

Abstract
Silencing mediator of retinoic acid and thyroid hormone receptor (SMRT), also known as nuclear corepressor 2 (NCOR2) is a transcriptional corepressor for multiple members of the nuclear receptor superfamily of transcription factors, including estrogen receptor-α (ERα). In the classical model of corepressor action, SMRT binds to antiestrogen-bound ERα at target promoters and represses ERα transcriptional activity and gene expression. Herein SMRT mRNA and protein expression was examined in a panel of 30 breast cancer cell lines. Expression of both parameters was found to vary considerably amongst lines and the correlation between protein and mRNA expression was very poor (R2 = 0.0775). Therefore, SMRT protein levels were examined by immunohistochemical staining of a tissue microarray of 866 patients with stage I–II breast cancer. Nuclear and cytoplasmic SMRT were scored separately according to the Allred score. The majority of tumors (67 %) were negative for cytoplasmic SMRT, which when detected was found at very low levels. In contrast, nuclear SMRT was broadly detected. There was no significant difference in time to recurrence (TTR) according to SMRT expression levels in the ERα-positive tamoxifen-treated patients (P = 0.297) but the difference was significant in the untreated patients (P = 0.01). In multivariate analysis, ERα-positive tamoxifen-untreated patients with high nuclear SMRT expression (SMRT 5-8, i.e., 2nd to 4th quartile) had a shorter TTR (HR = 1.94, 95 % CI, 1.24–3.04; P = 0.004) while there was no association with SMRT expression for ERα-positive tamoxifen-treated patients. There was no association between SMRT expression and overall survival for patients, regardless of whether they received tamoxifen. Thus while SMRT protein expression was not predictive of outcome after antiestrogen therapy, it may have value in predicting tumor recurrence in patients not receiving adjuvant tamoxifen therapy.
Keywords: SMRT, NCOR2, Corepressor, Tamoxifen, Estrogen receptor, Breast cancer
Introduction
One of the significant developments in our understanding of breast cancer is the recognition that this is a heterogeneous disease with multiple distinct molecular phenotypes. With a greater appreciation of the underlying gene expression profiles associated with these phenotypes, it is anticipated that pathways critical for breast tumorigenesis or progression will be identified that can be therapeutically targeted. On a more modest scale, single gene markers of breast tumor progression or phenotype can be exploited to identify patients at risk of progression, predict their response to current therapeutic agents or developed as new targets for therapeutic intervention.
A well characterized and exploited biomarker of breast cancer prognosis and response to antihormonal therapy is estrogen receptor-α (ERα). This member of the steroid receptor superfamily of transcription factors mediates programs of gene expression in response to estrogens that are important for breast cancer growth and survival. Moreover, one of this receptor’s target genes, progesterone receptor (PR) is also a transcription factor that may promote breast tumor development and/or progression [1–4]. Based on the recognized importance of estrogens and ERα to breast carcinogenesis, various therapeutic approaches are currently used to block this receptor’s action, including antiestrogens such as tamoxifen which competitively inhibit estrogen binding to receptor, and aromatase inhibitors which block estradiol synthesis. However, the presence of ERα within tumor tissue has limited power to predict which patients will respond positively to antihormonal therapies [5], indicating that further enhancement of our understanding of cellular factors that control estrogen receptor action and its response to antagonists would be of clear clinical benefit.
The expression of ERα target genes is dependent not only upon levels of the receptor and its ligand, but also on the presence of a cohort of coregulators that serve to either activate (coactivators) or repress (corepressors) the transcriptional activity of this receptor as well as a broad spectrum of other transcription factors including other members of the nuclear receptor superfamily. Coactivators interact with agonist-bound receptors at target genes, and as some of these positive coregulators possess intrinsic enzymatic functions such as histone acetyltransferase and other activities necessary for chromatin remodeling, increased coactivator expression and/or activity promotes ERα-dependent gene expression and consequently breast tumorigenesis [6]. In addition to regulating the activity of estrogen-bound ERα, coactivators also influence the transcriptional activity of ERα bound to antiestrogens such as tamoxifen. For instance, elevated SRC-1 coactivator expression promotes the estrogen-like properties of tamoxifen [7], and increased expression of the SRC-3 coactivator in conjunction with HER2 overexpression has been associated with poor outcome for tamoxifen-treated breast cancer patients [8, 9].
Conversely, classic nuclear receptor corepressors such as SMRT/NCOR2 bind to ERα when occupied by antiestrogens, and suppress the receptor’s transcriptional activity [10]. Formation of ERα-corepressor complexes competitively block coactivator interaction with the receptor while promoting the recruitment of negative chromatin regulators such as histone deacetylases to target genes; this reduces histone acetylation leading to a condensed chromatin structure that effectively attenuates ERα target gene expression. In this view, SMRT is predicted to be a positive indicator of response to tamoxifen which prompted interest in the relationship between SMRT expression and the responsiveness of breast cancers to antiestrogens.
Much of the prior work examined SMRT mRNA levels in breast cancer cells [11–15] and while several studies demonstrated no change in SMRT mRNA expression relative to changes in cellular response to ERα ligands [13, 16] other studies have demonstrated increased levels of SMRT mRNA in cells resistant to tamoxifen or another antiestrogen, toremifene, as well as cells whose growth has become estrogen-independent [11, 12]. Moreover, several small studies examining either SMRT protein or mRNA in human breast tumors failed to detect differences in expression between normal and tumor tissues [17], between tamoxifen-sensitive and tamoxifen-resistant tumors [16] or relative to clinico-pathological parameters [18]. The apparent lack of association between SMRT and breast cancer cell sensitivity to tamoxifen is mirrored in a clinical study in which no association between SMRT mRNA levels and clinical response to tamoxifen was detected [19].
Recent evidence, however, indicates that SMRT control of ERα function is more complex, as this coregulator can promote ERα transcriptional activity and estrogen-dependent expression of a cohort of ERα target genes such as cyclin D1 and PR which play important roles in breast cancer [20, 21]. Depletion of SMRT expression also reduces proliferation of ERα-positive MCF-7 breast cancer cells [20] raising the possibility that SMRT may contribute to breast tumorigenesis through amplifying the activity of estrogen-bound ERα. More broadly, SMRT enhances the intrinsic transcriptional activity of the SRC-3 coactivator which is a breast oncogene [21], suggesting that SMRT also could promote breast tumorigenesis independent of its effects on ERα. Interestingly, high levels of SMRT protein were associated with a poor clinical outcome in one study [22] but as the tumor characteristics (e.g., ERα positive and negative) and treatments (i.e., with respect to use of chemotherapy and antihormonal therapy) were heterogeneous, the significance of this is difficult to interpret, particularly in view of the other studies which did not detect a relationship between SMRT and breast cancer [16, 17, 19]. It should be noted, however, that one possible factor contributing to the apparent conflict between this result and other studies may be the result of assessing SMRT protein rather than mRNA.
In this study, we examined the relationship between SMRT protein and mRNA levels in a panel of breast cancer cell lines and found variable levels of SMRT mRNA that were not predictive of protein expression. Thus, to assess the role of SMRT in human breast tumors, an immunohistochemical approach was developed and employed to measure SMRT expression in ERα-positive breast tumors obtained from patients that received no further adjuvant treatment and in patients that received tamoxifen therapy. Expression was then compared to pathoclinical parameters and survival outcomes. We show that higher SMRT expression is associated with a poor time to recurrence in patients that did not receive adjuvant hormonal therapy, while there was no association with SMRT expression for patients that received tamoxifen. Thus, while SMRT expression was not predictive of response to antiestrogen therapy, these data are consistent with a role for SMRT in predicting breast cancer aggressiveness for patients not receiving antihormonal therapy.
Materials and methods
SMRT mRNA and protein measurements in breast cancer cell lines
The breast cancer cell lines were obtained as part of the National Cancer Institute ICBP45/ATCC cell line kit and cultured according to ATCC recommendations. For assessment of SMRT mRNA expression, RNA was isolated from breast cancer cell lines using QIAshredder and RNeasy RNA isolation kits (Qiagen). The RNA was reverse transcribed using Superscript II Reverse Transcriptase (Invitrogen) and random primers, followed by qPCR with Power SYBR Green Master Mix (Applied Biosystems) according to the manufacturer’s instructions. The following primers were used: SMRT-F—GGTCAAG TCCAAGAAGCAAGAGAT, SMRT-R—GCTTCTATAG GTCATAAGGCCTGTTC, β-actin-F—CCCTGGCACCC AGCAC, and β-actin-R—GCCGATCCACACGGAGTAC. Samples were run in a 7500 Fast Real-Time PCR System (Applied Biosystems). Expression data was analyzed by ΔΔCt method (each sample adjusted to β-actin and the average ΔCt for all samples).
For western blots, cell lysates were prepared by sonication in modified RIPA buffer, containing 10 mM sodium phosphate, 1 % NP-40, 150 mM NaCl, 50 mM sodium fluoride, 200 mM sodium orthovanadate, 2 mM EDTA, and 1 mM PMSF. Equal amounts of protein were separated by SDS-PAGE on 3–8 % gels (Invitrogen, NuPAGE), transferred to nitrocellulose membranes and immunoblotted with primary antibody overnight. After washing, the membrane was incubated with HRP-conjugated anti-mouse or anti-rabbit secondary antibody (GE Healthcare, Piscataway, NJ) as appropriate, and the target proteins were visualized by ECL Plus Western Blotting Detection System (GE Healthcare). The primary antibodies are anti-SMRT (611386, BD Transduction Laboratories) and anti-actin (MAB1501R, Millipore). For quantitative analysis, the films were scanned and analysis was performed using Image J software.
Tumor specimens and patient population
This study was approved by the Baylor College of Medicine Institutional Review Board. Samples obtained from a prospectively assembled bank of frozen tumor specimens (Tumor Bank and Data Network Core in the Lester and Sue Smith Breast Center at Baylor College of Medicine) were prepared as previously described [23]. Individual specimens were fixed for 8 h in 10 % neutral buffered formalin and processed to paraffin blocks. Thereafter, samples were arrayed (12 samples/array; diameter of each core = 5 mm). These tissue specimens have been evaluated in prior studies for breast cancer prognostic and predictive factors including ER [24] and PR [25]. The study population was composed of patients diagnosed between 1973 and 1998 with stage I and II primary breast cancer with no distant metastasis. They were treated with mastectomy or lumpectomy plus axillary node dissection, with or without post-operative radiation therapy. Complete data on tumor size, number of nodes, receptor status, S-phase fraction, ploidy, and use and type of adjuvant therapy were available. A total of 866 patients (330 tamoxifen-treated and 435 tamoxifen-untreated patients with ER-positive tumors and 101 tamoxifen-untreated patients with ER-negative tumors) were analyzed. Median follow-up was 86 months.
Immunohistochemistry (IHC)
MCF-7 breast cancer and HeLa cervical cancer cells with manipulated SMRT levels were used to validate the SMRT antibody for IHC. MCF-7 or HeLa cells were transfected with control or SMRT siRNA using Lipofectamine transfection reagent (Invitrogen). The pan-SMRT siRNA has been described previously [20] and Silencer #2 negative control (Ambion) was used as a nonspecific control. For overexpression of SMRT, HeLa cells were transfected with a pCR3.1-hSMRT plasmid [21] using Lipofectamine. After transfection, cells were washed with PBS, fixed (2 h) in 10 % formalin, washed again in PBS, and finally resuspended and pelleted in 4 % Agar to be further processed for cell line blocks. Levels of SMRT in a second set of cells transfected in parallel were assessed by western blot analyses performed essentially as described previously [26] using anti-SMRT (611386) and anti-actin (MAB1501R) as primary antibodies.
IHC was performed on tissue microarrays (TMAs) using a standard immunoperoxidase procedure. Antigen retrieval was performed by heating in a pressure cooker for 10 min in 0.1 M Tris–HCl buffer (pH 9.0). Slides were incubated with the SMRT primary antibody (Clone 44; Cat # 611387; BD Transduction Laboratories) at a dilution of 1:300 for 1 h at room temperature and then incubated with the secondary, biotinylated antibody for 30 min. Sections were then incubated with streptavidin-peroxidase for 30 min and the enzyme was visualized after 15 min of incubation with diaminobenzidine. Nuclei were counterstained with hematoxylin before mounting.
Scoring of immunohistochemistry
Nuclear and cytoplasmic SMRT were scored by two pathologists (IM, CG) who were blinded to the clinical data. Immunostained slides were evaluated for nuclear SMRT according to the Allred score [27]. Briefly, each entire core was evaluated by light microscopy. First, a proportion score was assigned, which represents the estimated proportion of positive-staining tumor cells (0, none; 1, <1/100; 2, 1/100 to 1/10; 3, 1/10 to 1/3; 4, 1/3 to 2/3; and 5 >2/3). Next, an intensity score was assigned, which represents the average intensity of positive tumor cells (0, none; 1, weak; 2, intermediate; 3, strong). The proportion and intensity scores were then added to obtain a total score, which ranged from 0 to 8. Cytoplasmic SMRT was evaluated based on the intensity of the staining (0 = none; 1 = weak; 2 = intermediate; 3 = strong).
Statistical analysis
Descriptive statistics were calculated as frequencies and proportions to summarize clinico-pathological characteristics in tamoxifen-treated and tamoxifen-untreated patients. In order to identify optimal cut points of nuclear and cytoplasmic SMRT, Allred scores, median, and quartiles were calculated and Martingale residual plots were generated to evaluate their functional forms. Nuclear and cytoplasmic SMRT were analyzed separately.
Correlations between SMRT and clinico-pathological characteristics were analyzed as continuous variables using Spearman rank correlation (r) and Hommel’s method for adjustment for multiple comparisons [28]. Univariate analysis of SMRT on time to recurrence (TTR) and overall survival (OS) was carried out using the Kaplan–Meier method and compared using the log-rank test. TTR was calculated from the time of diagnosis to the date of the first proven recurrence. Patients without recurrence were censored at last follow-up or death. OS was calculated from the time of diagnosis to death from any cause or censored at last follow-up. Follow-up was truncated at 168 months for purposes of plotting.
In multivariate analysis, Cox proportional hazards regression models with stepwise selection were used to explore the association of SMRT with TTR and OS. Tests for proportionality were performed by including a time dependent covariate in the Cox models. To evaluate whether ER status influenced TTR, Cox proportional hazards regression analysis stratified by ER status was carried out and tested using a likelihood ratio test. Clinico-pathological variables were categorized according to standard cutoffs. Analyses for TTR and OS were performed separately for Tam-treated and untreated patients.
Results
SMRT mRNA and protein expression in breast cancer cell lines
Our prior work indicated that SMRT could positively influence the proliferation of MCF-7 breast cancer cells, and analysis of a small number of human breast tumor lysates by western blot analysis indicated that SMRT expression was variable [21]. It is possible that this variability was due to differences in the cellular composition of the tumor specimens, and we therefore wanted to determine the relative expression of SMRT in uniform populations of breast cancer cells. To this end, SMRT mRNA levels were determined for 30 different breast cancer cell lines by RT-qPCR. The mRNA for SMRT was detected in all 30 breast cancer cell lines tested, but at differing levels (Fig. 1a). The highest levels of SMRT mRNA were detected for MDA-MB-134 VI cells; this was ~20 times greater than levels measured for CAMA-1 cells which had the lowest SMRT mRNA expression. Molecular profiling has defined distinct breast cancer phenotypes [29], and the 30 breast cancer cell lines are representative of basal A (HCC1954, HCC1937, HCC1187, HCC1569, MDA-MB-468, BT-20, and HCC1599), basal B (MDA-MB-231, BT-549, MCF-10A, MDA-MB-157, MDA-MB-436, and HCC1395), and luminal (CAMA-1, T-47D, AU565, HCC1428, UACC812, HCC1419, SKBR3, MCF-7, BT-474, UACC893, MBA-MB-361, HCC2218, HCC1500, MDA-MB-415, MDA-MB-134-VI, MDA-MB-174-V11, and DU4475) profiles. The average level of SMRT mRNA expression did not differ between each of these three types of breast cancer cells (Supplemental Fig. 1).
Fig. 1.

Expression of SMRT in breast cancer cell lines. a Levels of SMRT mRNA were measured by RT-qPCR. Inset Representative blot of SMRT protein expression as assessed by western blot analysis. Actin blots were employed as a loading control. b Comparison of SMRT protein levels determined by western blot and SMRT mRNA levels determined by RT-qPCR for the 30 breast cancer cell lines. c Average SMRT protein expression for breast cancer cell lines grouped according to luminal-basal subtype. Values are presented as mean ± SEM
To determine if SMRT protein expression mirrored expression of its mRNA, western blot analyses of the 30 cell lines were performed. As shown in a representative blot of lysates prepared from cells with similar SMRT mRNA levels, SMRT protein expression varied considerably from barely detected levels in HCC1419 cells to the high levels observed for BT-474 cells (Fig. 1a, inset). We had previously noted that MCF-7 cells expressed two major forms of SMRT, a full-length form termed SMRTα and a splice variant lacking the first repression domain, termed SMRTβ [20], and this was also observed for these breast cancer cell lines. Comparison of the levels of total SMRT protein and mRNA for all 30 breast cancer cell lines revealed no correlation (Fig. 1b), suggesting that the variation in expression of SMRT protein is largely achieved via post-transcriptional mechanisms. Levels of SMRT protein expression were not significantly different between the tested breast cancer cells of the basal A, basal B, or luminal phenotypes (Fig. 1c).
Collectively, these data revealed that SMRT protein expression can vary widely in different breast cancer cell lines and that measurement of SMRT mRNA levels is not a good surrogate for protein expression of this coregulator. This indicates that determinations of SMRT in human breast tumors specimens should focus on protein measurement. To ensure that the SMRT antibody selected for immunohistochemical measurements accurately detects SMRT, established approaches were employed to alter SMRT expression in two different cell lines. First, expression of SMRT was depleted in MCF-7 cells using a selective siRNA [20] and cells were either fixed and embedded for IHC or lysates were prepared for analysis by western blot. Both approaches revealed a marked decrease in the level of detectable SMRT protein (Fig. 2a, b, bottom panel). Second, HeLa cells were either transfected with the SMRT siRNA or transiently transfected with a human SMRT expression vector. As noted in prior western blot analyses [20], HeLa cells express very low to undetectable amounts of SMRTβ. The analyses of the HeLa cells indicates that the reduced IHC staining of SMRT observed for siRNA-treated cells and the strong staining obtained for cells expressing exogenous SMRT corresponds well to the changes in SMRT expression determined for parallel sets of cells analyzed by western blot (Fig. 2a, b, top panel). By these criteria, the specificity of the SMRT IHC procedure is suitable for assessment of SMRT in human breast tumor specimens.
Fig. 2.

Validation of SMRT immunohistochemical detection. a HeLa (top) and MCF-7 (bottom) cells were transfected with control siRNA, SMRT-specific siRNA (siSMRT) or SMRTα expression vector (SMRTα), as indicated, and subsequently embedded in paraffin for immunohistochemical detection of SMRT expression. b HeLa (top) and MCF-7 (bottom) cells transfected in parallel were processed for SMRT expression assessment by western blot analysis. Actin is shown as a loading control
Patients and tumor characteristics
A total of 866 patients from the Baylor Breast Cancer SPORE National Tissue Resource were studied including 330 ERα-positive (ER+) disease patients who were treated with adjuvant tamoxifen monotherapy, 435 ER+-disease patients who received no adjuvant hormonal therapy after their primary treatment and 101 ERα-negative (ER−) disease patients that did not receive adjuvant endocrine therapy. The distributions of the patient’s clinic-pathological characteristics are presented in Table 1. The majority of patients were older than 50 years of age. All patients had tumors less than 5 cm in diameter and the majority was node negative. Of the 765 ERα-positive tumors, nearly 70 % were PR positive while ERα-negative tumors were largely PR negative. Approximately 62 % of tumors were of low to intermediate S-phase, and nearly 60 % were aneuploid. The median follow-up was 86 months. The clinico-pathological characteristics of the tamoxifen-untreated ER+ versus tamoxifen-treated ER+ groups were similar with the exception that a greater percentage of tamoxifen-untreated patients were ≤50 years of age (20.9 vs 9.1 %) and node negative (82.8 vs 54.5 %), respectively. More differences were observed between the untreated ER+ and untreated ER− groups with the latter having a greater percentage of patients ≤50 years old (20.9 vs 40.6 %) with tumors of larger size (>2–5 cm; 55.5 vs 69.4 %), high S-phase (>10 %; 30.6 vs 74.1 %), aneuploidy (56.1 vs 80 %), and negative staining for PR (<5 fmol/mg; 32.7 vs 86.7 %).
Table 1.
Distribution of clinico-pathological characteristics of all patients
| All (n = 866) | ER+
|
ER− | P† | P‡ | ||
|---|---|---|---|---|---|---|
| −TAM (n = 435) | +TAM (n = 330) | −TAM (n = 101) | ||||
| Age, n (%) | ||||||
| ≤50 years | 162 (18.7) | 91 (20.9) | 30 (9.1) | 41 (40.6) | <0.0001 | <0.0001 |
| >50 years | 704 (81.3) | 344 (79.1) | 300 (90.9) | 60 (59.4) | ||
| Tumor size, n (%) | ||||||
| 0–2 cm | 351 (41.1) | 191 (44.5) | 130 (39.6) | 30 (30.6) | 0.178 | 0.012 |
| >2–5 cm | 504 (58.9) | 238 (55.5) | 198 (60.4) | 68 (69.4) | ||
| Missing | 11 | |||||
| Nodes, n (%) | ||||||
| Node negative | 629 (72.6) | 360 (82.8) | 180 (54.5) | 89 (88.1) | <0.0001 | 0.370 |
| Node positive | ||||||
| 1–3 | 143 (16.5) | 50 (11.5) | 84 (25.5) | 9 (8.9) | ||
| >3 | 94 (10.9) | 25 (5.7) | 66 (20.0) | 3 (3.0) | ||
| S-phase, n (%) | ||||||
| Low (0 to <6 %) | 230 (32.5) | 131 (36.8) | 88 (32.5) | 11 (13.6) | 0.525 | <0.0001 |
| Intermediate (≥6 to ≤10 %) | 219 (30.9) | 116 (32.6) | 93 (34.3) | 10 (12.3) | ||
| High ( >10 %) | 259 (36.6) | 109 (30.6) | 90 (33.2) | 60 (74.1) | ||
| Missing | 158 | |||||
| Ploidy, n (%) | ||||||
| Diploid | 288 (38.8) | 162 (43.9) | 109 (37.7) | 17 (20.0) | 0.110 | <0.0001 |
| Aneuploid | 455 (61.2) | 207 (56.1) | 180 (62.3) | 68 (80.0) | ||
| Missing | 123 | |||||
| PR, n (%) | ||||||
| Negative ( <5 fmol/mg) | 317 (37.7) | 137 (32.7) | 95 (29.4) | 85 (86.7) | 0.339 | <0.0001 |
| Positive (≥5 fmol/mg) | 523 (62.3) | 282 (67.3) | 228 (70.6) | 13 (13.3) | ||
| Missing | 26 | |||||
| Median follow-up time | ||||||
| Months | 86 | 86 | 85 | 83 | 0.394 | 0.438 |
Comparison of −Tam vs. +Tam ER+ patients; P values based on Chi-square test
Comparison of ER+ vs. ER−−TAM patients; P values based on Chi-square test
SMRT expression
All tumor sections were evaluated for SMRT IHC staining; representative cases are shown in Fig. 3. The majority of tumors (67 %) were negative for cytoplasmic SMRT expression (Table 2), and the average cytoplasmic expression of SMRT was very low for all patient groups (Supplemental Table 1). Allred scores were determined for nuclear SMRT expression and on this basis, tumors were grouped approximately by quartiles (1st ≤ 4; 2nd >4, ≤6; 3rd = 7; 4th = 8). The mean nuclear SMRT score of all tumors was 5.6 and distribution of scores were similar in different patient populations (Supplemental Table 1). The highest nuclear SMRT expression (Allred score = 8) was observed in 22 % of the specimens (Table 2).
Fig. 3.
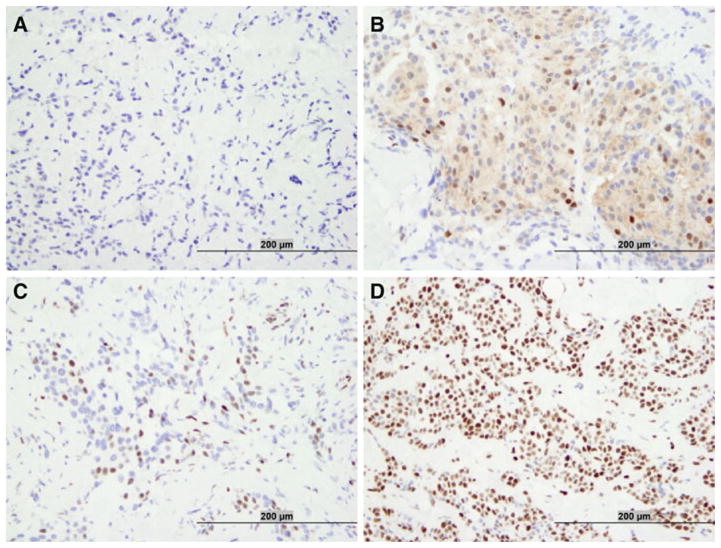
Representative photographs of immunohistochemical expression of SMRT in breast cancer specimens showing negative (a), predominantly cytoplasmic (b), low nuclear (c), and high nuclear (d) SMRT expression
Table 2.
Distribution of SMRT expression according to ER status and treatment
| All (n = 866) | ER+
|
ER− | P† | P‡ | ||
|---|---|---|---|---|---|---|
| −TAM (n = 435) | +TAM (n = 330) | −TAM (n = 101) | ||||
| Nuclear | ||||||
| 1st quartile (≤4) | 237 (27.4) | 131 (30.1) | 78 (23.6) | 28 (27.7) | 0.145 | 0.443 |
| 2nd quartile ( >4, ≤6) | 256 (29.6) | 132 (30.3) | 99 (30.0) | 25 (24.8) | ||
| 3rd quartile (= 7) | 181 (20.9) | 85 (19.5) | 70 (21.2) | 26 (25.7) | ||
| 4th quartile (= 8) | 192 (22.2) | 87 (20.0) | 83 (25.2) | 22 (21.8) | ||
| Cytoplasm | ||||||
| Negative (= 0) | 581 (67.1) | 289 (66.4) | 224 (67.9) | 68 (67.3) | 0.674 | 0.864 |
| Positive ( >0) | 285 (32.9) | 146 (33.6) | 106 (32.1) | 33 (32.7) | ||
Tam-treated vs. untreated ER+ patients; P values were based on Chi-square test or Wilcoxon rank-sum test
ER+ vs. ER− Tam-untreated patients; P values were based on Chi-square test or Wilcoxon rank-sum test
Correlation with clinico-pathological characteristics
Correlations between nuclear and cytoplasmic SMRT expression with various clinical and pathological characteristics were assessed for all patients (Table 3). Nuclear and cytoplasmic SMRT expression were slightly correlated with each other (r = 0.150, P <0.0001). We observed a very small negative correlation between nuclear SMRT and lymph node involvement (r = −0.073, P = 0.030) whereas very small positive correlations were observed between nuclear SMRT and ERα (r = 0.069, P = 0.043), S-phase (r = 0.087, P = 0.020) and ploidy (r = 0.093, P = 0.011). There also was a very small correlation between cytoplasmic SMRT and ERα (r = 0.089, P = 0.009) and S-phase (r = 0.131, P = 0.0005). There were no significant correlations between SMRT, either cytoplasmic or nuclear, and age, PR status or tumor size. The strongest of the correlations accounts for less than 2 % of the variation in the biomarker, and after adjustment for multiple comparisons using Hommel’s method, none are significant, except cytoplasmic SMRT and S-phase.
Table 3.
Correlation of nuclear and cytoplasm SMRT with clinico-pathological characteristics
| All (n = 866)
|
||
|---|---|---|
| Nuclear correlation (P)† | Cytoplasm correlation (P)† | |
| Nuclear SMRT‡ | 1 | 0.150 ( <0.0001) |
| Cytoplasmic SMRT‡ | 0.150 ( <0.0001) | 1 |
| Age | −0.025 (0.455) | −0.041 (0.223) |
| ERα | 0.069 (0.043) | 0.089 (0.009) |
| PR | 0.048 (0.167) | 0.040 (0.244) |
| Tumor size | −0.020 (0.563) | 0.040 (0.239) |
| Nodes | −0.073 (0.030) | −0.007 (0.840) |
| S-phase | 0.087 (0.020) | 0.131 (0.0005) |
| Ploidy | 0.093 (0.011) | 0.009 (0.804) |
Spearman rank correlation
Correlation between nuclear and cytoplasm
Univariate analysis
As SMRT is a regulator of ERα action, the influence of this coregulator on time to recurrence (TTR) in patients with ERα-positive tumors that received tamoxifen was examined. However, no significant differences relative to SMRT expression were observed (P = 0.297) (Fig. 4) suggesting that nuclear SMRT expression did not predict outcome after tamoxifen treatment. However, for patients not receiving tamoxifen with ERα-positive tumors, the time to recurrence varied depending on the level of SMRT expression in the primary tumor (P = 0.01; Fig. 5a). Specifically, in this subgroup we observed that patients with high nuclear SMRT expression (SMRT 5-8, i.e., 2nd to 4th quartile) had shorter TTR than those with nuclear SMRT ≤ 4 (i.e., 1st quartile) (P = 0.003).
Fig. 4.

Time to recurrence in ER+, tamoxifen-treated patients (n = 330) according to nuclear SMRT levels. P values based upon log-rank test
Fig. 5.

Time to recurrence in patients that did not receive tamoxifen whose tumors were a ER+ (n = 435), b ER− (n = 101), or c either ER+ or ER− (n = 536) according to nuclear SMRT levels. P values based upon log-rank test
The finding of an association between TTR and SMRT expression in patients not receiving adjuvant hormonal therapy raised the question of whether this apparent effect of SMRT was dependent on ERα function. A small number (n = 101) of ERα-negative tumors obtained from patients untreated with tamoxifen were analyzed for the effect of SMRT expression on TTR. There was a trend towards a significant difference in time to tumor recurrence relative to nuclear SMRT expression (P = 0.079; Fig. 5b). Analysis of SMRT expression in the pooled population of tamoxifen-untreated patients regardless of their ERα status revealed a significance difference in TTR with relation to nuclear SMRT expression (P = 0.007; Fig. 5c). In this subgroup, patients with high nuclear SMRT expression (SMRT 5-8, i.e., 2nd to 4th quartile) had worse TTR than those with nuclear SMRT ≤ 4 (i.e., 1st quartile) (P = 0.001). However, there were no significant associations between levels of nuclear SMRT expression and overall survival found for any of the tamoxifen-treated or untreated patients (Supplemental Fig. 2). Nor were there differences in overall survival or TTR found for positive versus negative cytoplasmic SMRT among untreated or treated patients (data not shown). Collectively, these results indicate that nuclear SMRT influenced tumor recurrence in patients that did not receive tamoxifen.
Multivariate analysis of patients treated or not with adjuvant hormonal therapy
The prognostic effects of clinico-pathological variables (nuclear SMRT, cytoplasmic SMRT, nodes) on TTR in tamoxifen-untreated, ERα-positive (n = 435) patients are shown in Table 4. In multivariate analysis, high nuclear SMRT expression (SMRT 5-8, i.e., 2nd to 4th quartile) was significantly associated with shorter time to recurrence (HR = 1.94; 95 % CI, 1.24–3.04; P = 0.004). Cytoplasmic SMRT expression was not associated with time to recurrence. As expected, lymph node positivity was also significantly associated with a greater risk for tumor recurrence (overall P <0.0001; 1–3 positive lymph nodes vs node negative: HR = 1.53; 95 % CI, 0.93–2.51; >3 positive lymph nodes versus node negative: HR = 3.42; 95 % CI, 1.94–6.06). There was no independent association between age or PR status and time to recurrence for patients with untreated ER+ tumors. Analysis of the combined ER+ and ER− tamoxifen-untreated patient populations (n = 536) yielded largely similar results as seen in the ER+ tamoxifen-untreated patients (data not shown). In addition, we performed stratified Cox proportional hazard regression analysis and obtained similar results as well. The likelihood ratio test was not significant, suggesting no evidence that the effect of SMRT differed by ER status.
Table 4.
Multivariate analysis of time to recurrence for tamoxifen-untreated, ER+ patients (n = 435)
| Variable | HR | 95 % CI | P |
|---|---|---|---|
| Nuclear | 0.004 | ||
| 1st quartile (≤4) | 1.00 | – | |
| Others (5–8) | 1.94 | 1.24–3.04 | |
| Cytoplasm | 0.259 | ||
| Negative (= 0) | 1.25 | 0.85–1.83 | |
| Positive ( >0) | 1.00 | – | |
| Node | <0.0001 | ||
| Node negative | 1.00 | – | |
| 1–3 | 1.53 | 0.93–2.51 | |
| >3 | 3.42 | 1.94–6.06 |
For ERα-positive patients treated with tamoxifen, there was no significant association between either cytoplasmic or nuclear SMRT and time to recurrence (Supplemental Table 2). However, age at diagnosis of ≤50 years had worse TTR (HR = 3.12; 95 % CI, 1.74–5.61, P <0.0001). Negative PR status, defined as <5 fmol/mg protein, also was associated with worse TTR for tamoxifen-treated patients (HR = 1.74; 95 % CI, 1.12–2.70, P = 0.013) as was lymph node positivity (overall P <0.0001; 1–3 positive lymph nodes versus node negative: HR = 1.16; 95 % CI, 0.65–2.07; >3 positive lymph nodes versus node negative: HR = 3.79; 95 % CI, 2.30–6.25).
Discussion
In the traditional model of corepressors and their regulation of ERα function, a corepressor such as SMRT is recruited to antagonist-bound ERα. This, in turn results in recruitment of additional corepressor molecules, such as HDAC3, TBL1, TBLR1, and GPS2, and collectively these molecules influence the biological activity of tamoxifen–ERα complexes such that they effectively execute an antiestrogen program of gene expression. This model predicts that the antagonist actions of tamoxifen, which include inhibiting the proliferation of ERα positive breast cancer cells, will be greatest in tissues expressing high levels of corepressor. As coregulators exert their transcriptional effect as proteins, we therefore established an IHC assay for SMRT protein in breast tumors. Surprisingly, our analyses failed to find an association between SMRT expression and recurrence for patients treated with tamoxifen. However, high SMRT expression was associated with shorter time to tumor recurrence for patients that did not receive tamoxifen adjuvant therapy. Thus, these results indicate that elevated SMRT expression may contribute to breast tumor progression for this group of patients independent of any effect it may have on promoting the antagonistic properties of tamoxifen hormonal therapy.
Expression of SMRT mRNA was detected in all cell lines examined by RT-qPCR, and this indicates that the SMRT gene is broadly expressed in breast cancer. However, levels varied extensively between different cell types suggesting that there are distinct cell-dependent differences in the regulation of SMRT mRNA synthesis or stability. Little is known about factors that regulate SMRT mRNA expression, but E2 or tamoxifen treatment of primary human skeletal muscle cells and MCF-7 cells reduces SMRT mRNA levels [30]. This was not, however, detected in a meta-analysis of microarray data obtained for E2-treated MCF-7 cells (http://www.nursa.org/gems/; Ref. [31]); the reason for this discrepancy is unknown, but may be related to the relative insensitivity of microarray detection in comparison to single gene assays. Expression of SMRT protein also was detected in most cell lines by western blot analyses, and as for the mRNA, there was significant variation in levels between different breast cancer cells. Remarkably, comparison of these parameters across all 30 lines yielded a poor correlation suggesting that post-transcriptional mechanisms exert a significant influence on SMRT protein expression. In support of this concept, it has been shown that phosphorylation of SMRT by Cdk2 leads to its association with Pin1 and decreased stability, thereby reducing SMRT protein expression [32].
Most of the breast tumors examined expressed SMRT protein, with greater than 70 % of tumors receiving a nuclear SMRT score of >4. This contrasts with a prior report in which nuclear SMRT protein was detected in only 16.7 % of breast tumors [22]. We also detected low levels of cytoplasmic SMRT in approximately one-third of the tumor specimens. Although SMRT, as a coregulator, is expected to be found in the nucleus, activation of IKK and MAPK signaling pathways has been shown to induce translocation of SMRT from the nucleus to the cytoplasm [33–35]. As some breast tumors would be expected to have elevated IKK and MAPK signaling, consequent to infiltration of macrophage producing pro-inflammatory cytokines or erbB2 overexpression, respectively [36, 37], the detection of some cytoplasmic SMRT is not surprising.
In the group of ER+ patients that did not receive adjuvant tamoxifen therapy higher levels of SMRT were associated with a shorter time to tumor recurrence which suggests that SMRT promotes breast cancer growth and/or progression. Prior work demonstrating the ability of SMRT to enhance the growth of ERα-positive MCF-7 breast cancer cells cultured in the presence of E2 and stimulate expression of the cell cycle regulator cyclin D1 is consistent with a role for the coregulator in promoting tumor recurrence in the absence of adjuvant therapy [20, 21]. It should be noted, however, that SMRT appeared to negatively impact the growth of MCF-7 cells in another study [38]. The reason for this discrepancy is unknown, but cell growth in the latter study was largely hormone insensitive suggesting the possibility of an ERα-independent effect. Whether the association between greater SMRT expression and reduced time to tumor recurrence reflects enhanced ERα-dependent growth is unknown. Nearly 80 % of the patients in the ERα+ untreated group are>50 years of age, suggesting that any estrogen/ER-dependent role of SMRT would be due to either an early, premenopausal promotion of E2-ERα activity or SMRT stimulation of ERα activity induced by the low levels of estrogen present following menopause.
Alternatively, elevated SMRT could promote recurrence independent of ERα via inhibition of the activity of other transcription factors such as other members of the nuclear receptor superfamily that serve to either maintain cell differentiation (e.g., RARα) or inhibit cell growth (e.g., VDR). Indeed, in prostate cancer, elevated levels of SMRT lead to altered VDR signaling and attenuation of vitamin D-induced growth inhibition [39, 40]. In an attempt to gain insight into whether the association of high SMRT and reduced time to tumor recurrence was dependent upon ERα expression, the ability of SMRT to impact breast cancer outcome was tested in a small group of patients (n = 101) with ERα-negative tumors that did not receive adjuvant therapy. Although the differences between groups did not reach statistical significance, there was a trend in this small group for higher levels of SMRT to be found in tumors of patients who experienced an early recurrence (P = 0.079). Combining the untreated, ERα-positive and ERα-negative patients for analysis of time to recurrence yielded a highly significant association with SMRT expression, further emphasizing that impact of elevated SMRT expression on tumor recurrence in tamoxifen-untreated patients independent of ERα expression. Consistent results were observed in the multivariate analysis.
In contrast to untreated patients, there was no relationship between SMRT expression and time to tumor recurrence for ER+ patients that received adjuvant tamoxifen therapy. This finding was somewhat surprising as classical models of the role of corepressors in regulating the activity of antagonist-bound ERα predict that high SMRT levels should promote the antiestrogen efficacy of tamoxifen. However, the role of SMRT in regulating proliferation of breast cancer cells in the presence of tamoxifen is controversial with several reports demonstrating increased proliferation in tamoxifen-treated, SMRT-depleted breast cancer cells [32, 38] while another paper revealed no difference in cell cycle progression of SMRT-depleted MCF-7 cells exposed to this antiestrogen [41]. This may reflect some redundancy between SMRT and NCoR as simultaneous depletion of both corepressors enhanced cell proliferation of tamoxifen-treated MCF-7 cells [41]. Thus, a simple interpretation of the data is that SMRT does not impact time to tumor recurrence for tamoxifen-treated patients. However, our findings of worse TTR in patients that did not receive tamoxifen with high SMRT levels suggest another possibility. Namely, the lack of discrimination found for tamoxifen-treated patients may represent a relative improvement in the response for patients with high SMRT such that their risk of tumor recurrence becomes similar to patients with low SMRT expression. It also is possible that the differences in SMRT expression present in primary breast tumors are not maintained in patients treated with tamoxifen as it has been demonstrated that tamoxifen treatment of MCF-7 cells significantly inhibits SMRT expression [30] raising the possibility that the absence of differences between groups reflects a general loss of SMRT in the remaining breast cancer cells. Finally, it must be noted that a greater percentage of the tamoxifen-treated patients had axillary node-positive disease, and lack of SMRT correlation with disease outcome may be a reflection of tumor differences between node-positive and node-negative patients.
There were no significant associations between SMRT and overall survival for any of the patient populations tested and these data suggest that while SMRT is associated with a shortened interval until tumor recurrence for a subset of patients, it does not have a significant effect on the aggressiveness of any resulting tumors. Two prior studies have examined SMRT expression in large predictive breast cancer studies. In the first, SMRT protein expression was determined by IHC in the primary tumors of patients that went on to receive a range of treatments including no adjuvant therapy, tamoxifen, gonadotropin-releasing hormone agonists or chemotherapy [22]. This report revealed that SMRT was an independent prognostic indicator of reduced overall survival and shorter disease free interval with a higher likelihood of local recurrence and distant metastasis. It is unknown why this study revealed an impact of SMRT expression on overall survival, whereas our study demonstrated an impact only on time to tumor recurrence, but it is possible that this reflects a difference in the patient populations and their treatments.
In contrast, in a subsequent report in which SMRT mRNA expression was determined by RT-qPCR, high SMRT mRNA levels were associated with a longer metastasis-free survival and overall survival [19] for patients that received surgery and no systemic therapy. This finding seemingly contrasts with our result and the report by Green et al. [22]. It is possible that differences in patient populations may contribute to this apparent discrepancy as the patients in the van Agthoven et al.’s report [19] were all lymph node negative whereas patients in the other two studies were a mixture of lymph node negative and positive. However, the very poor correlation observed for SMRT protein and mRNA expression in breast cancer cell lines provides support for another possibility; namely that the differing results arise from the assessment of SMRT mRNA rather than protein levels. Indeed, given this result and our findings in breast cancer cell lines, it could be argued that comparative analysis of SMRT protein and mRNA levels in human breast tumor specimens should be undertaken.
None of the two large published studies examining SMRT expression in human breast cancer [19, 22] nor the results presented herein, provide support for an association between SMRT expression and clinical benefit of tamoxifen, and this lends further strength to the conclusion that SMRT is not a major determinant of tamoxifen response in human breast cancer. Regardless, the clear association between SMRT and tumor recurrence in tamoxifen-untreated patients implicates a role for SMRT in breast tumorigenesis, and indicates that the actions of corepressors, like coactivators such as the SRC-3 oncogene, can contribute to disease pathogenesis.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (DK53002 to CLS, and a SPORE Pilot Project P50CA058183 to SO), the Department of Defense (W81XWH-08-1-0586 to CLS), the American Cancer Society (PF-07-233-01-TBE to MCP), the Proteomics and Biostatistics & Informatics Cores of the NCI Cancer Center Support Grant (P30CA125123) and the Breast Cancer SPORE Grant (P50CA058183).
Footnotes
Conflict of interest The authors declare that they have no conflict of interest.
Electronic supplementary material The online version of this article (doi: 10.1007/s10549-012-2262-7) contains supplementary material, which is available to authorized users.
Contributor Information
Carolyn L. Smith, Email: carolyns@bcm.edu, Department of Molecular & Cellular Biology, BCM130, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA. Department of Urology, Baylor College of Medicine, Houston, TX 77030, USA
Ilenia Migliaccio, Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, TX 77030, USA.
Vaishali Chaubal, Department of Molecular & Cellular Biology, BCM130, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA.
Meng-Fen Wu, Dan L. Duncan Cancer Center, Baylor College of Medicine, Houston, TX 77030, USA.
Margaret C. Pace, Department of Molecular & Cellular Biology, BCM130, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA
Ryan Hartmaier, Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, TX 77030, USA.
Shiming Jiang, Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, TX 77030, USA.
Dean P. Edwards, Department of Molecular & Cellular Biology, BCM130, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA. Department of Pathology & Immunology, Baylor College of Medicine, Houston, TX 77030, USA
M. Carolina Gutiérrez, Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, TX 77030, USA. Department of Pathology & Immunology, Baylor College of Medicine, Houston, TX 77030, USA.
Susan G. Hilsenbeck, Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, TX 77030, USA. Dan L. Duncan Cancer Center, Baylor College of Medicine, Houston, TX 77030, USA
Steffi Oesterreich, Department of Molecular & Cellular Biology, BCM130, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030, USA. Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, TX 77030, USA. The Women’s Cancer Research Center, University of Pittsburgh Cancer Institute, Pittsburgh, PA 15213, USA.
References
- 1.Ross RK, Paganini-Hill A, Wan PC, Pike MC. Effect of hormone replacement therapy on breast cancer risk: estrogen versus estrogen plus progestin. J Natl Cancer Inst. 2000;92(4):328–332. doi: 10.1093/jnci/92.4.328. [DOI] [PubMed] [Google Scholar]
- 2.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J Writing Group for the Women’s Health Initiative Investigators. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288(3):321–333. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 3.Anderson E. The role of oestrogen and progesterone receptors in human mammary development and tumorigenesis. Breast Cancer Res. 2002;4:197–201. doi: 10.1186/bcr452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clarke CA, Glasser SL, Uratsu CS, Selby JV, Kushi LH, Herrington LJ. Recent declines in hormone therapy utilization and breast cancer incidence: clinical and population-based evidence. J Clin Oncol. 2006;24:e49–e50. doi: 10.1200/JCO.2006.08.6504. [DOI] [PubMed] [Google Scholar]
- 5.Symmans WF, Hatzis C, Sotiriou C, Andre F, Peintinger F, Regitnig P, Daxenbichler G, Desmedt C, Domont J, Marth C, Delaloge S, Bauernhofer T, Valero V, Booser DJ, Hortobagyi GN, Pusztai L. Genomic index of sensitivity to endocrine therapy for breast cancer. J Clin Oncol. 2010;27:4111–4119. doi: 10.1200/JCO.2010.28.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu J, Wu RC, O’Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer. 2009;9:615–630. doi: 10.1038/nrc2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295(5564):2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- 8.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SAW, Wong J, Allred DC, Clark GM, Schiff R. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. J Natl Cancer Inst. 2003;95(5):353–361. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 9.Shou J, Massarweh S, Osborne CK, Wakeling AE, Ali S, Weiss H, Schiff R. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96(12):926–935. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 10.Smith CL, Nawaz Z, O’Malley BW. Coactivator and core-pressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol. 1997;11(6):657–666. doi: 10.1210/mend.11.6.0009. [DOI] [PubMed] [Google Scholar]
- 11.Scott DJ, Parkes AT, Ponchel F, Cummings M, Poola I, Speirs V. Changes in expression of steroid receptors, their downstream target genes and their associated co-regulators during the sequential acquisition of tamoxifen resistance in vitro. Int J Oncol. 2007;31(3):557–565. doi: 10.3892/ijo.31.3.557. [DOI] [PubMed] [Google Scholar]
- 12.Sarvilinna N, Eronen H, Miettinen S, Vienonen A, Ylikomi T. Steroid hormone receptors and coregulators in endocrine-resistant and estrogen-independent breast cancer cells. Int J Cancer. 2006;118(4):832–840. doi: 10.1002/ijc.21431. [DOI] [PubMed] [Google Scholar]
- 13.Chan CMW, Martin L-A, Johnston SRD, Ali S, Dowsett M. Molecular changes associated with the acquisition of oestrogen hypersensitivity in MCF-7 breast cancer cells on long-term oestrogen deprivation. Steroid Biochem Mol Biol. 2002;81(4–5):333–341. doi: 10.1016/s0960-0760(02)00074-2. [DOI] [PubMed] [Google Scholar]
- 14.Thenot S, Charpin M, Bonnet S, Cavailles V. Estrogen receptor cofactors expression in breast and endometrial human cancer cells. Mol Cell Endocrinol. 1999;156(1–2):85–93. doi: 10.1016/s0303-7207(99)00139-2. [DOI] [PubMed] [Google Scholar]
- 15.Magklara A, Brown TJ, Diamandis EP. Characterization of androgen receptor and nuclear receptor co-regulator expression in human breast cancer cell lines exhibiting differential regulation of kallikreins 2 and 3. Int J Cancer. 2002;100(5):507–514. doi: 10.1002/ijc.10520. [DOI] [PubMed] [Google Scholar]
- 16.Chan CMW, Lykkesfeldt AE, Parker MG, Dowsett M. Expression of nuclear receptor interacting proteins TIF-1, SUG-1, receptor interacting protein 140, and corepressor SMRT in tamoxifen-resistant breast cancer. Clin Cancer Res. 1999;5(11):3460–3467. [PubMed] [Google Scholar]
- 17.Kurebayashi J, Otsuki T, Kunisue H, Tanaka K, Yamamoto S, Sonoo H. Expression levels of estrogen receptor-α, estrogen receptor-β, coactivators, and corepressors in breast cancer. Clin Cancer Res. 2000;6(2):512–518. [PubMed] [Google Scholar]
- 18.Fleming FJ, Hill AD, McDermott EW, O’Higgins NJ, Young LS. Differential recruitment of coregulator proteins steroid receptor coactivator-1 and silencing mediator for retinoid and thyroid receptors to the estrogen receptor-estrogen response element by β-estradiol and 4-hydroxytamoxifen in human breast cancer. J Clin Endocrinol Metab. 2004;89(1):375–383. doi: 10.1210/jc.2003-031048. [DOI] [PubMed] [Google Scholar]
- 19.van Agthoven T, Sieuwerts AM, Veldscholte J, Meijervan Gelder ME, Smid M, Brinkman A, den Dekker AT, Leroy IM, van Ijcken WFJ, Sleijfer S, Foekens JA, Dorssers LCJ. CITED2 and NCOR2 in antioestrogen resistance and progression of breast cancer. Brit J Cancer. 2009;101(11):1824–1832. doi: 10.1038/sj.bjc.6605423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peterson TJ, Karmakar S, Pace MC, Gao T, Smith CL. The silencing mediator of retinoic acid and thyroid hormone receptor (SMRT) corepressor is required for full estrogen receptor-α transcriptional activity. Mol Cell Biol. 2007;27(17):5933–5948. doi: 10.1128/MCB.00237-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karmakar S, Gao T, Pace MC, Oesterreich S, Smith CL. Cooperative activation of cyclin D1 and progesterone receptor gene expression by the SRC-3 coactivator and SMRT corepressor. Mol Endocrinol. 2010;24:1187–1202. doi: 10.1210/me.2009-0480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Green AR, Burney C, Granger CJ, Paish EC, El-Sheikh S, Rakha EA, Powe DG, Macmillin RD, Ellis IO, Stylianou E. The prognostic significance of steroid receptor co-regulators in breast cancer: co-repressor NCOR2/SMRT is an independent indicator of poor outcome. Breast Cancer Res Treat. 2008;110(3):427–437. doi: 10.1007/s10549-007-9737-y. [DOI] [PubMed] [Google Scholar]
- 23.Migliaccio I, Wu M-F, Gutierrez C, Malorni L, Mohsin SK, Allred DC, Hilsenbeck SG, Osborne CK, Weiss H, Lee AV. Nuclear IRS-1 predicts tamoxifen response in patients with early breast cancer. Breast Cancer Res Treat. 2009;123(3):651–660. doi: 10.1007/s10549-009-0632-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harvey JM, Clark GM, Osborne CK, Allred DC. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. J Clin Oncol. 1999;17(5):1474–1481. doi: 10.1200/JCO.1999.17.5.1474. [DOI] [PubMed] [Google Scholar]
- 25.Mohsin SK, Clark GM, Havighurst T, Weiss H, Berardo M, Roanh LD, To TV, Zhang Q, Love RR, Allred DC. Progesterone receptor by immunohistochemistry and clinical outcome in breast cancer: a validation study. Modern Pathol. 2004;17(12):1545–1554. doi: 10.1038/modpathol.3800229. [DOI] [PubMed] [Google Scholar]
- 26.Karmakar S, Foster EA, Smith CL. Unique roles of p160 coactivators for regulation of breast cancer cell proliferation and estrogen receptor-α transcriptional activity. Endocrinology. 2009;150:1588–1596. doi: 10.1210/en.2008-1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allred DC, Harvey JM, Bernardo M, Clark GM. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod Pathol. 1998;11(2):155–168. [PubMed] [Google Scholar]
- 28.Hommel G. A stagewise rejective multiple test procedure based on a modified Bonferroni test. Biometrika. 1988;75:383–386. [Google Scholar]
- 29.Kao J, Salari K, Bocanegra M, Choi Y-L, Girard L, Gandhi J, Kwei KA, Hernandez-Boussard T, Wang P, Gazdar AF, Minna JD, Pollack JR. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS ONE. 2009;4(7):e6146. doi: 10.1371/journal.pone. 0006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dieli-Conwright CM, Spektor TM, Rice JC, Schroeder ET. Oestradiol and SERM treatments influence oestrogen receptor coregulator gene expression in human skeletal muscle cells. Acta Physiol. 2009;197(3):187–196. doi: 10.1111/j.1748-1716.2009.01997.x. [DOI] [PubMed] [Google Scholar]
- 31.Ochsner SA, Steffen DL, Hilsenbeck SG, Chen ES, Watkins CM, McKenna NJ. GEMS (Gene Expression MetaSignatures), a web resource for querying meta-analysis of expression microarray datasets: 17β-estradiol in MCF-7 cells. Canc Res. 2009;69(1):23–26. doi: 10.1158/0008-5472.CAN-08-3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stanya KJ, Liu Y, Means AR, Kao H-Y. Cdk2 and Pin1 negatively regulate the transcriptional corepressor SMRT. J Cell Biol. 2008;183(1):49–61. doi: 10.1083/jcb.200806172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoberg JE, Yeung F, Mayo MW. SMRT derepression by the IκB kinase α: a prerequisite to NF-κB transcription and survival. Mol Cell. 2004;16(2):245–255. doi: 10.1016/j.molcel.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 34.Jonas BA, Privalsky ML. SMRT and N-CoR corepressors are regulated by distinct kinase signaling pathways. J Biol Chem. 2004;279(52):54676–54686. doi: 10.1074/jbc.M410128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jonas BA, Varlakhanova N, Hayakawa F, Goodson M, Privalsky ML. Response of SMRT (Silencing Mediator of Retinoic Acid and Thyroid Hormone Receptor) and N-CoR (Nuclear Receptor Corepressor) corepressors to mitogen-activated protein kinase kinase kinase cascades is determined by alternative mRNA splicing. Mol Endo. 2007;21(8):1924–1939. doi: 10.1210/me.2007-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu E, Thor A, He M, Marcos M, Ljung BM, Benz C. The HER2 (c-erbB-2) oncogene is frequently amplified in in situ carcinomas of the breast. Oncogene. 1992;7:1027–1032. [PubMed] [Google Scholar]
- 37.Leek RD, Lewis CE, Whitehouse R, Greenall M, Clarke J, Harris AL. Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 1996;56:4625–4629. [PubMed] [Google Scholar]
- 38.Cheng X, Kao H-Y. G protein pathway suppressor 2 (GPS2) is a transcriptional corepressor important for estrogen receptor α-mediated transcriptional regulation. J Biol Chem. 2009;284:36395–36404. doi: 10.1074/jbc.M109.062109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khanim FL, Gommersall LM, Wood VHJ, Smith KL, Montalvo L, O’Neill LP, Xu Y, Peehl DM, Stewart PM, Turner BM, Campbell MJ. Altered SMRT levels disrupt vitamin D3 receptor signalling in prostate cancer cells. Oncogene. 2004;23:6712–6725. doi: 10.1038/sj.onc.1207772. [DOI] [PubMed] [Google Scholar]
- 40.Ting H-J, Bao B-Y, Reeder JE, Messing EM, Lee Y-F. Increased expression of corepressors in aggressive androgen-independent prostate cancer cells results in loss of 1α, 25-di-hydroxyvitamin D3 responsiveness. Mol Cancer Res. 2007;5:967–980. doi: 10.1158/1541-7786.MCR-06-0318. [DOI] [PubMed] [Google Scholar]
- 41.Keeton EK, Brown M. Cell cycle progression stimulated by tamoxifen-bound estrogen receptor-α and promoter-specific effects in breast cancer cells deficient in N-CoR and SMRT. Mol Endocrinol. 2005;19(6):1543–1554. doi: 10.1210/me.2004-0395. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.