

Abstract
Identification of vitamin D as a potent anti-rachitic factor almost a century ago, prompted investigations aimed at addressing its mechanism of action and key target tissues. Studies in vitamin D deficiency models and in kindreds with impaired hormone activation and function were critical in identifying key steps in the vitamin D signaling pathway. Studies in humans with vitamin D receptor mutations provided a tremendous amount of information regarding the role of this receptor in calcium and skeletal homeostasis. The availability of mouse models of vitamin D receptor ablation provided an important tool for detailed molecular analyses of the pathophysiologic basis for the skeletal, parathyroid and cutaneous phenotypes observed in mice and humans with impaired vitamin D receptor function. These investigations revealed that a critical action of the liganded receptor is the promotion of intestinal calcium absorption. Bypassing this defect by dietary or transgenic rescue prevents the severe skeletal phenotype of the vitamin D receptor ablated mice, as well as the development of hyperparathyroidism. In contrast, intestine specific ablation of the receptor results in marked skeletal pathology. Like their human counterparts, vitamin D receptor knockout mice develop alopecia. Studies in these mice demonstrated that the actions of the vitamin D receptor required for cyclical regeneration of the hair follicle and prevention of alopecia were shown independent of 1,25-dihydroxyvitamin D demonstrating that the unliganded receptor has an important role in the cutaneous homeostasis.
Keywords: Rickets, osteomalacia, alopecia, ligand-independent
Like humans affected by Vitamin D receptor (VDR) mutations, mice with targeted ablation of the VDR are indistinguishable from their normal littermates at birth[1–4]. Abnormalities in growth and mineral ion homeostasis are not observed until the third week of life[2], consistent with the observation that intestinal calcium absorption is largely vitamin D independent in rodents until this age[5]. The critical role of the VDR and its ligand in intestinal calcium absorption at later stages of development was confirmed by studies in 10 week old VDR knockout mice, which demonstrated a dramatic decrease in duodenal calcium absorption[3]. This was associated with impaired expression of TRPV5 and TRPV6, the calcium channels that are thought to regulate calcium entry into the enterocyte, as well as suppressed levels of mRNA encoding Calbindin D9K, a 1,25-dihydroxyvitamin D inducible gene whose expression correlates with active intestinal calcium transport. Interestingly, ablation of both TRPV6 and Calbindin D9K leads to mild hyperparathyroidism, but normal serum calcium and persistent active transport of calcium in the intestine, suggesting that additional genes play a significant role in active intestinal transport of calcium [6]. Correlating with the presence of increased renal calcium clearance, calbindin D9K mRNA levels are also decreased in the kidneys of the VDR null mice, although levels of TRPV5 and TRPV6 are not altered.
Secondary hyperparathyroidism in the VDR null mice is seen by 21 days of age[2] and initially, PTH-mediated bone resorption is able to maintain normocalcemia in the presence of impaired intestinal calcium absorption. However, after approximately 4 weeks of age, calcium levels in the VDR null mice decline and stabilize at levels approximately 30% lower than those of their heterozygous and wildtype littermates. This secondary hyperparathyroidism results in hypophosphatemia due to increased renal phosphate losses and is accompanied by growth retardation, which persists throughout life[2]. Analyses of the skeletal phenotype of the VDR null mice demonstrate progressive expansion of the growth plate within 3 days of development of hypophosphatemia (Figure 1). Histologically, the reserve and proliferative chondrocyte layers of the growth plate are not significantly different from those of wildtype mice; however, there is a dramatic increase in the length of the hypertrophic chondrocyte layer of the growth plate, secondary to impaired apoptosis of these cells[7]. Analyses of the molecular basis for this impaired apoptosis demonstrate that phosphate activates the mitochondrial apoptotic pathway in hypertrophic, but not proliferative chondrocytes, and that this process is impaired by hypophosphatemia[8]. Thus, the receptor-dependent actions of 1,25-dihydroxyvitamin D in chondrocytes are not required for normal growth plate maturation. However, investigations in mice with chondrocyte-specific ablation of the VDR demonstrate delayed vascular invasion associated with a reduction in VEGF mRNA levels as well as a decrease in FGF23 expression by osteoblasts, leading to increased circulating phosphate and 1,25-dihydroxyvitamin D levels[9]. In an analogous fashion, chondrocyte-specific ablation of Cyp27b1 led to an increase in the hypertrophic chondrocyte layer of embryonic bones, decreased RANKL expression, impaired osteoclastogenesis and decreased VEGF expression in the neonatal growth plate [10], demonstrating a role for the VDR and its ligand in skeletal development. The receptor-dependent actions of 1,25-dihydroxyvitamin D also play a critical role in preserving normal growth plate morphology in the hypophosphatemic Npt2a knockout mice [11], demonstrating that, while the vitamin D signaling pathway is not essential for normal growth plate maturation, it can compensate for the effects of hypophosphatemia in this setting.
Fig. 1.

Skeletal phenotype of VDR null mice at 35 days of age. Radiographs of the tibia and fibula of Wildtype (A) and VDR null mice (B) are shown. Arrow heads point to the cortical bone, which is undermineralized, thus thinner in the VDR null mouse. Arrow points to the expanded, radiolucent growth plate, classic for rickets. Von kossa staining demonstrates a decrease in mineral in the cortical and trabecular bone of the VDR null mouse (D), relative to its wildtype littermate, as well as expansion of the growth plate. Modified from [2]. Copyright 1997, National Academy of Sciences, U.S.A.
In addition to a rachitic growth plate, VDR null mice also develop osteomalacia(Figure 1). This is manifested by a 30 fold increase in osteoid volume at 70 days of age in the VDR null mice, relative to that of their wildtype littermates[12]. This increase in osteoid is largely responsible for the increase in trabecular bone volume and trabecular thickness observed in the VDR null mice. It also leads to the reduced stiffness of the bones observed on biomechanical testing, and decreased strength[12]. Osteoblast numbers are increased, presumably due to the marked increase in circulating parathyroid hormone. Despite these high parathyroid hormone levels, osteoclast number is not significantly increased, suggesting that absence of a functional VDR leads to impaired osteoclast differentiation. However, osteoclast-spleen co-cultures using cells isolated from VDR null and wildtype mice demonstrate that, in the absence of the VDR, parathyroid hormone can promote osteoclastogenesis, likely reflecting the redundant actions of these two hormones on induction of RANK ligand expression[13]. Thus, the inappropriately normal number of osteoclasts observed in the absence of a functional VDR presumably reflects the inability of these cells to bind to and resorb unmineralized matrix, rather than a defect in osteoclast differentiation. Interestingly, intestine-specific ablation of the VDR leads to normal calcium levels, but enhanced osteoclastogenesis, and high levels of 1,25-dihydroxyvitamin D that lead to osteopenia and impaired matrix mineralization [14].
The absence of a functional VDR also leads to dental pathology. VDR knockout mice have both enamel and dentin abnormalities, associated with dental root resorption. Whereas the latter phenotype is reversed by maintaining normal mineral ion levels, enamel and dentin integrity is only partially restored[15].
Investigations in humans with VDR mutations have demonstrated that intravenous calcium infusions, which bypass the defect in intestinal calcium absorption, profoundly ameliorate their skeletal phenotype[16]. Similarly, institution of a calcium and phosphate rich diet, supplemented with lactose (which promotes intestinal calcium absorption in rodents) prevents the development of abnormal mineral ion homeostasis in the VDR null mice[17] as does transgenic overexpression of the VDR in the intestine of VDR null mice[18,19]. These observations demonstrate that VDR-dependent intestinal calcium absorption is the major physiological action by which the VDR contributes to normal mineral ion homeostasis. They also provided a mouse model in which to identify which physiological consequences of impaired vitamin D action are a reflection of impaired mineral ion homeostasis, impaired ligand-dependent actions of the VDR, and impaired ligand-independent effects.
VDR null mice weaned onto this calcium and phosphate rich diet by day 18 of life do not develop hyperparathyroidism[17]. Unlike the VDR null mice with abnormal mineral ion homeostasis that exhibit an increase in parathyroid cell proliferation accompanied by a 10 fold increase in parathyroid glandular volume and a 16 fold increase in circulating parathyroid hormone levels by 70 days of age, the parathyroid glands of the VDR null mice with normal mineral ion homeostasis are indistinguishable from those of their wildtype and heterozygous littermates. This suggests that parathyroid cellular proliferation is predominantly influenced by calcium and that, in the presence of normal circulating calcium levels, the role of the VDR in parathyroid homeostasis is redundant. This is supported by the phenotype of mice with parathyroid-specific ablation of the VDR. These mice do not demonstrate an increase in parathyroid cellular proliferation, but do exhibit a decrease in expression of the parathyroid calcium sensor, resulting in a mild increase in circulating PTH levels despite normocalcemia [20]. Similar to normalization of parathyroid function in normocalcemic VDR null mice, the skeletal consequences of VDR ablation are prevented by maintaining normal mineral ion homeostasis: rickets is not observed, nor is osteomalacia. The bones of the VDR null mice with normal mineral ion homeostasis are histologically, histomorphometrically and biomechanically indistinguishable from those of their wildtype littermates[12], demonstrating that the physiological actions of the VDR on the skeleton are largely indirect, and primarily reflect its critical role in intestinal calcium absorption.
The VDR null mice have been shown to have higher blood pressure than wildtype mice. This is associated with an increase in renin mRNA and plasma angiotensin II levels[21]. While saline suppression decreased renin mRNA levels in the VDR null mice, levels remained high compared to wildtype mice subjected to similar salt loading. Thus, the renin-angiotensin system retains its responsiveness to salt in the absence of a functional VDR; however, the VDR null mice exhibit an increase in basal renin activity. Consistent with the ligand-dependence of the effects of the VDR in regulating the renin-angiotensin system, a 50% increase in renin mRNA levels was observed in vitamin D deficient wildtype mice. In vitro studies were able to identify regulatory regions in the renin gene that mediate transcriptional repression in response to 1,25-dihydroxyvitamin D[21].
Independently of the abnormalities in the renin-angiotensin system, the VDR plays a role in regulation of cardiac function. Mice with cardiomyocyte-specific deletion of the VDR exhibit left ventricular hypertrophy and a reduction in end-diastolic and end-systolic volume. Molecular investigations suggest that cardiac hypertrophy in the VDR null mice results from impaired ligand-dependent inhibition of the pro-hypertrophic calcineurin/NFATc1 pathway[22]. In contrast, vitamin D receptor ablation resulted in small and variably sized skeletal muscle fibers. This was accompanied by persistent expression of early myogenic markers, although overall myocyte differentiation was found to be normal [23].
While many epidemiological studies suggest an inverse correlation between vitamin D status and malignancies in humans, no spontaneous tumors are observed in the VDR null mice. However, these mice do exhibit hyperproliferation of colonic cells [24] as well as increased breast[25] and skin[26] cancer in response to chemical carcinogens. Analyses of mammary gland maturation demonstrate an increased number of secondary branch points and terminal end buds, as well as an increased proliferative response to estrogen and progesterone[27]. In vitro studies demonstrating suppression of sex steroid induced branching by 1,25-dihydroxyvitamin D suggest that the ligand-dependent effects of the VDR serve to attenuate mammary gland proliferation and branching.
The effect of VDR ablation on immune function has been extensively investigated. Although defects in T cell and macrophage function were observed, these were reversed by maintaining normocalcemia in the VDR null mice, suggesting that these abnormalities were secondary to impaired mineral ion homeostasis[28]. However, normocalcemic VDR null mice have decreased IFN production by T helper cells, thought to be secondary to impaired macrophage production of IL-18[29]. VDR null mice exhibit enhanced sensitivity to induction of inflammatory bowel disease[30], but are less susceptible to experimental autoimmune encephalomyelitis[31] and airway inflammation[32], suggesting that the VDR does modulate immune function, but its effects vary depending on the model and perhaps the underlying genetic background of the mice being studied.
Similar to some human kindreds affected by VDR mutations[33], mice lacking functional VDRs develop alopecia[2] (Figure 2). The first coat of hair develops normally, however, after the morphogenic period, which ends day 14 of postnatal life in mice, the VDR null mice are unable to establish new hair follicles, resulting in progressive hair loss[34]. Histologically, the skin of the VDR null mice demonstrates expansion of the sebocyte component, the presence of lipid laden dermal cysts and patulous hair follicle remnants (Figure 3). Unlike the skeletal and parathyroid abnormalities observed in the VDR null mice, the cutaneous phenotype is not prevented by maintaining normal mineral ion levels and is particularly intriguing since it is not observed in vitamin D deficiency nor is it seen in mice or humans lacking Cyp27B1, the enzyme responsible for conversion of 25-hydroxyvitamin D to its active metabolite, 1,25-dihydroxyvitamin D[35]. This suggested that the VDR was required to maintain normal hair, and that its actions in the hair follicle do not require 1,25-dihydroxyvitamin D.
Fig. 2.

Alopecia and growth retardation in Vitamin D receptor null mice. Appearance of wildtype (left), heterozygous (middle) and homozygous (right) vitamin D receptor knockout littermates at 3.5 months of age. Reproduced from [2]. Copyright 1997, National Academy of Sciences, U.S.A.
Fig. 3.
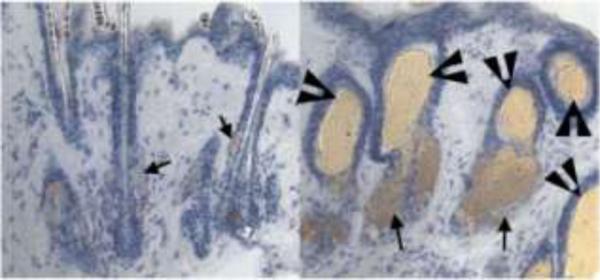
Skin phenotype of VDR-null mice. Skin from 8-month-old VDR-null mice (B) demonstrates lipid-laden dermal cysts (arrowheads) and an increase in sebaceous activity (arrows), relative to that seen in wildtype mice (A). Sections were stained for lipid with oil-red-O and counterstained with hematoxylin. Modified from [42]. Copyright 2007, National Academy of Sciences, U.S.A.
In mice, hair follicles begin to form day 14 of embryonic development, via interactions between the epidermal placode and underlying mesodermal condensate. Reciprocal signaling between these two compartments results in the formation of a mature hair follicle, with the epidermal component giving rise to the keratinocyte component of the hair follicle, and the mesodermal condensation giving rise to the dermal papilla. Postnatally, the hair follicle goes through periods of growth (anagen), regression (catagen) and rest (telogen) [36]. During this latter phase of the hair cycle, the dermal papilla is approximated to the bulge region of the hair follicle which contains keratinocyte stem cells. This is thought to permit reciprocal signaling between these two cell compartments that result in induction of a new anagen phase of the hair cycle, during which a new hair shaft is formed.
Investigations aimed at defining the pathophysiologic basis for the alopecia observed in the VDR null mice demonstrated that, after d 14 of life which marks the end of the morphogenic period, the VDR null mice are unable to initiate new hair cycles[34]. Hair reconstitution assays, in which neonatal keratinocytes and dermal papilla cells of differing genotypes are implanted into a nude mouse host, demonstrated that expression of the VDR in the dermal papilla component was not required for postmorphogenic hair cycling, whereas expression in keratinocytes was critical[37]. This was further confirmed by investigations demonstrating that targeting VDR expression to the keratinocytes of VDR null mice corrected the hair cycle defect, regardless of mineral ion status[38]. These observations led to investigations aimed at identifying the regions of the VDR required for the maintenance of post-morphogenic hair cycles. Investigations in VDR null mice with targeted ablation of the first zinc finger of the DNA binding domain reveal a surrogate initiator methionine, resulting in the expression of a VDR protein lacking the AF1 domain and the first zinc finger[1]. These mice are a phenocopy of mice that do not express any VDR protein, demonstrating that this amino terminal region of the VDR plays a critical role in post-morphogenic hair cycles. Keratinocyte-specific expression of a VDR with a mutation that prevents ligand binding and ligand-dependent transactivation also prevents the hair cycle defect, confirming that the actions of the VDR that regulate the post-morphogenic hair cycle are ligand independent[39].
These observations were particularly intruiging, given the results of studies in keratinocytes isolated from VDR null mice. 1,25-dihydroxyvitamin D treatment of wildtype keratinocytes had been shown to attenuate keratinocyte proliferation and to promote keratinocyte differentiation[40]. Its actions in this respect were redundant with those of calcium. In vitro studies demonstrated that, while 1,25-dihydroxyvitamin D could not exert a pro-differentiation and anti-proliferative effect in VDR null keratinocytes, the effects of calcium were indistinguishable from those observed in wildtype keratinocytes[34]. Furthermore, in vivo studies revealed a defect in keratinocyte differentiation in the skin of VDR null mice that was normalized by maintenance of normal mineral ion homeostasis[41]. These observations, which were at odds with the observation that normal mineral ion levels could not prevent alopecia in the VDR null mice, suggested that the VDR acted in a unique population of keratinocytes to maintain normal post-morphogenic hair cycling.
The keratinocytes that reside in the bulge region of the hair follicle contain a population of keratinocyte stem cells that are responsible for regeneration of the hair follicle during postmorphogenic hair cycles and can contribute to the sebocytes of the sebaceous gland. While this bulge niche forms normally in the absence of the VDR, by 9 months of age, it is no longer apparent in the VDR null mice[42]. Like other stem cells, when plated in culture at low density, keratinocyte stem cells give rise to large colonies (CFUs). While cells isolated from the morphogenic period form large colonies regardless of VDR status, cells isolated from 4 week old VDR null mice do not. This abnormality in colony formation is rescued by targeting expression of the VDR to the keratinocytes of VDR null mice, demonstrating that CFU formation is VDR dependent[42].
Bulge keratinocyte stem cells can be isolated by flow cytometry using the cell surface markers CD34 and alpha 6-integrin. These investigations demonstrated a normal number of keratinocyte stem cells in 4 week old VDR null mice, but revealed a progressive decline with ageing, a phenotype that was prevented by the keratinocyte-specific VDR transgene, suggesting a self-renewal defect in keratinocyte stem cells. In addition, the observation that keratinocyte stem cell number is normal at 4 weeks of age, yet these cells are unable to reconstitute a hair follicle in vivo or to form colonies in vitro, suggest a lineage progression defect as well.
While the molecular partners that interact with the unliganded VDR to promote keratinocyte stem cell lineage progression and self renewal have yet to be identified, alopecia is also a feature of mouse models in which transcriptional co-factors that associate with the VDR have been deleted. Mutation of the Hairless gene in mice and humans leads to alopecia associated with sebaceous cysts, similar to those observed in VDR null mice[43,44]. Impaired canonical Wnt signaling leads to defects in hair follicle morphogenesis and cycling[45]. Ablation of RXR-alpha in keratinocytes leads to a phenotype more severe than that observed with ablation of the VDR, suggesting that its interactions with other nuclear receptors in keratinocytes contribute to cutaneous homeostasis[46]. Interestingly, the interaction of the VDR with modulators of these pathways involves sequences outside the activation factor (AF1 and AF2) domains that bind classic nuclear receptor co-modulators. Thus identification of the molecular partners of the VDR that are required to maintain normal keratinocyte stem cell function are likely to reveal novel functions of this nuclear receptor.
Acknowledgements
This work was supported by a grant from the National Institutes of Health R01-DK-46974.
Footnotes
Disclosures. None
The author has stated that there is no conflict of interest.
References
- 1.Erben RG, Soegiarto DW, Weber K, Zeitz U, Lieberherr M, Gniadecki R, Moller G, Adamski J, Balling R. Deletion of deoxyribonucleic acid binding domain of the vitamin D receptor abrogates genomic and nongenomic functions of vitamin D. Mol Endocrinol. 2002;16(7):1524–1537. doi: 10.1210/mend.16.7.0866. [DOI] [PubMed] [Google Scholar]
- 2.Li YC, Pirro AE, Amling M, Delling G, Baron R, Bronson R, Demay MB. Targeted ablation of the vitamin D receptor: an animal model of vitamin D-dependent rickets type II with alopecia. Proc Natl Acad Sci U S A. 1997;94(18):9831–9835. doi: 10.1073/pnas.94.18.9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Cromphaut SJ, Dewerchin M, Hoenderop JG, Stockmans I, Van Herck E, Kato S, Bindels RJ, Collen D, Carmeliet P, Bouillon R, Carmeliet G. Duodenal calcium absorption in vitamin D receptor-knockout mice: functional and molecular aspects. Proc Natl Acad Sci U S A. 2001;98(23):13324–13329. doi: 10.1073/pnas.231474698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshizawa T, Handa Y, Uematsu Y, Takeda S, Sekine K, Yoshihara Y, Kawakami T, Alioka K, Sato H, Uchiyama Y, Masushige S, Fukamizu A, Matsumoto T, Kato S. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat Genetics. 1997;16(4):391–396. doi: 10.1038/ng0897-391. [DOI] [PubMed] [Google Scholar]
- 5.Halloran BP, DeLuca HF. Appearance of intestinal cytosolic receptor for 1,25-dihydroxyvitamin D3 during neonatal development in the rat. J Biol Chem. 1981;256:7338–7342. [PubMed] [Google Scholar]
- 6.Benn BS, Ajibade D, Porta A, Dhawan P, Hediger M, Peng JB, Jiang Y, Oh GT, Jeung EB, Lieben L, Bouillon R, Carmeliet G, Christakos S. Active intestinal calcium transport in the absence of transient receptor potential vanilloid type 6 and calbindin-D9k. Endocrinology. 2008;149(6):3196–3205. doi: 10.1210/en.2007-1655. doi:10.1210/en.2007-1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Donohue MM, Demay MB. Rickets in VDR null mice is secondary to decreased apoptosis of hypertrophic chondrocytes. Endocrinology. 2002;143(9):3691–3694. doi: 10.1210/en.2002-220454. [DOI] [PubMed] [Google Scholar]
- 8.Sabbagh Y, Carpenter TO, Demay M. Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci. 2005;102:9637–9642. doi: 10.1073/pnas.0502249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Masuyama R, Stockmans I, Torrekens S, Van Looveren R, Maes C, Carmeliet P, Bouillon R, Carmeliet G. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J Clin Invest. 2006;116(12):3150–3159. doi: 10.1172/JCI29463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naja RP, Dardenne O, Arabian A, St Arnaud R. Chondrocyte-specific modulation of Cyp27b1 expression supports a role for local synthesis of 1,25-dihydroxyvitamin D3 in growth plate development. Endocrinology. 2009;150(9):4024–4032. doi: 10.1210/en.2008-1410. [DOI] [PubMed] [Google Scholar]
- 11.Miedlich SU, Zhu ED, Sabbagh Y, Demay MB. The receptor-dependent actions of 1,25-dihydroxyvitamin D are required for normal growth plate maturation in NPt2a knockout mice. Endocrinology. 2010;151(10):4607–4612. doi: 10.1210/en.2010-0354. doi:en.2010-0354 [pii] 10.1210/en.2010-0354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amling M, Priemel M, Holzmann TCK, Rueger JM, Baron R, Demay MB. Rescue of the Skeletal Phenotype of Vitamin D Receptor Ablated Mice in the Setting of Normal Mineral Ion Homeostasis: Formal Histomorphometric and Biomechanical Analyses. Endocrinology. 1999;140:4982–4987. doi: 10.1210/endo.140.11.7110. [DOI] [PubMed] [Google Scholar]
- 13.Takeda S, Yoshizawa T, Nagai Y, Yamato H, Fukumoto S, Sekine K, Kato S, Matsumoto T, Fujita T. Stimulation of Osteoclast Formation by 1,25-dihydroxyvitamin D requires its binding to Vitamin D receptor (VDR) in osteoblastic cells: Studies using VDR Knockout mice. Endocrinology. 1999;140:1005–1008. doi: 10.1210/endo.140.2.6673. [DOI] [PubMed] [Google Scholar]
- 14.Lieben L, Masuyama R, Torrekens S, Van Looveren R, Schrooten J, Baatsen P, Lafage-Proust MH, Dresselaers T, Feng JQ, Bonewald LF, Meyer MB, Pike JW, Bouillon R, Carmeliet G. Normocalcemia is maintained in mice under conditions of calcium malabsorption by vitamin D-induced inhibition of bone mineralization. J Clin Invest. 2012;122(5):1803–1815. doi: 10.1172/JCI45890. doi:10.1172/JCI45890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Descroix V, Kato S, Lezot F, Berdal A. Physiopathology of dental rickets in vitamin D receptor-ablated mice. Journal of dental research. 2010;89(12):1427–1432. doi: 10.1177/0022034510379603. doi:10.1177/0022034510379603. [DOI] [PubMed] [Google Scholar]
- 16.Balsan S, Garabedian M, Larchet M, Gorski A-M, Cournot G, Tau C, Bourdeau A, Silve C, Ricour C. Long-term nocturnal calcium infusions can cure rickets and promote normal mineralization in hereditary resistance to 1,25-dihydroxyvitamin D. J Clin Invest. 1986;77:1661–1667. doi: 10.1172/JCI112483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li YC, Amling M, Pirro AE, Priemel M, Meuse J, Baron R, Delling G, Demay MB. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology. 1998;139(10):4391–4396. doi: 10.1210/endo.139.10.6262. [DOI] [PubMed] [Google Scholar]
- 18.Marks HD, Fleet JC, Peleg S. Transgenic expression of the human Vitamin D receptor (hVDR) in the duodenum of VDR-null mice attenuates the age-dependent decline in calcium absorption. J Steroid Biochem Mol Biol. 2007;103(3–5):513–516. doi: 10.1016/j.jsbmb.2006.11.014. doi:10.1016/j.jsbmb.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 19.Xue Y, Fleet JC. Intestinal vitamin D receptor is required for normal calcium and bone metabolism in mice. Gastroenterology. 2009;136(4):1317–1327. e1311–1312. doi: 10.1053/j.gastro.2008.12.051. doi:10.1053/j.gastro.2008.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meir T, Levi R, Lieben L, Libutti S, Carmeliet G, Bouillon R, Silver J, Naveh-Many T. Deletion of the vitamin D receptor specifically in the parathyroid demonstrates a limited role for the receptor in parathyroid physiology. Am J Physiol Renal Physiol. 2009;297(5):F1192–1198. doi: 10.1152/ajprenal.00360.2009. doi:10.1152/ajprenal.00360.2009. [DOI] [PubMed] [Google Scholar]
- 21.Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP. 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest. 2002;110(2):229–238. doi: 10.1172/JCI15219. doi:10.1172/JCI15219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen S, Law CS, Grigsby CL, Olsen K, Hong TT, Zhang Y, Yeghiazarians Y, Gardner DG. Cardiomyocyte-specific deletion of the vitamin d receptor gene results in cardiac hypertrophy. Circulation. 2011;124(17):1838–1847. doi: 10.1161/CIRCULATIONAHA.111.032680. doi:10.1161/CIRCULATIONAHA.111.032680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Endo I, Inoue D, Mitsui T, Umaki Y, Akaike M, Yoshizawa T, Kato S, Matsumoto T. Deletion of vitamin D receptor gene in mice results in abnormal skeletal muscle development with deregulated expression of myoregulatory transcription factors. Endocrinology. 2003;144(12):5138–5144. doi: 10.1210/en.2003-0502. doi:10.1210/en.2003-0502. [DOI] [PubMed] [Google Scholar]
- 24.Tong WM, Kallay E, Hofer H, Hulla W, Manhardt T, Peterlik M, Cross HS. Growth regulation of human colon cancer cells by epidermal growth factor and 1,25-dihydroxyvitamin D3 is mediated by mutual modulation of receptor expression. European journal of cancer. 1998;34(13):2119–2125. doi: 10.1016/s0959-8049(98)00267-6. [DOI] [PubMed] [Google Scholar]
- 25.Zinser GM, Welsh J. Vitamin D receptor status alters mammary gland morphology and tumorigenesis in MMTV-neu mice. Carcinogenesis. 2004;25(12):2361–2372. doi: 10.1093/carcin/bgh271. doi:10.1093/carcin/bgh271. [DOI] [PubMed] [Google Scholar]
- 26.Zinser GM, Sundberg JP, Welsh J. Vitamin D(3) receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis. 2002;23(12):2103–2109. doi: 10.1093/carcin/23.12.2103. [DOI] [PubMed] [Google Scholar]
- 27.Zinser G, Packman K, Welsh J. Vitamin D(3) receptor ablation alters mammary gland morphogenesis. Development. 2002;129(13):3067–3076. doi: 10.1242/dev.129.13.3067. [DOI] [PubMed] [Google Scholar]
- 28.Mathieu C, Van Etten E, Gysemans C, Decallonne B, Kato S, Laureys J, Depovere J, Valckx D, Verstuyf A, Bouillon R. In vitro and in vivo analysis of the immune system of vitamin D receptor knockout mice. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2001;16(11):2057–2065. doi: 10.1359/jbmr.2001.16.11.2057. doi:10.1359/jbmr.2001.16.11.2057. [DOI] [PubMed] [Google Scholar]
- 29.O'Kelly J, Hisatake J, Hisatake Y, Bishop J, Norman A, Koeffler HP. Normal myelopoiesis but abnormal T lymphocyte responses in vitamin D receptor knockout mice. J Clin Invest. 2002;109(8):1091–1099. doi: 10.1172/JCI12392. doi:10.1172/JCI12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Froicu M, Weaver V, Wynn TA, McDowell MA, Welsh JE, Cantorna MT. A crucial role for the vitamin D receptor in experimental inflammatory bowel diseases. Molecular endocrinology. 2003;17(12):2386–2392. doi: 10.1210/me.2003-0281. doi:10.1210/me.2003-0281. [DOI] [PubMed] [Google Scholar]
- 31.Meehan TF, DeLuca HF. CD8(+) T cells are not necessary for 1 alpha,25-dihydroxyvitamin D(3) to suppress experimental autoimmune encephalomyelitis in mice. Proc Natl Acad Sci U S A. 2002;99(8):5557–5560. doi: 10.1073/pnas.082100699. doi:10.1073/pnas.082100699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wittke A, Chang A, Froicu M, Harandi OF, Weaver V, August A, Paulson RF, Cantorna MT. Vitamin D receptor expression by the lung micro-environment is required for maximal induction of lung inflammation. Archives of biochemistry and biophysics. 2007;460(2):306–313. doi: 10.1016/j.abb.2006.12.011. doi:10.1016/j.abb.2006.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malloy PJ, Pike JW, Feldman D. The vitamin D receptor and the syndrome of hereditary 1,25-dihydroxyvitamin D-resistant rickets. Endocr Rev. 1999;20(2):156–188. doi: 10.1210/edrv.20.2.0359. [DOI] [PubMed] [Google Scholar]
- 34.Sakai Y, Demay MB. Evaluation of keratinocyte proliferation and differentiation in vitamin D receptor knockout mice. Endocrinology. 2000;141(6):2043–2049. doi: 10.1210/endo.141.6.7515. [DOI] [PubMed] [Google Scholar]
- 35.Dardenne O, Prud'homme J, Arabian A, Glorieux FH, St-Arnaud R. Targeted inactivation of the 25-hydroxyvitamin D(3)-1(alpha)-hydroxylase gene (CYP27B1) creates an animal model of pseudovitamin D-deficiency rickets. Endocrinology. 2001;142(7):3135–3141. doi: 10.1210/endo.142.7.8281. [DOI] [PubMed] [Google Scholar]
- 36.Paus R, Cotsarelis G. The biology of hair follicles. N Engl J Med. 1999;341(7):491–497. doi: 10.1056/NEJM199908123410706. [DOI] [PubMed] [Google Scholar]
- 37.Sakai Y, Kishimoto J, Demay M. Metabolic and cellular analysis of alopecia in vitamin D receptor knockout mice. J Clin Invest. 2001;107:961–966. doi: 10.1172/JCI11676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen C, Sakai Y, Demay M. Targeting expression of the human vitamin D receptor to the keratinocytes of vitamin D receptor null mice prevents alopecia. Endocrinol. 2001;142:5386–5389. doi: 10.1210/endo.142.12.8650. [DOI] [PubMed] [Google Scholar]
- 39.Skorija K, Cox M, Sisk JM, Dowd DR, MacDonald PN, Thompson CC, Demay MB. Ligand-independent actions of the vitamin D receptor maintain hair follicle homeostasis. Mol Endocrinol. 2005;19(4):855–862. doi: 10.1210/me.2004-0415. [DOI] [PubMed] [Google Scholar]
- 40.Bikle DD, Oda Y, Xie Z. Calcium and 1,25(OH)2D: interacting drivers of epidermal differentiation. J Steroid Biochem Mol Biol. 2004;89–90(1–5):355–360. doi: 10.1016/j.jsbmb.2004.03.020. [DOI] [PubMed] [Google Scholar]
- 41.Xie Z, Komuves L, Yu QC, Elalieh H, Ng DC, Leary C, Chang S, Crumrine D, Yoshizawa T, Kato S, Bikle DD. Lack of the vitamin D receptor is associated with reduced epidermal differentiation and hair follicle growth. J Invest Dermatol. 2002;118(1):11–16. doi: 10.1046/j.1523-1747.2002.01644.x. [DOI] [PubMed] [Google Scholar]
- 42.Cianferotti L, Cox M, Skorija K, Demay MB. Vitamin D receptor is essential for normal keratinocyte stem cell function. Proc Natl Acad Sci U S A. 2007;104(22):9428–9433. doi: 10.1073/pnas.0702884104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ahmad W, Faiyaz ul, Haque M, Brancolini V, Tsou HC, ul HS, Lam H, Aita VM, Owen J, deBlaquiere M, Frank J, Cserhalmi FP, Leask A, McGrath JA, Peacocke M, Ahmad M, Ott J, Christiano AM. Alopecia universalis associated with a mutation in the human hairless gene. Science. 1998;279(5351):720–724. doi: 10.1126/science.279.5351.720. [DOI] [PubMed] [Google Scholar]
- 44.Potter GB, Beaudoin GM, 3rd, DeRenzo CL, Zarach JM, Chen SH, Thompson CC. The hairless gene mutated in congenital hair loss disorders encodes a novel nuclear receptor corepressor. Genes Dev. 2001;15(20):2687–2701. doi: 10.1101/gad.916701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W. beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105(4):533–545. doi: 10.1016/s0092-8674(01)00336-1. [DOI] [PubMed] [Google Scholar]
- 46.Li M, Chiba H, Warot X, Messaddeq N, Gerard C, Chambon P, Metzger D. RXR alpha ablation in skin keratinocytes results in alopecia and epidermal alterations. Development. 2001;128:675–688. doi: 10.1242/dev.128.5.675. [DOI] [PubMed] [Google Scholar]