

Introduction
Frontotemporal lobar degeneration (FTLD) is a clinically and genetically diverse dementia syndrome, with phenotypes of behavioral variant frontotemporal dementia (bvFTD), semantic dementia (SD) and progressive non-fluent aphasia (PNFA). A proportion of patients with FTLD also develop amyotrophic lateral sclerosis (ALS). Common genetic causes of FTLD, i.e. mutations in microtubule-associated protein tau (MAPT) and progranulin (PGRN) are rare in Finnish patients with FTLD (Supplemental Digital Content 1, references S1 and S2), whereas the recently discovered hexanucleotide repeat expansion within C9ORF72 explains nearly 50% of Finnish familial FTLD and ALS.1
Neuronal ubiquitin-positive cytoplasmic inclusions are the pathological hallmark of the most common FTLD subtype (FTLD-TDP) and ALS. TDP-43 protein is the major component of these inclusions in both diseases, and it has provided evidence that the two diseases belong to a clinicopathological spectrum of overlapping neurodegenerative disorders.2 The pathogenic role of TDP-43 in neurodegeneration has been further strengthened by the identification of mutations within the coding gene, TAR DNA-binding protein (TARDBP) gene, in patients with familial and sporadic ALS (Supplemental Digital Content 1, reference S3). Mutations in TARDBP have been evaluated in several FTLD cohorts, but pathogenic TARDBP mutations have been detected only in five cases with FTLD, with or without ALS (Table 1).3–6
Table 1.
Characteristics of the patients with FTLD ± ALS phenotype carrying TARDBP mutations.
| Samplea | Nucleotide changeb | Phenotype
|
Family history
|
Ethnic origin | Reference | ||
|---|---|---|---|---|---|---|---|
| Presenting phenotype | Other symptoms | FTLD/dementia | ALS | ||||
| Ex6 K263E | c.787A>G | bvFTD | Supranuclear palsy, chorea | No | No | Hungarian | 6 |
| Ex6 N267S | c.800A>G | bvFTD | - | No | No | Italian | 4 |
| Ex6 S292del* | c.876-878delCAG | bvFTD | - | Yes | No | Finnish | Present study |
| Ex6 G295S | c.883G>A | bvFTD | ALS | No | No | French | 3 |
| Ex6 G295S | c.883G>A | SD | ALS | No | Yes | French | 3 |
| Ex6 M359V | c.1075A>G | bvFTD | - | No | NA | Italian | 5 |
Numbering according to GenPept accession number NP_031401.1.
Numbering according to GenBank accession number NM_007375.3 starting at the translation initiation codon.
Pathogenic nature unclear.
ALS indicates amyotrophic lateral sclerosis; bvFTD, behavioral variant frontotemporal dementia; FTLD, frontotemporal lobar degeneration; NA, data not available; SD, semantic dementia.
Given the reports linking TDP-43 in ALS-FTLD spectrum, we aimed at further investigating the prevalence and clinical features of TARDBP mutations in a cohort of Finnish patients with FTLD.
Methods
Patients and Controls
The study group consisted of 77 patients (47% men; mean age at onset 58.5 ± 7.2 y, range 38–79 y) meeting the clinical criteria for FTLD and recruited from the Memory Clinic at the Oulu University Hospital, Finland, during the years 1999–2010. BvFTD was the most common clinical phenotype (63%), with PNFA and SD in 25% and 12% of cases, respectively. Concomitant ALS was present in nine (12%) patients. There were 30 (39%) patients with familial presentation, and in those with familial presentation, there was a pair of siblings from three different families. Mutations in MAPT and PGRN were previously excluded. As a part of the recent mutation discovery study, the C9ORF72 expansion was screened in 75 out of 77 patients included in this series and identified in 22 (29%) patients.1
Control samples were obtained from 27 cognitively healthy elderly people (mean age 79.4 ± 7.2 y, range 67–93 y), and 130 self-reported healthy, anonymous middle-aged volunteers (mean age at blood collection 52.3 ± 5.3 y, range 45–64 y) as part of blood donations at Finnish Red Cross offices in Northern Finland. The research protocols were approved by the Ethics Committees of the Northern Ostrobothnia Hospital District and the Finnish Red Cross. Written informed consent was obtained from all the patients or their guardians.
Genetic Analyses
All the patients were screened for the exons 1–6 and flanking intronic regions of TARDBP. Genomic DNA was amplified by polymerase chain reaction (PCR) and sequenced with the ABI3130xl Genetic Analyzer (Applied Biosystem, Foster City, CA) using relevant specific genomic primers. Obtained sequences were compared with the genomic DNA sequence of TARDBP (GenBank Accession Number NG_008734.1). Nucleotide changes were numbered corresponding to the largest TARDBP transcript (NM_007375.3) starting at the translation initiation codon. Protein numbering was relative to the largest TDP-43 isoform (NP_031401.1). A novel TARDBP c.876_878delCAG variant in exon 6 was screened in 157 controls by PCR using mismatch primers (forward 5′-TCAGGGTGGATTTGGTAAT:::AGAG -3′ [: indicating the deletion CAG] and reverse 5′-GCATGTAGACAGTATTCCTATGGC -3′) and confirmed by direct sequencing.
To investigate whether the three p.Ser292del carriers are descendants of a common founder, allele sharing study was performed with seven microsatellite markers flanking 6.7 Mb around the TARDBP gene (see Supplemental Digital Content 2 for detailed methods).
Related protein sequences were searched with protein BLAST (http://blast.ncbi.nlm.nih.gov/) with human TDP-43 (Q13148.1) as the query sequence. Multiple alignment of protein sequences was done with ClustalW 2.1 with default parameters (http://www.ebi.ac.uk). Phosphorylation probabilities were analyzed with NetPhos 2.0 (http://www.cbs.dtu.dk), hydrophobicity with ProtScale (http://web.expasy.org) with Kyte & Doolittle amino acid scale and 5-residue window size.
Results
Mutation screening of TARDBP did not reveal any definitely pathogenic mutations. Instead, we found a novel heterozygous sequence variation; a trinucleotide deletion c.874-878del3 in the exon 6, resulting in a deletion of the serine residue 292. The sequence of Ser292-Arg293 in TARDBP is …-AGC-AGA-…; deletion of -AGC- (c.874_876del), GC-A (c.875_877del) and C-AG (c.876_878del) at DNA level all three result at protein level in p.Ser292del. It is not possible to determine exactly which of the positions is deleted, so the variant was arbitrarily named c.876-878delCAG (p.Ser292del) according to the most 3′ position (Fig. 1A).
Figure 1. Identification of the novel p.Ser292del variant of TARDBP in a family with frontotemporal dementia.
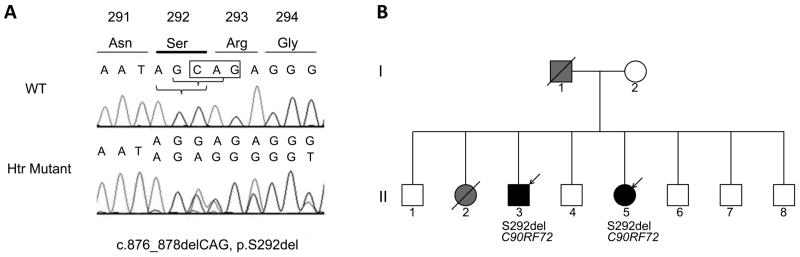
A. DNA sequence electropherograms of the TARDBP gene. The chromatogram of the patient II-5 (lower panel) displays a heterozygous trinucleotide deletion of CAG at position c.876-878 (c.876-878delCAG), resulting in a serine deletion at position 292 (p.Ser292del). The upper panel shows a wild type (WT) nucleotide and amino acid sequence of the same region in a normal control DNA. The position of the deleted CAG is indicated in box. The complementary DNA is numbered according to the largest TARDBP transcript (NM_007375.3) starting at the translation initiation codon. Protein numbering is relative to the largest TDP-43 isoform (NP_031401.1). B. Pedigree of the family with the TARDBP p.Ser292del and C9ORF72 repeat expansion. Square indicates male; circle, female; diagonal lines, deceased; open symbols, unaffected; black symbol, FTLD phenotype; grey symbol, unknown dementia syndrome; arrow, the probands.
The p.Ser292del variant was identified in two siblings, II-3 and II-5 (Fig. 1B). Both patients also carried the C9ORF72 expansion. At the age of 54 years, patient II-3 developed compulsive thoughts and stereotyped behavior relating to food. Patient II-5 presented with decline in social conduct, disinhibition, depressive symptoms and mild memory problems at age 47 years. Subsequently, both siblings developed severe neuropsychiatric symptoms. No signs of ALS were detected. With problems only in executive functions, the cognition of the patient II-3 remained quite stable for nearly 10 years, while in the patient II-5, marked deterioration was seen in three years. Their father (I-1) had died of acute myocardial infarction at the age of 60 years. In his last years, he had suffered aggressive and impulsive behavior, but diagnosis of dementia was not made. The mother (I-2), aged 94 years, is alive and has developed mild memory problems after the age of 90 years. One sister (II-2) died at the age of 67 years, and she is reported to have suffered from progressive dementia. There was no family history of ALS. The p.Ser292del was also detected in heterozygous form in one healthy control (0.6%). The control carrying the variant is a male and his age at blood collection was 46 years. He reported himself healthy when participating in blood donation. Both patients with the p.Ser292del shared the 6.7 Mb haplotype around the TARDBP gene. The control with the p.Ser292del variant shared the same haplotype as the two patients, except for the nearest 5′ marker, D1S2736 (see Supplemental Digital Content 2 for the detailed results of haplotype analysis).
P.Ser292 is highly conserved among animals but not in human heterogeneous nuclear ribonucleoprotein (hnRNP) paralogs (see Supplemental Digital Content 3 illustrating the conservation of amino acid residues). There is a conserved series of glycine residues in human hnRNPs and TDP-43, which in TDP-43 are targets of pathogenic mutations in ALS/FTLD. P.Ser292 is an improbable phosphorylation target (17.9 %, NetPhos 2.0), and deletion of this residue does not significantly change hydrophobicity of this region of the protein (not shown).
In addition, a novel intronic TARDBP variant was detected in one patient: insertion NG_008734.1:g. 14428_14429insT (NM_007375.3:c.715-74insT) located upstream of the exon 6.
Discussion
Mutations in TARDBP are mainly associated with ALS. Today, there are also a few reports on TARDBP mutations in patients with other neurodegenerative phenotypes as well, including FTLD (Table 1).3–6 We aimed at defining the role of TARDBP in the cohort of Finnish FTLD patients. No evident pathogenic mutations were identified. However, we detected a novel TARDBP deletion variant, p.Ser292del, in a pair of siblings. This is the first description of a deletion variant in the coding region of TARDBP. The variant is located in exon 6 that seems to represent a hotspot for ALS-linked TARDBP mutations, suggesting a pathogenic role of the variant. The same residue, p.Ser292, has previously been linked to Chinese patients with familial and sporadic ALS carrying a likely pathogenic missense mutation p.Ser292Asn (c.875G>A).7,8 Interestingly, both patients with the p.Ser292del of TARDBP also carried the C9ORF72 expansion. The repeat expansion has been found to be the most common cause of familial FTLD and ALS,1 and it is assumed as the disease causing mutation also in our siblings. Owing to the absence of DNA from family members, we cannot determine whether the TARDBP deletion variant segregates with the disease.
The fact that TARDBP p.Ser292del was detected in one healthy middle-aged control speaks against the pathogenicity of the variant. However, we cannot exclude if he has developed any cognitive or neuropsychiatric symptoms later on. The variant p.Ser292del may have some role in neurodegeneration. Based on our in silico analyses, the variant is unlikely to affect the phosphorylation status of the protein or local hydrophobicity. However, there is a conserved pattern of glycine residues spanning the affected p.Ser292 site that is even more prominent in human hnRNP paralogs than in TDP-43 proteins of different species (Supplemental Digital Content 3). As glycine-rich domains in hnRNPs A1 and A2/B1, as well as in TDP-43, are known to have a role in protein-protein interactions (Supplemental Digital Content 1, reference S4) and small amino acids are important on protein interaction surfaces, we propose that this protein region might function in protein-protein interaction function, which is important for neuronal cell. The p.Ser292del changes the spacing of these glycine residues, and therefore, has more prominent effect for this presumed interaction than any missense mutation affecting only one of the glycine residues.
Interestingly, there are now two studies reporting a combination of the pathogenic C9ORF72 expansion and a missense mutation of another gene: one study describing two Sardinian probands with ALS-FTLD carrying the C9ORF72 expansion and TARDBP p.Ala382Thr mutation, and the other reporting two expansion cases with FTLD carrying two novel missense mutations in PGRN (p.Tyr294Cys) and in PSEN2 (p.Ile146Val).9,10 The ALS-FTLD proband with paternal C9ORF72 expansion and maternal TARDBP mutation had a much younger age at disease onset than the parents, suggesting genetic burden in the proband.9 On the contrary, the phenotypes and the age at onset of our two siblings did not differ from typical bvFTD, and no signs of ALS were detected.
In conclusion, mutations in TARDBP are a rare cause of FTLD. In the present family, the C9ORF72 expansion is the probable disease-causing factor. Our data does not support a direct pathogenic role of the p.Ser292del variant in TARDBP. Yet, we cannot fully exclude that the p.Ser292del variant would not have any role in the disease, for example as a disease-modifying factor. In genetic screening studies, cases with known or suspected pathogenic mutations are often excluded in the study design. This report emphasizes that exclusion of those cases may lead to missing other genetic factors and may blur the broad genetic background behind neurodegenerative diseases.
Supplementary Material
Acknowledgments
Source of funding: This work was supported in part by the Finnish Medical Foundation, the Health Care Foundation of Northern Finland, the clinical EVO grants from Oulu University Hospital, and the Intramural Research Programs of the NIH, National Institute on Aging (Z01-AG000949-02).
The authors thank Ms. Anja Heikkinen and Ms. Pirjo Keränen for technical assistance, as well as Laura Kytövuori, MSc, for assistance with the genetic analyses.
Footnotes
Conflicts of interest: Dr. Traynor has a patent pending on the diagnostic and therapeutic uses based on the discovery of the hexanucleotide repeat expansion in C9ORF72. The remaining authors disclose no conflicts of interest.
References
- 1.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 3.Benajiba L, Le Ber I, Camuzat A, et al. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65:470–473. doi: 10.1002/ana.21612. [DOI] [PubMed] [Google Scholar]
- 4.Borroni B, Bonvicini C, Alberici A, et al. Mutation within TARDBP leads to frontotemporal dementia without motor neuron disease. Hum Mutat. 2009;30:E974–83. doi: 10.1002/humu.21100. [DOI] [PubMed] [Google Scholar]
- 5.Borroni B, Archetti S, Del Bo R, et al. TARDBP mutations in frontotemporal lobar degeneration: frequency, clinical features, and disease course. Rejuvenation Res. 2010;13:509–517. doi: 10.1089/rej.2010.1017. [DOI] [PubMed] [Google Scholar]
- 6.Kovacs GG, Murrell JR, Horvath S, et al. TARDBP variation associated with frontotemporal dementia, supranuclear gaze palsy, and chorea. Mov Disord. 2009;24:1843–1847. doi: 10.1002/mds.22697. [DOI] [PubMed] [Google Scholar]
- 7.Xiong HL, Wang JY, Sun YM, et al. Association between novel TARDBP mutations and Chinese patients with amyotrophic lateral sclerosis. BMC Med Genet. 2010;11:8. doi: 10.1186/1471-2350-11-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zou ZY, Peng Y, Wang XN, et al. Screening of the TARDBP gene in familial and sporadic amyotrophic lateral sclerosis patients of Chinese origin. Neurobiol Aging. 2012 doi: 10.1016/j.neurobiolaging.2012.03.014. [In press, available online 8 May 2012] [DOI] [PubMed] [Google Scholar]
- 9.Chio A, Restagno G, Brunetti M, et al. ALS/FTD phenotype in two Sardinian families carrying both C9ORF72 and TARDBP mutations. J Neurol Neurosurg Psychiatry. 2012;83:730–733. doi: 10.1136/jnnp-2012-302219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferrari R, Mok K, Moreno JH, et al. Screening for C9ORF72 repeat expansion in FTLD. Neurobiol Aging. 2012;33:1850.e1–1850.e11. doi: 10.1016/j.neurobiolaging.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.