype in several polyQ disorders, such as SCA6 [50], [51], or HD [52]. In the case of SCA7, while our data suggest that altered atxn7 function plays a role in the disease phenotype, this hypothesis was challenged by the observation that transgenic mice homozygous for an expanded atxn7266Q allele, SCA7266Q/266Q mice, but not hemizygous SCA7266Q/− mice, displayed a worsened phenotype compared with SCA7266Q/+ animals [33]. However, it is important to keep in mind that such huge expansions in SCA7 (>200 residues), which have been described in only very few cases, induce pathologies that can no longer be classified as SCA, the disease, manifest from the first weeks post-pregnancy onward, affecting several non-neuronal tissues, such as the heart, kidneys and liver, and causing early lethality during the very first years of life [53]–[56]. In this context, whether partial depletion of the non-expanded protein participated in the pathophysiology of SCA7 remains an open question; further studies are required to evaluate the importance of altered atxn7 function in the disease process.
In the case of zebrafish atxn7, in close agreement with the results observed in mammals [16], [31], [32], [57], the gene is expressed throughout embryonic development and in several neuronal populations, including the cerebellum, spinal cord, optic tectum and telencephalon. However, we observed that only the differentiation of photoreceptors and cerebellar neurons was impaired following moderate zebrafish atxn7 depletion. The fact that differentiation of photoreceptors was compromised following partial loss-of-function of the zebrafish protein was consistent with data from the analysis of several SCA7 mouse models. Indeed, it has been shown that selective vulnerability of photoreceptors in SCA7 is related to interference of the mutant protein with CRX [27], [29], [30], a homeobox transcription factor crucial for photoreceptors differentiation through transcriptional regulation of photoreceptor-specific genes [58]–[61] and a direct partner of atxn7 [27]. By analogy, our results suggest that atxn7-SAGA might also be a partner of a transcription factor crucial for either differentiation and maintenance of cerebellar neurons or proper expression of another factor essential for cerebellar neuron differentiation and physiology. RORα, a transcription factor belonging to the family of retinoid-related orphan nuclear-receptors and crucial for proper differentiation of Purkinje cells [62], [63], appears as a good candidate. Indeed, rorα was also down-regulated in a knock-in transgenic mouse model of SCA1 (atxn182Q/82Q), but not in atxn1−/− mice [64], [65] and partial loss of function of the rorα gene enhanced the pathogenicity of atxn182Q [64]. All these observations raised the question of whether partial atxn7/SAGA-mediated RORα loss of function also underlay Purkinje cell loss in MOzatxn7 morphant embryos and also possibly in SCA7 patients. The finding that down-regulation of rorα or partial loss of RORα activity plays a role in the pathophysiology of SCA7 would be an important step toward the development of new therapeutic strategies. Further work is now required to evaluate the role of RORα in cerebellar neuron degeneration in SCA7.
Blast analysis identified 4 atxn7 paralogs in zebrafish, zatxn7, zatxn7l2a and l2b, and zatxn7l3, which are the orthologs of the human atxn7, atxn7L2 and atxn7L3 genes, respectively. Although our data show a specific requirement for zebrafish atxn7 in embryonic development and differentiation of photoreceptors and cerebellar neurons, the identification of atxn7L2 and atxn7L3 as components of the SAGA complex [66], [67], suggests a possible functional redundancy of these proteins.
Finally, the ability of human atxn7 to compensate the loss of the zebrafish protein in D. rerio embryos emphasizes the conservation of the function of this protein during evolution and thus, the interest of this fish as model to test therapeutic hypotheses.
Materials and Methods
Animals
Zebrafish were maintained at 28°C in a standard zebrafish facility (Aquatic Habitats, Apopka, FL. U.S.A.) as described in Westerfield [68]. Developmental stages were determined as hours post-fertilization (hpf) as described by Kimmel et al. [69]. Wild-type embryos were from the AB and TL strains. For in situ hybridization and immunohistochemistry, embryos were treated with 0.005% phenylthiourea from 20 hpf onward to prevent pigmentation.
Validation of the Structure and Sequences of the Zebrafish atxn7 Gene
Zebrafish atxn7 sequences were found in Ensembl (ENSDARG00000074804). To confirm the in silico data, the coding sequence of the gene was amplified from reverse-transcribed adult zebrafish cDNA, using overlapping zebrafish atxn7-specific primer sets (available from the authors on request) and directly sequenced using the BigDye technology (Applied Biosystem) in an ABI3730 automated sequencer. Experimental sequences were subsequently aligned with in silico predictions using Autoassembler (Applied Biosystems), and the consensus sequence was then analysed by UCSC Blat (http://genom.ucsc.edu/) to find the exon-intron boundaries and splice site locations. The zebrafish atxn7 protein sequence was then aligned with human and mouse counterparts using Align (http://www.ebi.ac.uk/Tools/emboss/align/) to determine domain conservation.
In situ Hybridization
The in situ detection of zebrafish atxn7 transcripts on dissected brains was carried out as described in Ayari et al. [70]. In situ hybridization on whole-mount embryos was performed principally as described in Yanicostas et al. [71]. The in situ hybridization of adult brain sections was done according to Bae et al. [34].
Immunohistochemistry
Immunohistochemistry on either dissected brains or brain sections was carried out as previously described in Ayari et al., [70]. Primary antibodies anti-paravalbumin7 (anti-Pavlb7, 1/1000, mouse ascites), anti-carbonic 8 (anti-Ca8, 1/100, mouse hybridoma supernatant), anti-vesicular1 glutamate transporter (anti-Vglut1, 1/1000, rabbit purified antibody), anti-zebrin II (1/200, hybridoma supernatant) [72] were used as described in Bae et al. [34]. The rabbit anti-GFAP (DAKO, used at 1∶1000 dilution), and human anti-HuC antibodies (kindly provided to us by Jean-Yves Delattre, used at 1∶4000 dilution). Eye cryosections were incubated with anti-rhodopsin rho4D2 antibodies (generous gift of Drs Serge Picaud and David Hicks, used at 1∶500 dilution) [73]. Primary antibodies were detected using fluorescently labelled secondary antibodies; Alexa 488-coupled goat-anti-rabbit antibodies (Molecular Probes, used at 1∶250 dilution), or with the corresponding biotinylated anti-human antibodies (MP Biomedicals Cappel, used at 1∶500 dilution) diluted in blocking solution. Secondary biotinylated antibodies were visualized by incubation with Alexa 555-coupled streptavidin (Molecular Probes, used at 1∶700 dilution) diluted in PBS. Following immunostaining, dissected brains were mounted on 1% agarose in PBS and photographed using an epifluorescent AXIO imager Z.1 microscope (Zeiss) equipped with an ApoTome system (Zeiss).
Morpholino-mediated Gene Inactivation
All morpholinos (MO) were designed by and obtained from GeneTools. To inactivate the translation of zebrafish atxn7 RNAs, we designed a morpholino-oligonucleotide, MOzatxn7AUG (5′-CGTCATCATCGGCCCTTTCCGACAT-3′), which is complementary to the sequence flanking the translation initiating codon of the messenger RNA (underlined). We also designed a second morpholino, MOzatxn7SPL (5′-ATCAAAACACACATACACACCTCTC-3′) that targets the donor site of the fourth intron of zebrafish atxn7 mRNA to impair proper splicing of this intron. As a control, we first designed a morpholino oligonucleotide derived from MOzatxn7AUG but comprising five mismatching bases (lower case letters), mmMOzatxn7AUG, (5′-CcTgATCATcGcCCCTTTgCGACAT-3′). We also used a non-specific morpholino oligonucleotide, MO control (5′-CCTCTTACCTCAGTTACAATTTATA-3′). For morpholino-mediated transcript inactivation experiments, 2 nl of 0.15, 0.3 or 0.5 mM solutions, and corresponding to 0.3, 0.6, and 1 pmol of the different morpholinos, respectively, were injected in 1- to 2-cell stage embryos using standard protocols.
RT-PCR Analysis of MOzatxnSPL Splice-blocking Activity
To check the efficiency of MOzatxn7SPL-mediated splice inhibition, a reverse transcription polymerase chain reaction (RT-PCR) was performed using RNAs, which were extracted from embryos that had received 0.3, 0.6, and 1 pmol MOzatxn7SPL. RNAs were isolated using the RNeasy mini Kit (Qiagen, Germantown, MD, USA) and then reverse transcribed as cDNA using the SuperScript II Reverse Transcriptase (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. A cDNA fragment of zebrafish atxn7 encompassing exons 3 to 5 was PCR amplified using the zatxn7-forw (5′-GGCCTTCCAAGCACATTAC-3′) and zatxn7-rev (5′-GTCATATCCATAACCCCAC-3′) primers.
Phenotypic Rescue
For repscue experiments, 2 nl of a mix containing MOzatxn7SPL (0,15 mM) and human atxn7 mRNA (1 µM), was injected into embryos at the one- to two-cell stage according to standard protocols, and the phenotypes were analysed at the indicated stages.
TUNEL Assay
For the detection of apoptotic cells, terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) assay was performed on dissected brains according to Yabu et al. [74].
Phalloidin-rhodamine Staining
Trunk muscles were visualized by phalloidin-rhodamine staining of F-actin. Briefly, 48 hpf embryos were anesthetized in tricaine, and fixed by an o/n incubation in 4% PFA in PBST (PBS, 0.1% Triton X-100) at 4°C, followed by three washes in PBST. Embryos were then incubated for 30 minutes in phalloidin-rhodamine (at 1/100) dissolved in PBS, washed three times in PBST and mounted in 1% low-melting agarose and imaged using a fluorescent microscope equipped with an ApoTome system (Zeiss).
Ethics Statement
All procedures involving animal handling in this study complied with the guidelines of the French Animal Ethics Committee and was approved by the same committee under the ethics statement: 2012-15/676-0069.
Supporting Information
Identification and sequence of the zebrafish atxn7 gene. Molecular phylogeny of the human (hatxn7 or ENSG00000163635; hatxn7L1 or ENSG00000146776; hatxn7L2 or ENSG00000162650; hatxn7L3 or ENSG00000087152; and hatxn7L3B or ENSG00000253719) and zebrafish (zatxn7 or ENSDARG00000074804; zatxn7l2a or ENSDARG00000055300; zatxn7l2b or ENSDARG00000056268; and zatxn7l3 or ENSDARG00000029331) atxn7 paralogs (A). RT-PCR analysis of the transcription of the zatxn7l2a, zatxn7l2b and zatxn7l3 genes in zebrafish embryos aged 24, 48 and 72 hpf (B). Sequence alignment of the human (H.s., ENSG00000163635), mouse (M.m., ENSMUSG00000021738), and zebrafish (D.r., ENSDARG00000074804) atxn7 protein sequences (C). All the sequences were obtained from the Ensembl data base (http://www.ensembl.org). Molecular phylogeny was determined using ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Peptidic sequences were aligned using Align (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Colour code for amino acids: identical amino acids, red; similar amino acids, blue. Abbreviations: Homo sapiens, H.s.; Mus musculus, M.m.; Danio rerio, D.r.
(DOCX)
Mild zebrafish atxn7 depletion impairs retina differentiation. 48 hpf 1 pmol mmMOzatxn7AUG (A) and 0.3 pmol MOzatxn7AUG morphant embryos (B). Eye of a 48 hpf 1 pmol mmMOzatxn7AUG morphant (C) and partially depigmented retinas of 48 hpf 0.3 pmol MOzatxn7AUG (D) and 0.3 pmol MOzatxn7SPL (E and F) morphant embryos.
(TIF)
Partial zebrafish atxn7 depletion impairs Purkinje cell differentiation. Frontal sections of dissected brains of 5 dpf 1 pmol mmMOzatxn7AUG (A-A’’) and 0.3 pmol MOzatxn7SPL morphant embryos (B-B’’). RORα immunostaining of Purkinje cells (A and B) and DAPI staining (A’ and B’). Merge images of the photographs A and A’ (A’’) and B and B’ (B’’).
(TIF)
Moderate zebrafish atxn7 depletion does not induce cerebellar neuron apoptosis. Dorsal views of dissected brains from DNase-treated non-injected control (A and B) and 1 pmol mmMOzatxn7AUG (C and D) and 0.3 pmol MOzatxn7SPL morphant embryos (E and F). Anterior is to the left. DAPI staining (A, C and E) and TUNEL labelling of apoptotic cells (B, D and F). Abbreviations: TeO, tectum optic; Cer, cerebellum.
(TIF)
Mild zebrafish atxn7 depletion does not impair overall brain organization. Dorsal view of dissected brains from 5 dpf 1 pmol mmMOzatxn7AUG (A-C) and 0.3 pmol MOzatxn7SPL morphant embryos (D-F). Anterior is to the left. GFAP immunostaining of glial cells (A and D) and HuC immunostaining of neuronal cells (B and E). Merge images of the photographs A and B (C) and D and E (F).
(TIF)
Partial zebrafish atxn7 depletion does not impair spinal cord differentiation. Lateral views of 48 hpf Tg[NBT:MAPT-GFP]zc1 transgenic embryos following injection of 1 pmol mmMOzatxn7AUG (A) and 0.3 pmol MOzatxn7SPL (B). Anterior is to the left.
(TIF)
Moderate zebrafish atxn7 depletion does not impair the differentiation of trunk muscles. Lateral views of 48 hpf 1 pmol mmMOzatxn7AUG (A) and 0.3 pmol MOzatxn7SPL morphant embryos (B) following rhodamine-coupled phalloidin labelling of muscle F-actin. Anterior is to the left.
(TIF)
Phenotypes of zebrafish atxn7 knockdown embryos.
(DOCX)
Acknowledgments
The authors are grateful to Dr. M. Latouche for helpful discussions, to Drs S. Picaud and D. Hicks for the anti-rhodopsin antibody and to J.C. Fernandez for technical assistance.
Funding Statement
This work was supported by the Institut National de la Santé et de la Recherche Médicale (Inserm), Avenir programs (Nos. R04190SP and R05245DS), the Association Française contre les Myopathies (AFM) and the Agence Nationale de la Recherche (ANR, Grant “SCA7”). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. David G, Abbas N, Stevanin G, Dürr A, Yvert G, et al. (1997) Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat Genet 17: 65–70. [DOI] [PubMed] [Google Scholar]
- 2. La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH (1991) Androgen receptor gene mutation in X-linked spinal and bulbar muscular atrophy. Nature 352: 77–79. [DOI] [PubMed] [Google Scholar]
- 3. Huntington’s Disease Collaborative Research Group (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72: 971–983. [DOI] [PubMed] [Google Scholar]
- 4. Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, et al. (1994) Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat Genet 6: 9–13. [DOI] [PubMed] [Google Scholar]
- 5. Orr HT, Chung MY, Banfi S, Kwiatkowski TJ Jr, Servadio A, et al. (1993) Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 4: 221–226. [DOI] [PubMed] [Google Scholar]
- 6. Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, et al. (1994) CAG expansion in a novel gene for Machodo-Joseph disease at chromosome 14q32.1. Nat Genet 8: 221–228. [DOI] [PubMed] [Google Scholar]
- 7. Imbert G, Saudou F, Yvert G, Devys D, Trottier Y, et al. (1996) Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat Genet 14: 285–291. [DOI] [PubMed] [Google Scholar]
- 8. Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, et al. (1996) Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 14: 269–276. [DOI] [PubMed] [Google Scholar]
- 9. Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, et al. (1996) Identification of the spinocerebellar ataxia type 2 gene using a direct identification of repeat expansion and cloning technique DIRECT. Nat Genet 14: 277–284. [DOI] [PubMed] [Google Scholar]
- 10. Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, et al. (1997) Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansion in the α1A-voltage-dependent calcium channel. Nat Genet 15: 62–69. [DOI] [PubMed] [Google Scholar]
- 11. Koide R, Kobayashi S, Shimohata T, Ikeuchi T, Maruyama M, et al. (1999) A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: a new polyglutamine disease ? Hum Mol Genet 8: 2047–2053. [DOI] [PubMed] [Google Scholar]
- 12. Fujigasaki H, Martin JJ, De Deyn PP, Camuzat A, Deffond D, et al. (2001) CAG repeat expansion in the TATA box-binding gene causes autosomal dominant cerebellar ataxia. Brain 124: 1939–1947. [DOI] [PubMed] [Google Scholar]
- 13. Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, et al. (2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10: 1441–1448. [DOI] [PubMed] [Google Scholar]
- 14. Dürr A, Stevanin G, Cancel G, Duyckaerts C, Abbas N, et al. (1996) Spinocerebellar ataxia 3 and Machado-Joseph disease: clinical, molecular, and neuropathological features. Ann Neurol 39: 490–499. [DOI] [PubMed] [Google Scholar]
- 15. Martin JJ, Van Regemorter N, Krols L, Brucher JM, de Barsy T, et al. (1994) On an autosomal dominant form of retinal-cerebellar degeneration: an autopsy study of five patients in one family. Acta Neuropathol 88: 277–286. [DOI] [PubMed] [Google Scholar]
- 16. Cancel G, Duyckaerts C, Holmberg M, Zander C, Yvert G, et al. (2000) Distribution of ataxin-7 in normal brain and retina. Brain 123: 2519–2530. [DOI] [PubMed] [Google Scholar]
- 17. Stevanin G, Dürr A, Brice A (2000) Clinical and molecular advances in autosomal dominant cerebellar ataxias: from genotype to phenotype and physiopathlogy. Eur J Hum Genet 8: 4–18. [DOI] [PubMed] [Google Scholar]
- 18. Lebre AS, Brice A (2003) Spinocerebellar ataxia 7 (SCA7). Cytogenet Genome Res 100: 154–163. [DOI] [PubMed] [Google Scholar]
- 19. Taroni F, DiDonato S (2004) Pathways to motor incoordination: the inherited ataxias. Nat Rev Neurosci 5: 641–655. [DOI] [PubMed] [Google Scholar]
- 20. Orr HT, Zoghbi HY (2007) Trinucleotide repeat disorders. Annu Rev Neurosci 30: 575–621. [DOI] [PubMed] [Google Scholar]
- 21. Garden GA, La Spada AR (2008) Molecular pathogenesis and cellular pathology of spinocerebellar ataxia type 7 neurodegeneration. Cerebellum 7: 138–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Paulson HL (2009) The spinocerebellar ataxias. J Neuroophtalmol 29: 227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Helmlinger D, Hardy S, Sasorith S, Klein F, Robert F, et al. (2004) Ataxin-7 is a subunit of GCN5 histone acetyltransferase-containing complexes. Hum Mol Genet 13: 1257–1265. [DOI] [PubMed] [Google Scholar]
- 24. Helmlinger D, Hardy S, Abou-Sleymane G, Eberlin A, Bowman AB, et al. (2006) Glutamine-expanded ataxin-7 alters TFTC/STAGA recruitment and chromatin structure leading to photoreceptor dysfunction. PLoS Biol 4(3): e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McMahon SJ, Pray-Grant MG, Schieltz D, Yates JR III, Grant PA (2005) Polyglutamine-expanded spinocerebellar ataxia-7 protein disrupts normal SAGA and SILK histone acetyltransferase activity. Proc Natl Acad Sci USA 102: 8478–8482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Palhan VB, Chen S, Peng GH, Tjernberg A, Gamper AM, et al. (2005) Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci USA 102: 8472–8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. La Spada AR, Fu YH, Sopher BL, Libby RT, Wang X, et al. (2001) Polygutamine-expanded ataxin-7 antagonizes CRX function and induces cone-rod dystrophy in a mouse model of SCA7. Neuron 31: 913–927. [DOI] [PubMed] [Google Scholar]
- 28. Aleman TS, Cideciyan AV, Volpe NJ, Stevanin G, Brice A, et al. (2002) Spinocerebellar ataxia type 7 (SCA7) shows a cone-rod dystrophy phenotype. Exp Eye Res 74: 737–745. [DOI] [PubMed] [Google Scholar]
- 29. Chen S, Peng GH, Wang X, Smith AC, Grote SK, et al. (2004) Interference of CRX-dependent transcription by ataxin-7 involves interaction between the glutamine regions and requires the ataxin-7 carboxy-terminal region for nuclear localization. Hum Mol Genet 13: 53–67. [DOI] [PubMed] [Google Scholar]
- 30. Abou-Sleymane G, Chalmel F, Helmlinger D, Lardenois A, Thibault C, et al. (2006) Polyglutamine expansion causes neurodegeneration by altering the neuronal differentiation program. Hum Mol Genet 15: 691–703. [DOI] [PubMed] [Google Scholar]
- 31. Jonasson J, Ström AL, Hart P, Brännström T, Forsgren L, et al. (2002) Expression of ataxin-7 in CNS and non-CNS tissue of normal and SCA7 individuals. Acta Neuropathol 104: 29–37. [DOI] [PubMed] [Google Scholar]
- 32. Ström AL, Jonasson J, Hart P, Brännström T, Forsgren L, et al. (2002) Cloning and expression analysis of the murine homolog of the spinocerebellar ataxia type 7 (SCA7) gene. Gene 285: 91–99. [DOI] [PubMed] [Google Scholar]
- 33. Yoo SY, Pennesi ME, Weeber EJ, Xu B, Atkinson R, et al. (2003) SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron 37: 383–401. [DOI] [PubMed] [Google Scholar]
- 34. Bae YK, Kani S, Shimizu T, Tanabe K, Nojima H, et al. (2009) Anatomy of zebrafish cerebellum and screen for mutations affecting its development. Dev Biol 330: 406–426. [DOI] [PubMed] [Google Scholar]
- 35. Garden GA, Libby RT, Fu YH, Kinoshita Y, Huang J, et al. (2002) Polyglutamine-expanded ataxin-7 promotes non-cell-autonomous Purkinje cell degeneration and displays proteolytic cleavage in ataxic transgenic mice. J Neurosci 22: 4897–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang HL, Yeh TH, Chou AH, Kuo YL, Luo LJ, et al. (2006) Polyglutamine-expanded ataxin-7 activates mitochondrial apoptotic pathway of cerebellar neurons by upregulating Bax and downregulating Bcl-xL . Cell Signal 18: 541–552. [DOI] [PubMed] [Google Scholar]
- 37. Latouche M, Lasbleiz C, Martin E, Monnier V, Debeir T, et al. (2007) A conditional pan-neuronal Drosophila model of spinocerebellar ataxia 7 with a reversible adult phenotype suitable for identifying modifier genes. J Neurosci 27 2483–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garden GA, Ellerby LM, La Spada AR (2006) Genetic instabilities and neurological diseases. In Wells AT, eds (ed.), Spinocerebellar ataxia type 7: clinical features to cellular pathogenesis, San Diego, CA, USA, 399–416.
- 39. Nielsen AL, Jorgensen AL (2003) Structural and functional characterization of the zebrafish gene for glial fibrillary acidic protein, GFAP. Gene 310: 123–132. [DOI] [PubMed] [Google Scholar]
- 40. Mueller T, Wullimann MF (2002) BrdU-, neuroD (nrd)- and Hu-studies reveal unusual non-ventricular neurogenesis in the postembryonic zebrafish forebrain. Mech Dev 117: 123–135. [DOI] [PubMed] [Google Scholar]
- 41. Tilton F, Tanguay RL (2008) Exposure to sodium metam during zebrafish somitogenesis results in early transcriptional indicators of the ensuing neuronal and muscular dysfunction. Toxicol Sci 106: 103–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. De Strooper B (2007) Loss-of-function presenilin mutations in Alzheimer disease. Taking point on the role of presenilin mutations in Alzheimer disease. EMBO Rep 8: 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen Q, Nakajima A, Choi SH, Xiong X, Tang YP (2008) Loss of presenilin function causes Alzheimer’s disease-like neurodegeneration in the mouse. J Neurosci Res 86: 1615–1625. [DOI] [PubMed] [Google Scholar]
- 44. Zander C, Takahashi J, El Hachimi KH, Fujigasaki H, Albanese V, et al. (2001) Similarities between spinocerebellar ataxia type 7 (SCA7) cell models and human brain: proteins recruited in inclusions and activation of caspase 3. Hum Mol Genet 10: 2569–2579. [DOI] [PubMed] [Google Scholar]
- 45. Scholefield J, Greenberg LJ, Weinberg MS, Arbuthnot PB, Abdelgany A, et al. (2009) Design of RNAi hairpins for mutation-specific silencing of ataxin-7 and correction of a SCA7 phenotype. PLoS One 4: e7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ikeda H, Yamaguchi M, Sugai S, Aze Y, Narumiya S, et al. (1996) Expanded polyglutamine in the Machado-Joseph disease protein induces cell death in vitro and in vivo. Nat Genet 13: 196–202. [DOI] [PubMed] [Google Scholar]
- 47. Ordway JM, Tallaksen-Greene S, Gutekunst CA, Bernstein EM, Cearley JA, et al. (1997) Ectopically expressed CAG repeats cause intranuclear inclusions and a progressive late onset neurological phenotype in the mouse. Cell 91: 753–763. [DOI] [PubMed] [Google Scholar]
- 48. Marsh JL, Walker H, Theisen H, Zhu ZY, Fielder T, et al. (2000) Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila. Hum Mol Genet 9: 13–25. [DOI] [PubMed] [Google Scholar]
- 49. McLeod CJ, O’Keefe LV, Richards RI (2005) The pathogenic agent in Drosophila models of ‘polyglutamine’ diseases. Hum Mol Genet 14: 1041–1048. [DOI] [PubMed] [Google Scholar]
- 50. Jun K, Piedras-Renteria ES, Smith SM, Wheeler DB, Lee SB, et al. (1999) Ablation of P/Q-type Ca2+ channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the α1A-subunit. Proc Natl Acad Sci USA 96: 15245–15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Miyasaki T, Hashimoto K, Shin HS, Kano M, Watanabe M (2004) P/Q-type CA2+ channel α1A regulates synaptic competition on developing cerebellar Purkinje cells. J Neurosci 24: 1734–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nasir J, Floresco SB, O’Kusky JR, Diewert VM, Richman JM, et al. (1995) Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioural and morphological changes in heterozygotes. Cell 81: 811–823. [DOI] [PubMed] [Google Scholar]
- 53. Johansson J, Forsgren L, Sandgren O, Brice A, Holmgren G, et al. (1998) Expanded CAG repeats in Swedish spinocerebellar ataxia type 7 (SCA7) patients: effect of CAG repeat length on the clinical manifestation. Hum Mol Genet 7: 171–176. [DOI] [PubMed] [Google Scholar]
- 54. Benton CS, de Silva R, Rutledge SL, Bohlega S, Ashizawa T, et al. (1998) Molecular and clinical studies in SCA7 define a broad clinical spectrum and infantile phenotype. Neurology 51: 1081–1086. [DOI] [PubMed] [Google Scholar]
- 55. Van de Warrenburg BPC, Frenken CWGM, Ausems MGEM, Kleefstra T, Sinke RJ, et al. (2001) Striking anticipation in spinocerebellar ataxia type 7: the infantile phenotype. J Neurol 248: 911–914. [DOI] [PubMed] [Google Scholar]
- 56. Whitney A, Lim M, Kanabar D, Lin JP (2007) Massive SCA7 expansion detected in a 7-month-old male with hypotonia, cardiomegaly, and renal compromise. Dev Med Child Neurol 49: 140–143. [DOI] [PubMed] [Google Scholar]
- 57. Einum DD, Townsend JJ, Ptacek LJ, Fu YH (2001) Ataxin-7 expression analysis in controls and spinocerebellar ataxia type 7 patients. Neurogenetics 3: 83–90. [DOI] [PubMed] [Google Scholar]
- 58. Furukawa T, Morrow EM, Cepko CL (1997) Crx, a novel otx-like homeobox gene, shows photoreceptor-specific expression and regulates photoreceptor differentiation. Cell 91: 531–541. [DOI] [PubMed] [Google Scholar]
- 59. Freund CL, Gregory-Evans CY, Furukawa T, Papaioannou M, Looser J, et al. (1997) Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 91: 543–553. [DOI] [PubMed] [Google Scholar]
- 60. Freund CL, Wang QL, Chen S, Muskat BL, Wiles CD, et al. (1998) De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat Genet 18: 311–312. [DOI] [PubMed] [Google Scholar]
- 61. Swain PK, Chen S, Wang QL, Affatigato LM, Coats CL, et al. (1997) Mutations in the cone-rod homeobox gene are associated with the cone-rod dystrophy photoreceptor degeneration. Neuron 19: 1329–1336. [DOI] [PubMed] [Google Scholar]
- 62. Landis DM, Sidman RL (1978) Electron microscopic analysis of postnatal histogenesis in the cerebellar cortex of staggerer mutant mice. J Comp Neurol 179: 831–863. [DOI] [PubMed] [Google Scholar]
- 63. Hatten ME, Messer A (1978) Postnatal cerebellar cells from staggerer mutant mice express embryonic cell surface characteristic. Nature 276: 504–506. [DOI] [PubMed] [Google Scholar]
- 64. Serra HG, Duvick L, Zu T, Carlson K, Stevens S, et al. (2006) RORα-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell 127: 697–708. [DOI] [PubMed] [Google Scholar]
- 65. Crespo-Barreto J, Fryer JD, Shaw CA, Orr HT, Zoghbi HY (2010) Partial loss of ataxin-1 function contributes to transcriptional dysregulation in spinocerebellar ataxia type 1 pathogenesis. PLoS Genet 6: e1001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sowa ME, Bennett EJ, Gygi SP, Harper JW (2009) Defining the human deubiquinating enzyme interaction landscape. Cell 138: 389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Krebs AR, Demmers J, Karmodiya K, Chang NC, Chang AC, et al. (2010) ATAC and mediator coactivators form a stable complex and regulate a set of non-coding RNA genes. EMBO reports 11: 541–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Westerfield M (2000) The zebrafish book. A guide for the laboratory use of zebrafish (Danio rerio). 4th ed. Univ. of Oregon Press, Eugene, Oregon. United States of America.
- 69. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF (1995) Stages of embryonic development of the zebrafish. Dev Dyn 233: 253–310. [DOI] [PubMed] [Google Scholar]
- 70. Ayari B, El Hachimi KH, Yanicostas C, Landoulsi A, Soussi-Yanicostas N (2010) Prokineticin 2 expression is associated with neural repair of injured adult zebrafish telencephalon. J Neurotrauma 27: 959–972. [DOI] [PubMed] [Google Scholar]
- 71. Yanicostas C, Ernest S, Dayraud C, Petit C, Soussi-Yanicostas N (2008) Essential requirement for zebrafish anosmin-1a in the migration of the posterior lateral line primordium. Dev Biol 320: 469–479. [DOI] [PubMed] [Google Scholar]
- 72. Lannoo MJ, Ross L, Maler L, Hawkes R (1991) Development of the cerebellum and its extracerebellar Purkinje cell projection in teleost fishes as determined by zebrin II immunocytochemistry. Prog Neurobiol 37: 329–363. [DOI] [PubMed] [Google Scholar]
- 73. Hicks D, Molday RS (1986) Differential immunogold-dextran labeling of bovine and frog rod and cone cells using monoclonal antibodies against bovine rhodopsin. Exp Eye Res 42: 55–71. [DOI] [PubMed] [Google Scholar]
- 74. Yabu T, Todoriki S, Yamashita M (2001) Stress-induced apoptosis by heat shock, UV and γ-ray irradiation in zebrafish embryos detected by increased caspase activity and whole mount TUNEL staining. Fish Sci 67: 333–340. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Identification and sequence of the zebrafish atxn7 gene. Molecular phylogeny of the human (hatxn7 or ENSG00000163635; hatxn7L1 or ENSG00000146776; hatxn7L2 or ENSG00000162650; hatxn7L3 or ENSG00000087152; and hatxn7L3B or ENSG00000253719) and zebrafish (zatxn7 or ENSDARG00000074804; zatxn7l2a or ENSDARG00000055300; zatxn7l2b or ENSDARG00000056268; and zatxn7l3 or ENSDARG00000029331) atxn7 paralogs (A). RT-PCR analysis of the transcription of the zatxn7l2a, zatxn7l2b and zatxn7l3 genes in zebrafish embryos aged 24, 48 and 72 hpf (B). Sequence alignment of the human (H.s., ENSG00000163635), mouse (M.m., ENSMUSG00000021738), and zebrafish (D.r., ENSDARG00000074804) atxn7 protein sequences (C). All the sequences were obtained from the Ensembl data base (http://www.ensembl.org). Molecular phylogeny was determined using ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Peptidic sequences were aligned using Align (http://www.ebi.ac.uk/Tools/msa/clustalw2/). Colour code for amino acids: identical amino acids, red; similar amino acids, blue. Abbreviations: Homo sapiens, H.s.; Mus musculus, M.m.; Danio rerio, D.r.
(DOCX)
Mild zebrafish atxn7 depletion impairs retina differentiation. 48 hpf 1 pmol mmMOzatxn7AUG (A) and 0.3 pmol MOzatxn7AUG morphant embryos (B). Eye of a 48 hpf 1 pmol mmMOzatxn7AUG morphant (C) and partially depigmented retinas of 48 hpf 0.3 pmol MOzatxn7AUG (D) and 0.3 pmol MOzatxn7SPL (E and F) morphant embryos.
(TIF)
Partial zebrafish atxn7 depletion impairs Purkinje cell differentiation. Frontal sections of dissected brains of 5 dpf 1 pmol mmMOzatxn7AUG (A-A’’) and 0.3 pmol MOzatxn7SPL morphant embryos (B-B’’). RORα immunostaining of Purkinje cells (A and B) and DAPI staining (A’ and B’). Merge images of the photographs A and A’ (A’’) and B and B’ (B’’).
(TIF)
Moderate zebrafish atxn7 depletion does not induce cerebellar neuron apoptosis. Dorsal views of dissected brains from DNase-treated non-injected control (A and B) and 1 pmol mmMOzatxn7AUG (C and D) and 0.3 pmol MOzatxn7SPL morphant embryos (E and F). Anterior is to the left. DAPI staining (A, C and E) and TUNEL labelling of apoptotic cells (B, D and F). Abbreviations: TeO, tectum optic; Cer, cerebellum.
(TIF)
Mild zebrafish atxn7 depletion does not impair overall brain organization. Dorsal view of dissected brains from 5 dpf 1 pmol mmMOzatxn7AUG (A-C) and 0.3 pmol MOzatxn7SPL morphant embryos (D-F). Anterior is to the left. GFAP immunostaining of glial cells (A and D) and HuC immunostaining of neuronal cells (B and E). Merge images of the photographs A and B (C) and D and E (F).
(TIF)
Partial zebrafish atxn7 depletion does not impair spinal cord differentiation. Lateral views of 48 hpf Tg[NBT:MAPT-GFP]zc1 transgenic embryos following injection of 1 pmol mmMOzatxn7AUG (A) and 0.3 pmol MOzatxn7SPL (B). Anterior is to the left.
(TIF)
Moderate zebrafish atxn7 depletion does not impair the differentiation of trunk muscles. Lateral views of 48 hpf 1 pmol mmMOzatxn7AUG (A) and 0.3 pmol MOzatxn7SPL morphant embryos (B) following rhodamine-coupled phalloidin labelling of muscle F-actin. Anterior is to the left.
(TIF)
Phenotypes of zebrafish atxn7 knockdown embryos.
(DOCX)