

Abstract
Aims
For the characterization of endothelial progenitor cells (EPCs), commonly the markers CD34 and KDR have been used. CD133+/CD34−/KDR+ cells may represent more immature ‘early’ progenitors. In patients with coronary artery disease (CAD), a large fraction of EPCs carry the osteoblastic marker osteocalcin (OCN), which may mediate vascular calcification and abnormal repair. The aim of this study was to evaluate the expression of OCN+ ‘early’ EPCs in patients with risk factors (RFs) and a history of stable (history of stenting/coronary artery bypass grafting) or unstable CAD (myocardial infarction).
Methods and results
Medical history and blood samples from 282 patients (age 58 ± 16 years) with CAD or at least one RF (mean 2.5 ± 1.5) were analysed. For the analysis of EPC markers (CD133, CD34, KDR) and OCN, the flow cytometry of peripheral blood mononuclear cells was performed. Circulating OCN+/CD133+/CD34−/KDR+ cells (median counts [interquartile range] per 100 000 events) were 15 [4–41] in patients with RF (n = 199), 26 [1–136] in those with a history of stable (n = 57), and 246 [105–308] in those with a history of unstable CAD (n = 26; P < 0.001). The association with unstable CAD remained highly significant even after multivariate adjusting for RFs and the different characteristics of the groups. Osteocalcin positive ‘early’ EPCs trend to predict further events [HR for each doubling of the cell number: 1.20 (95% CI: 1.00–1.46), P = 0.06].
Conclusion
Circulating OCN+ ‘early’ EPCs are strongly associated with unstable CAD. Therefore, this particular subset of EPCs could mediate abnormal vascular repair and may help identifying patients with a more unstable phenotype of atherosclerosis.
Keywords: Coronary artery disease, Arthrosclerosis, Endothelial progenitor cells, Marker, Osteocalcin
Introduction
Atherosclerosis is a chronic disease, characterized by continuous vascular injury and repair. The role of endothelial progenitor cells (EPCs) in this process continues to emerge. In tissue ischaemia and endothelial damage, EPCs are mobilized from the bone marrow into the circulation and home to the affected areas to mediate tissue recovery.1 EPCs have been recently defined by cell surface markers, and their characterization has been done using CD34, CD133, KDR (kinase insert domain receptor), and others. The stem cell marker CD133 is highly expressed on immature cells and lost during maturation. Friedrich et al.2 demonstrated the existence of functionally highly active ‘early’ CD133+/CD34−/KDR+ cells, which differentiate to CD133+/CD34+/KDR+ cells over time. Experimentally, these ‘early’ CD133+/CD34−/KDR+ EPCs home to the site of ischaemia, are found in larger numbers in human unstable coronary lesions, and promote re-endothelization and a reduction in lesion size following vascular injury.2
We recently reported that coronary atherosclerosis is characterized by an increased proportion in EPCs carrying the osteoblastic marker osteocalcin (OCN); these ‘osteogenic’ EPCs may mediate abnormal repair and vascular calcification.3 Moreover, its performance as a peripheral marker of early atherosclerosis exceeded the commonly used systemic marker, highly sensitive C-reactive protein (hsCRP).3 In a further study, we demonstrated that in patients with early coronary atherosclerosis, circulating EPCs with an osteoblastic phenotype are retained within the coronary circulation, suggesting their functional role in the vascular repair process.4 Thus, vascular injury is likely associated with the activation of osteoblastic genes by EPCs, especially considering the overlap between endothelial and osteoblastic lineages.5
The role of this particular combination—the more ‘early’, highly active EPC phenotype (CD34−/CD133+) with osteogenic potential (OCN+)—in patients with cardiovascular (CV) risk factors (RFs) or coronary artery disease (CAD) is not yet known.
The aim of the current study was, therefore, to evaluate the expression and prognostic role of these cells in patients with CV RFs when compared with patients with CV disease (CVD).
Methods
Patient selection
We evaluated the medical history and analysed blood samples of a cohort of 282 patients with a blood draw for the EPC measurement. All patients were part of studies approved by the Mayo Clinic Institutional Review Board, and informed consent was obtained from all participants. All patients included had at least one CV RF (hypertension, family history of CVD, diabetes mellitus, dyslipidemia, smoking, and obesity) or established CVD. All blood examinations were performed after an overnight fast in the morning. Follow-up data were collected for each patient using the Mayo Clinic electronic medical record. Follow-up was recorded [median follow-up 519 (179 of 917) days].
Assessment of patient characteristics
Characteristics and CV RFs were obtained from the patients. Additional measurements related to coronary risk were evaluated, such as mean arterial blood pressure, heart rate, pulse pressure, body mass index (BMI), glomerular filtration rate (GFR) using the modification of diet in renal disease formula, lipid profile, glucose, and glycosylated haemoglobin. Blood samples were tested for red and white cell counts, electrolytes, and hsCRP. In addition, in a subset of patients, soluble intercellular adhesion molecule-1 ((ICAM-1) n = 51), soluble vascular cell adhesion molecule-1 (sVCAM-1; n = 53), interleukin-6 (IL-6; n = 53), tumour necrosis factor-α (TNF-α; n = 52), and lipoprotein-associated phospholipase A2 [Lp-PLA2 mass (n = 66) and Lp-PLA2 activity (n = 64)] were measured.
Risk factors were defined as follows: (i) systemic hypertension [arterial blood pressure >140/90 mmHg or the use of antihypertensive therapy (except if used for secondary prevention solely)], (ii) family history of CVDs in first-degree male relatives <55 or <65 years (female), (iii) diabetes mellitus (patients history and/or need for insulin or oral hypoglycaemic agents), (iv) dyslipidemia [patients history and/or total serum cholesterol level >240 mg/dL or treatment with lipid-lowering drugs (any patient taking lipid-lowering drugs was considered having dyslipidemia, even though it was for secondary prevention only)], (v) smoking (history of smoking), and (vi) obesity (BMI > 30).
For the analysis, the patients were divided into following groups: (I) patients with CV RFs (at least one CV RF) and absent criteria for Group II or III, (II) patients with a history of stable CAD (defined by a history of any stenting or coronary artery bypass grafting), and (III) patients with a history unstable CAD (any history of ST or non-ST-elevation myocardial infarction).
Flow cytometry
Flow cytometry was performed as described previously.4,6 In brief, peripheral blood mononuclear cells were isolated from fresh blood samples using a Ficoll density gradient, and immunofluorescent cell staining was performed using the following fluorescent conjugated antibodies: CD34-PerCP Cy 5.5 (Beckton-Dickinson), CD133-phycoerythrin (Miltenyi Biotec GmbH), and kinase insert domain receptor KDR-APC (R&D Systems) and the appropriate isotype controls. In addition, OCN+ cells were identified using an anti-human OCN antibody (Santa Cruz Biotechnology) and a fluorescein isothiocyanate secondary antibody (Jackson ImmunoResearch), as described previously. Cell fluorescence was measured immediately after staining (Becton Dickinson, FACS Calibur), and data were analysed using CellQuest software (Becton Dickinson). A total of 150 000 events were counted and final data were obtained within the lymphocyte gate. The frequency of positive cells was measured as the percentile of gated cells in fluorescent channels with activities above 99.7% for CD34, CD133, and KDR and above 99% for OCN of the corresponding isotype controls, thus including backgrounds below 0.3–1%. The results are expressed as counts per 100 000 events. The person doing the cell analysis was not aware of the results of the patient classification. Two operators analysed flow specimen (M.G. and A.J.F.).
Invasive endothelial function testing
A subset of patients (n = 37) had invasive endothelial function testing at the time of blood draw for EPCs. This procedure has previously been described.4,7 In brief, 5000 units of heparin were given intravenously, and a Doppler guidewire (Flowire, Volcano Inc.) within a coronary-infusion catheter (Ultrafuse, SciMed Life System) was positioned into the mid-portion of the left anterior descending coronary artery. Acetylcholine at increasing concentrations (10−6–10−4) was infused into the left anterior descending coronary artery to assess endothelium-dependent vasoreactivity. Coronary artery diameter was measured by an independent investigator in the segment 5 mm distal to the tip of the Doppler wire using a computer-based image analysis system. Average peak velocity (APV) was derived from the Doppler flow velocity spectra and coronary blood flow (CBF) was determined as π(coronary artery diameter/2)2 × (APV/2). Epicardial and microvascular endothelial function were assessed by the change in epicardial diameter and the change in CBF, respectively, in response to the maximal dose of acetylcholine (10−4 M).
Statistical analyses
Non-normally distributed data, including all EPC values, are presented as median [interquartile (25th–75th percentiles) range], and normally distributed variables are presented as mean ± standard deviation (SD). Discrete variables were summarized as frequencies and percentages. Differences between the different groups were compared using the Wilcoxon/Kruskal–Wallis (rank-sum) test. For correlation, the non-parametric Spearman method was used. For multivariate analysis, the numbers of ‘early’ OCN + EPC were log-transformed and a linear regression model was used. The Kaplan–Meier estimates were used to describe the event-free survival on follow-up with the log-rank test employed to test group differences. To further evaluate the prognostic value of these cells, a Cox proportional hazard analysis was performed, taking into account the higher risk for events in patients with a history of unstable CAD. All statistical tests were two-sided and P-values of <0.05 was considered to be statistically significant. JMP software 9.0.1, SAS Institute, was used as statistical software.
Results
Patient characteristics
Medical history and blood samples from 282 patients (age 58 ± 16.39 years, 61% male) were analysed. All patients had at least one classical RF (mean 2.5 ± 1.5) or established CVD. Clinical characteristics are presented in Table 1, and concomitant medical therapy is shown in Table 2. Laboratory data are displayed in Table 3.
Table 1.
Clinical characteristics
| Study population (n = 282) | Group I (n = 199) | Group II (n = 57) | Group III (n = 26) | P-value | |
|---|---|---|---|---|---|
| Age | 58 ± 16.4 | 54.1 ± 16.3 | 69.6 ± 11.4 | 65.5 ± 12.1 | <0.001 |
| BMI | 29.2 ± 5.6 | 29.0 ± 6.0 | 29.5 ± 4.6 | 29.5 ± 4.3 | 0.8 |
| sBP | 120.8 ± 19.4 | 119.4 ± 19.0 | 124.9 ± 20.3 | 122.1 ± 19.7 | 0.15 |
| dBP | 69.1 ± 10.5 | 69.8 ± 9.9 | 66.3 ± 10.9 | 70.5 ± 12.8 | 0.06 |
| Heart rate | 66.3 ± 10.8 | 67.2 ± 10.9 | 63.1 ± 9.5 | 66.5 ± 12.5 | 0.04 |
| Number of RF | 2.5 ± 1.5 | 2.1 ± 1.4 | 3.2 ± 1.2 | 3.5 ± 1.1 | <0.001 |
| Number of CV drugs | 2.8 ± 2.2 | 2.0 ± 1.9 | 4.2 ± 1.9 | 5.7 ± 1.5 | <0.001 |
| Male gender (%) | 61 | 50.5 | 87.7 | 84.6 | <0.001 |
| CV family history (%) | 41 | 38 | 48 | 48 | 0.3 |
| Dyslipidemia (%) | 72 | 61 | 94 | 100 | <0.001 |
| Hypertension (%) | 54 | 44 | 74 | 81 | <0.001 |
| Diabetes mellitus (any; %) | 13 | 7 | 28 | 31 | <0.001 |
| Obesity (BMI > 30; %) | 40 | 37 | 46 | 42 | 0.51 |
| Smoking history (%) | 27 | 21 | 35 | 54 | <0.001 |
Group I, patients with at least one CV RF; Group II, patients with a history of significant but stable CAD (history of stenting or coronary artery bypass grafting); Group III, patients with a history of myocardial infarction. sBP, systolic blood pressure; dBP, diastolic blood pressure; RF, risk factor; CV, cardiovascular; BMI, body mass index. P-value for Groups I–III.
Table 2.
Medications
| Study population (%) | Group I (%) | Group II (%) | Group III (%) | P-value | |
|---|---|---|---|---|---|
| ACE-inhibitors | 28 | 20 | 32 | 85 | <0.001 |
| Angiotensin receptor blocker | 10 | 7 | 19 | 12 | 0.05 |
| Aspirin | 57 | 45 | 84 | 88 | <0.001 |
| β-Blocker | 46 | 34 | 67 | 92 | <0.001 |
| Calcium channel-blocker | 19 | 18 | 23 | 19 | 0.69 |
| Clopidogrel | 13 | 5 | 25 | 42 | <0.001 |
| Digoxin | 5 | 4 | 5 | 8 | 0.71 |
| Diuretic (any) | 30 | 22 | 42 | 62 | <0.001 |
| Insulin | 4 | 2 | 9 | 11 | 0.01 |
| Multivitamins | 40 | 38 | 52 | 35 | 0.15 |
| Nitrates | 16 | 9 | 28 | 46 | <0.001 |
| Omega fatty acids | 21 | 18 | 32 | 19 | 0.08 |
| Statin | 48 | 36 | 74 | 77 | <0.001 |
| Other lipid-lowering drugs | 14 | 6 | 30 | 38 | <0.001 |
| Spironolactone or eplerenone | 4 | 1 | 4 | 16 | 0.01 |
| Oral anticoagulation | 14 | 14 | 13 | 15 | 0.94 |
Group I, patients with at least one CV RF; Group II, patients with significant CAD (history of stenting or CABG); Group III, patients with a history of myocardial infarction. P-value for Groups I–III.
Table 3.
Laboratory characteristics
| Study population | Group I | Group II | Group III | P-value | |
|---|---|---|---|---|---|
| Haemoglobin (g/dL) | 13.8 ± 1.3 | 14.0 ± 1.2 | 13.5 ± 1.3 | 13.4 ± 1.2 | 0.01 |
| WBC (×109/L) | 7.3 ± 3.8 | 7.2 ± 4.2 | 7.5 ± 2.8 | 7.2 ± 2.9 | 0.59 |
| Neutrophils (×109/L) | 4.3 ± 1.8 | 4.2 ± 1.8 | 4.6 ± 1.8 | 4.5 ± 1.6 | 0.47 |
| Eosinophils (×109/L) | 0.18 ± 0.12 | 0.17 ± 0.12 | 0.18 ± 0.12 | 0.21 ± 0.14 | 0.45 |
| Lymphocytes (×109/L) | 1.7 [1.3–2.2] | 1.7 [1.3–2.2] | 1.5 [1.2–2.1] | 1.9 [1.5–2.2] | 0.19 |
| Monocytes (×109/L) | 0.60 ± 0.24 | 0.58 ± 0.22 | 0.60 ± 0.21 | 0.69 ± 0.28 | 0.17 |
| Basophilic (×109/L) | 0.04 ± 0.02 | 0.04 ± 0.03 | 0.03 ± 0.01 | 0.03 ± 0.01 | 0.23 |
| Thrombocytes (×109/L) | 210.9 ± 59.9 | 216.7 ± 60.3 | 204.4 ± 57.0 | 189.1 ± 58.9 | 0.06 |
| Sodium (mmol/L) | 139.9 ± 2.5 | 140.2 ± 2.3 | 139.8 ± 2.4 | 138.9 ± 3.5 | 0.06 |
| Potassium (mmol/L) | 4.4 ± 0.4 | 4.4 ± 0.4 | 4.4 ± 0.5 | 4.2 ± 0.4 | 0.06 |
| GOT (U/L) | 31.3 ± 29.6 | 29.2 ± 11.4 | 38.1 ± 57.1 | 28.5 ± 11.1 | 0.33 |
| GPT (U/L) | 32.4 ± 14.4 | 33.4 ± 15.1 | 30.5 ± 9.9 | 28.1 ± 18.2 | 0.56 |
| GFR (mL/min/BSA) | 74.5 ± 18.1 | 77.0 ± 16.8 | 69.1 ± 18.8 | 73.7 ± 20.8 | 0.02 |
| Glucose (mg/dL) | 103.8 ± 25.4 | 100.4 ± 15.2 | 108.1 ± 31.5 | 121.7 ± 58.1 | 0.01 |
| HbA1c (%) | 5.9 ± 0.9 | 5.7 ± 0.7 | 6.4 ± 1.1 | 6.6 ± 1.0 | <0.001 |
| hsCRP (mg/L) | 0.45 [0.11–1.93] | 0.39 [0.09–1.9] | 0.76 [0.24–2.35] | 0.46 [0.14–1.22] | 0.71 |
| Cholesterol (mg/dL) | 175.7 ± 43.3 | 181.3 ± 41.2 | 166.9 ± 43.8 | 161.1 ± 49.5 | 0.02 |
| LDL (mg/dL) | 100.5 ± 35.8 | 104.1 ± 32.8 | 94.3 ± 4.8 | 92.2 ± 46.5 | 0.10 |
| HDL (mg/dL) | 48.6 ± 14.1 | 50.1 ± 14.7 | 45.4 ± 10.8 | 45.1 ± 14.4 | 0.04 |
| TG (mg/dL) | 126.8 ± 67.3 | 125.3 ± 66.2 | 134.7 ± 77.5 | 120.2 ± 49.7 | 0.57 |
Numbers expressed as means ± SD or medians [interquartile ranges]. Group I, patients with at least one CV RF; Group II, patients with a history of significant, but stable CAD (history of stenting or CABG); Group III, patients with a history of myocardial infarction. WBC, white blood count; GOT, glutamic oxaloacetic transaminase; GPT, glutamic pyruvic transaminase; BSA, body surface area; hsCRP, high sensitivity C-reactive protein; LDL, low-density lipoprotein; HDL, high-density lipoprotein; TG, triglycerides. P-value for trend groups I–III.
Osteocalcin positive ‘early’ endothelial progenitor cells in coronary artery disease
Unadjusted numbers of circulating OCN+ ‘early’ (CD133+/CD34−/KDR+) EPCs are displayed in Table 4. Circulating OCN+ ‘early’ EPCs are higher in patients with a history of stable and unstable CAD. Fifteen [4–41] counts—per 100 000 gated events—in patients with only RF (Group I), 26 [1–136] in patients with a history of stable, significant CAD (Group II), and 246 [105–308] in patients with a history of unstable CAD (Group III) (P < 0.001; P = 0.02 between Groups I and II, P < 0.001 between Groups I and III, and P < 0.001 between Groups II and III, Figure 1).
Table 4.
Circulating endothelial progenitor cells
| Study population | Group I | Group II | Group III | P-value | |
|---|---|---|---|---|---|
| ‘Early’ EPCs (CD133+/CD34−/KDR+) | 28 [5–89] | 22 [5–66] | 28 [0–92] | 381 [105–552] | <0.001 |
| ‘Early’ EPC and OCN+ | 18 [6–62] | 15 [4–41] | 26 [1–136] | 246 [108–308] | <0.001 |
| % of ‘early’ EPC which are OCN+ (%) | 58.6 | 54 | 55 | 72 | 0.58 |
Data presented as counts per 100 000 gated events (medians and 25th and 75th quartiles). Group I, patients with at least one CV RF; Group II, patients with a history of significant but stable CAD (history of stenting or CABG); Group III, patients with a history of myocardial infarction. EPC, endothelial progenitor cell; OCN, osteocalcin. P-value for Groups I–III.
Figure 1.
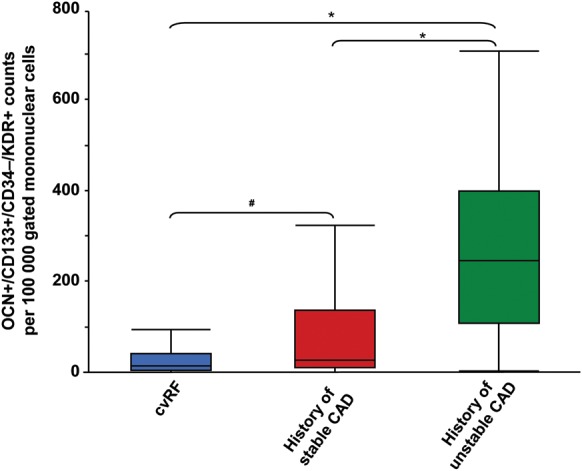
Osteocalcin positive ‘early’ endothelial progenitor cells. Figure depicts the counts (per 100 000 events) of ‘early’ osteocalcin positive endothelial progenitor cells (OCN+/CD133+/CD34−/KDR+). Blue: Group I, patients with risk factor but absent history of stent implantation, coronary artery bypass grafting, or myocardial infarction. Red: Group II, patients with a history of stable coronary artery disease (history of coronary artery bypass grafting or stenting). Green: Group III, patients with a history of unstable coronary artery disease (myocardial infarction). *P < 0.001, #P = 0.02.
A history of smoking and hypertension (P = 0.026 and 0.013, respectively) as well as the current use of statins, ACE inhibitors, diuretics, nitrates, and spironolactone/eplerenone (P-value all <0.05) was also associated with a larger number of OCN+ ‘early’ EPCs. Furthermore, there were significant correlations between OCN+ ‘early’ EPCs and age (r = 0.18, P = 0.004, n = 259), systolic blood pressure (r = 0.123, P = 0.048, n = 258), and the number of RFs (r = 0.17, P = 0.01, n = 259).
In a multivariate model adjusting for classical CV RFs (hypertension, smoking, diabetes, dyslipidemia, family history, obesity, gender, as well as age), the association between OCN+ ‘early’ EPC patients with a history of unstable CAD (Group III) remained highly significant (Group II: β = −0.40, P = 0.06; Group III: β = 1.52, P < 0.001). In another model, we included all variables with a significant association between OCN+ ‘early’ EPCs in the study population (hypertension, smoking, current use of statins, ACE-inhibitors, diuretics, nitrates and aldosterone antagonists, age, systolic blood pressure, and the number or RFs). Also, this model demonstrates a highly independent significant association of OCN+ ‘early’ EPC with unstable CAD (Group II: β = −0.60, P = 0.01; Group III: β = 1.58, P < 0.001). In both models, the additional inclusion of OCN negative early EPCs did not change the results. In a third model, we tried to take into account the different characteristics of the three study groups and included age, the number or RF, the number of CV medications, gender, haemoglobin, glomerular filtration rate, HbA1c, and high-density lipoprotein (HDL) into the analysis. Again, the association remained significant (Group II: β = −0.59, P = 0.11; Group III: β = 1.62, P < 0.001).
Prognostic impact of osteocalcin positive ‘early’ endothelial progenitor cells
To illustrate the potential prognostic value of OCN positive ‘early’ EPCs per se, a Kaplan–Meier estimate of event-free survival is shown in Figure 2. For this purpose, patients were divided into those with high and low cell counts (median split). Event-free survival was lower in patients with higher OCN+ ‘early’ EPC counts when compared with those with lower count (median split, P = 0.036). During the median follow-up of 519 (179 of 917) days, 17 events (4 death, 8 angina, 4 acute coronary syndrome (ACS), and 1 transient ischemic attack (TIA)) occurred in patients with the higher half of OCN+ ‘early’ EPCs and 5 (2 death, 2 TIA, and 1 angina) in those with the lower half. Events stratified according to the Groups I, II, and II were as follows: five (one death, one angina, and three TIA), nine (one death, six angina, and two ACS), and eight events (four deaths, two ACS, and two angina), respectively.
Figure 2.
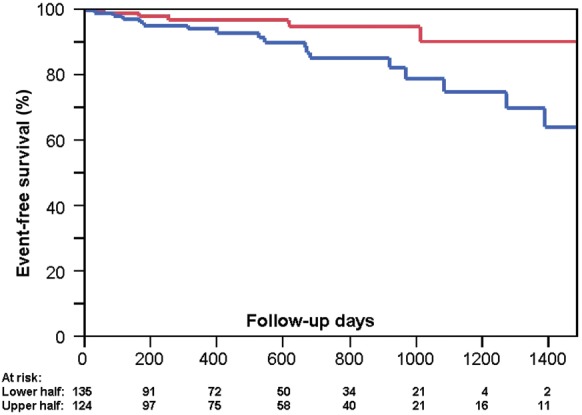
Event-free survival according to the level of osteocalcin positive ‘early’ endothelial progenitor cells. The Kaplan–Meier event-free survival according to a median split of osteocalcin positive ‘early’ endothelial progenitor cells. Values representing exactly the median value were attributed to the lower half group. Red: lower half of osteocalcin positive early endothelial progenitor cell count, n = 135, 5 events; Blue: upper half of osteocalcin positive early endothelial progenitor cell counts, n = 124, 17 events. P = 0.036 (logrank). Composite endpoint: death, MI, ACS, unstable angina pectoris, TIA, or stroke.
Because OCN+ ‘early’ EPCs are highly associated with the presence or the absence of a history of unstable CAD, a potentially strong confounder in the Kaplan–Meier analysis, the prognostic value was additionally assessed in a simple Cox proportional model. There ‘log2 OCN+ “early” EPCs’ have a tendency to predict further events [HR for doubling of the cell number: 1.20 (95% CI: 1.00–1.46), β = 0.18, P = 0.06; the absence of an unstable CAD history: β = −0.33, P = 0.22].
Osteocalcin positive ‘early’ endothelial progenitor cells and coronary endothelial function
In a subgroup of patients (n = 37), coronary vascular function testing was performed at the time of blood sampling for EPC measurements. The average change in coronary blood flow to acetylcholine was 102.5 ± 126.7%. Similar as we have shown previously,4 OCN+ ‘early’ EPCs negatively correlated with coronary microvascular function (r = −0.48, P = 0.003, Figure 3). Weaker correlations were also found with OCN+ CD34+/CD133+/KDR+ cells (r = −0.37, P = 0.02) but not with OCN+ CD34+/CD133−/KDR+ cells. Remarkably, there is no correlation with CD133, CD34, KDR, or OCN alone. Epicardial endothelial function did not correlate significantly with ‘early’ OCN+ EPCs [but with OCN+ CD34+/CD133+/KDR+ cells (r = −0.37, P = 0.02) and CD133 (r = −0.41, P = 0.01)].
Figure 3.
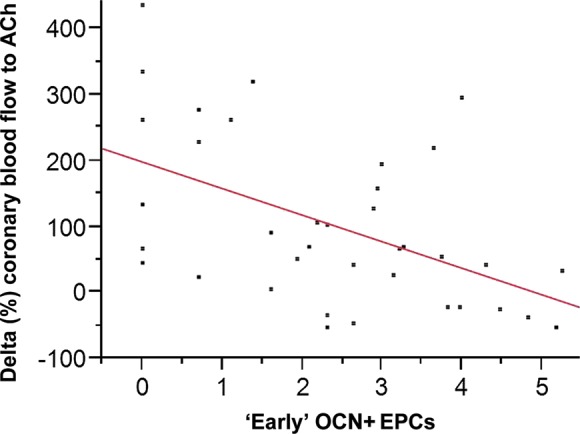
Correlation between ‘early’ osteocalcin positive endothelial progenitor cells and coronary microvascular endothelial function. The x-axis depicts ‘early’ osteocalcin positive endothelial progenitor cells (after log transformation) and the y-axis the % change in coronary blood flow with the maximal dose of acetylcholine. Spearman's correlation coefficient = −0.48, P = 0.003, n = 37.
Osteocalcin positive ‘early’ endothelial progenitor cells and inflammatory markers
Additionally to hsCRP, in a subgroup of patients, several inflammatory markers were measured. The average ICAM-1, VCAM-1, IL-6, and TNF-α serum levels were 186.3 ± 38.7 ng/mL, 557.6 ± 123.9 ng/mL, 1.1 [0.8–1.5] pg/mL, and 1.1 [0.7–1.5] pg/mL, respectively. Serum levels of Lp-PLA2 mass and activity were 242.2 ± 44.2 and 146.3 ± 37.1 ng/mL, respectively. Although there was no association of OCN+ ‘early’ EPCs with hsCRP, VCAM-1, IL-6, and TNF-α, OCN+ ‘early’ EPCs significantly correlated with ICAM-1 (r = 0.31, P = 0.025). Interestingly, we found a highly significant correlation of Lp-PLA2 mass with CD133+ cells (r = 0.41, P = 0.001), as well as with OCN+ CD133+/CD34+/KDR+ (r = 0.26, P = 0.04) and CD133+/CD34−/OCN+ (r = 0.28, P = 0.03) cells.
Discussion
In this study, we demonstrate for the first time an independent highly significant association between patients with a history of myocardial infarction and the circulating OCN+ CD133+/CD34−/KDR+ (‘early’) EPCs, a particular subset of vascular progenitor cells. This cell marker combination shows the best association with unstable CAD (history of myocardial infarction). Additionally, these cells seem to be linked to endothelial dysfunction and vascular-specific inflammation and might even be linked to the prognostication of future evens. Thus, the results of the current study may potentially hint towards a possible role of these cells in development, progression, and complications of coronary atherosclerosis and might serve as a marker for patients with an unstable phenotype of arthrosclerosis.
The exact role of EPCs in the development of atherosclerosis and in mediating ischaemic vascular disease is still unknown. Furthermore, counting the number of EPCs in blood does not take into account their functional status in vivo. Many ‘classical’ studies—after the first description of EPCs by Asahara8—demonstrated an inverse correlation with CV RFs,9 and a negative prognostic impact on event-free survival in patients with lower counts.10 However, recent research has provided conflicting results. In a population-based study, EPC numbers were positively correlated with CV RFs as expressed by Framingham risk score.11 One reason for these discrepancies could lie in the lack of an exact definition for EPCs as most studies used CD34 and KDR as surface markers.
In our study, we found CD133+ and KDR+, but CD34− and OCN+ circulating cells to be best associated with an unstable phenotype of CAD. One explanation for this finding, although speculative, might be the characteristic role of CD133+ cells, which represent ‘early’ progenitors for both endothelial cells and the haematopoietic system.12 CD133+ cells have the potential to differentiate also into mast cells,13 express cell surface proteins for adhesion molecules and receptors involved in transmigration. As typical for human mast cells, cultured CD133+ cells produce tryptase,14 an enzyme able to activate endothelial cell protease-activated receptor-2 expressed on the endothelial cell surface15 and to increase calcium-independent phospholipase A2 activity on human coronary artery endothelial cells, thus potentially playing an important role in propagating inflammatory response.16 Indeed, we here demonstrate a highly significant correlation of CD133+ cells with serum levels of Lp-PLA2, an enzyme particularly expressed by inflammatory cells in atherosclerotic, rupture-prone plaques17,18 with a high burden of macrophages.19 Similar to CRP,20 Lp-PLA2 is associated with CV outcomes,21 but is likely a better biomarker for vascular-specific inflammation.22 Interestingly, specific humeral factors released by vascular, but not systemic inflammation, lead to the recruitment of vascular progenitor cells.23 These cells, CD133+ in particular,24 may on one hand be responsible for vascular repair,25 but on the other hand their inflammatory paracrine activity may be harmful in certain microenvironmental conditions.26
As demonstrated by Friedrich et al.,2 there is an important population of CD133+/CD34−/KDR+ cells which have been shown to be highly active ‘early’ EPCs which may be able to differentiate to more ‘classical’ CD133+/CD34+/KDR+ EPCs. Experimentally, these ‘early’ EPCs home to the site of ischaemia, are found in larger numbers in human unstable coronary lesions, and promote re-endothelization and reduce lesion size following vascular injury.2,27 Interestingly, CD133+ EPCs have been demonstrated to be increased shortly after acute myocardial infarctions.28 Furthermore, in patients undergoing coronary artery stenting, those with restenosis in the follow-up have significantly larger numbers of CD133/KDR EPC than controls, an association not seen with CD34/KDR,29 a finding pointing towards a potential important role of this subset of EPCs.
The role of the expression of OCN on EPCs in CVD is continuing to emerge. Recently, increased bone turnover has been associated with vascular calcification and increased CV mortality,30–33 and EPCs are a potential candidate for providing a link between bone metabolism and the vascular system, especially since CD133+ and CD34+ cells are not only capable of differentiating into mature endothelial cells but also into osteoblastic cells.34,35 Furthermore, vascular calcification is an important process in atherosclerosis, and intimal microcalcification indeed contributes to destabilizing plaques.36 We recently demonstrated an increase in OCN expressing CD34+/KDR+ EPCs in patients with atherosclerosis as defined by endothelial dysfunction or multivessel disease.3 In a subsequent study in patients with early coronary atherosclerosis, circulating EPCs with the osteoblastic phenotype were shown to be retained within the coronary artery circulation, thus suggesting a functional role of these cells in the vascular repair process.4 Importantly, circulating cells expressing OCN found in peripheral blood have been shown to be able to calcify in vitro and in vivo.6 Recently, Fadini et al.37 demonstrated that calcifying progenitor cells (OCN and bone alkaline phosphatase positive progenitors) form ectopic calcification in vivo are over-represented in atherosclerotic lesions. Interestingly, these cells are increased in diabetic patients, especially in the presence of CVD.37
One could, therefore, speculate that in our patients with a history of myocardial infarction, the release of OCN+ early EPCs from the bone marrow is increased and the cells home to the sites of ischaemic vascular disease mediating repair and calcification. This might be supported by the observation that patients with a vascular injury or an acute myocardial infarction do mobilize EPCs.27,38
In the light of these studies and our results, one might conclude that the larger number of OCN+ early EPCs is an expression of arthrosclerosis, unstable in particular, possibly due to or augmented by microvascular dysfunction. Indeed, in a subgroup of patients in our study, where coronary microvascular function was available, we found a highly significant negative correlation of coronary endothelial microvascular function with OCN+ early EPCs. Low-grade ischaemic conditions might also contribute, as experimentally induced ischaemia by a simple exercise test in patients with symptomatic CAD demonstrated an increase in the number of circulating EPCs.39 Low-grade inflammation might be another mechanism behind increased EPC numbers in patients with atherosclerosis. Although hsCRP was not correlated with early OCN + EPC counts, interestingly, the more vascular-specific inflammation marker ICAM did correlated with ‘early’ EPC OCN counts in our study.
The interplay between OCN + EPC and CAD might involve bone morphogenetic proteins (BMPs), which are found in human calcified atherosclerotic plaques. They play a crucial role in atherosclerotic calcifications,40,41 are important in osteogenic cell signalling, and might directly programme vascular progenitors into an osteogenic direction.42 Interestingly, inflammation as well as oxidative stress stimulates the endothelium to up-regulate the secretion of BMPs,43 which again trigger the release of bone marrow-derived osteogenetic progenitor cells.44
Taken together, the increase in circulating ‘early’ EPCs might be a consequence of vascular disease and the fact that OCN is co-expressed in a very high percentage of these cells might point towards a more functional aspect of theses cells, mediating vascular calcification.
There are several limitations of our study. This is an analysis in patients of the whole spectrum of atherosclerosis. Thus, certain selection and treatment bias has to be taken into account, although we tried to correct for the later statistically and therefore believe that this does not jeopardize our results. Our finding of a potential prognostic role of the OCN expressing ‘early’ EPCs is not definitive and is limited by potential confounding and by the small numbers of outcome events, not justifying further multivariate adjustment. Thus, further studies should clarify the prognostic role of this particular subset EPCs. Furthermore, we have measured the amount of EPCs in the circulating blood; thus, we can only speculate about their functional role in pathophysiology. While we think that this study provides a strong rationale to evaluate OCN+ ‘early’ EPCs in future functional cell culture experiments, the strength of our results lies in the recognition of a potentially strong marker for an unstable phenotype of atherosclerosis.
In conclusion, we demonstrate a significant increase in ‘early’ OCN co-expressing EPCs in patients with CAD, particularly in those with a history of myocardial infarction, when compared with patients with CV RFs but no established coronary disease. Impaired coronary microvascular dysfunction and vascular-specific inflammation might be mechanistic explanations for these findings. These cells may play a role in the pathogenesis of unstable CAD and may potentially serve as a marker for prognostication.
Funding
This work was supported by the National Institute of Health (NIH) (HL-92954 and AG-31750 to A.L., and DK-73608, HL-77131, and HL-085307 to L.O.L.). A.J.F. is supported by the Walter and Gertrud Siegenthaler Foundation, the young academics Support Committee of the University of Zurich, and the Swiss foundation for medical-biological scholarships (SSMBS; SNSF No PASMP3_132551).
Conflict of interest: none declared.
References
- 1.Zampetaki A, Kirton JP, Xu Q. Vascular repair by endothelial progenitor cells. Cardiovasc Res. 2008;78:413–421. doi: 10.1093/cvr/cvn081. doi:10.1093/cvr/cvn081. [DOI] [PubMed] [Google Scholar]
- 2.Friedrich EB, Walenta K, Scharlau J, Nickenig G, Werner N. CD34-/CD133+/VEGFR-2+ endothelial progenitor cell subpopulation with potent vasoregenerative capacities. Circ Res. 2006;98:e20–e25. doi: 10.1161/01.RES.0000205765.28940.93. doi:10.1161/01.RES.0000205765.28940.93. [DOI] [PubMed] [Google Scholar]
- 3.Gossl M, Modder UI, Atkinson EJ, Lerman A, Khosla S. Osteocalcin expression by circulating endothelial progenitor cells in patients with coronary atherosclerosis. J Am Coll Cardiol. 2008;52:1314–1325. doi: 10.1016/j.jacc.2008.07.019. doi:10.1016/j.jacc.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gossl M, Modder UI, Gulati R, Rihal CS, Prasad A, Loeffler D, Lerman LO, Khosla S, Lerman A. Coronary endothelial dysfunction in humans is associated with coronary retention of osteogenic endothelial progenitor cells. Eur Heart J. 2010;31:2909–2914. doi: 10.1093/eurheartj/ehq373. doi:10.1093/eurheartj/ehq373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen JL, Hunt P, McElvain M, Black T, Kaufman S, Choi ES. Osteoblast precursor cells are found in CD34+ cells from human bone marrow. Stem Cells. 1997;15:368–377. doi: 10.1002/stem.150368. doi:10.1002/stem.150368. [DOI] [PubMed] [Google Scholar]
- 6.Eghbali-Fatourechi GZ, Lamsam J, Fraser D, Nagel D, Riggs BL, Khosla S. Circulating osteoblast-lineage cells in humans. N Engl J Med. 2005;352:1959–1966. doi: 10.1056/NEJMoa044264. doi:10.1056/NEJMoa044264. [DOI] [PubMed] [Google Scholar]
- 7.Hasdai D, Gibbons RJ, Holmes DR, Jr, Higano ST, Lerman A. Coronary endothelial dysfunction in humans is associated with myocardial perfusion defects. Circulation. 1997;96:3390–3395. doi: 10.1161/01.cir.96.10.3390. doi:10.1161/01.CIR.96.10.3390. [DOI] [PubMed] [Google Scholar]
- 8.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–967. doi: 10.1126/science.275.5302.964. doi:10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 9.Vasa M, Fichtlscherer S, Aicher A, Adler K, Urbich C, Martin H, Zeiher AM, Dimmeler S. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89:E1–7. doi: 10.1161/hh1301.093953. doi:10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- 10.Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Bohm M, Nickenig G. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353:999–1007. doi: 10.1056/NEJMoa043814. doi:10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- 11.Xiao Q, Kiechl S, Patel S, Oberhollenzer F, Weger S, Mayr A, Metzler B, Reindl M, Hu Y, Willeit J, Xu Q. Endothelial progenitor cells, cardiovascular risk factors, cytokine levels and atherosclerosis—results from a large population-based study. PLoS One. 2007;2:e975. doi: 10.1371/journal.pone.0000975. doi:10.1371/journal.pone.0000975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizrak D, Brittan M, Alison MR. CD133: molecule of the moment. J Pathol. 2008;214:3–9. doi: 10.1002/path.2283. doi:10.1002/path.2283. [DOI] [PubMed] [Google Scholar]
- 13.Dahl C, Hoffmann HJ, Saito H, Schiotz PO. Human mast cells express receptors for IL-3, IL-5 and GM-CSF; a partial map of receptors on human mast cells cultured in vitro. Allergy. 2004;59:1087–1096. doi: 10.1111/j.1398-9995.2004.00606.x. doi:10.1111/j.1398-9995.2004.00606.x. [DOI] [PubMed] [Google Scholar]
- 14.Rastogi P, White MC, Rickard A, McHowat J. Potential mechanism for recruitment and migration of CD133 positive cells to areas of vascular inflammation. Thromb Res. 2008;123:258–266. doi: 10.1016/j.thromres.2008.03.020. doi:10.1016/j.thromres.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molino M, Barnathan ES, Numerof R, Clark J, Dreyer M, Cumashi A, Hoxie JA, Schechter N, Woolkalis M, Brass LF. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J Biol Chem. 1997;272:4043–4049. doi: 10.1074/jbc.272.7.4043. doi:10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- 16.Meyer MC, Kell PJ, Creer MH, McHowat J. Calcium-independent phospholipase A2 is regulated by a novel protein kinase C in human coronary artery endothelial cells. Am J Physiol Cell Physiol. 2005;288:C475–C482. doi: 10.1152/ajpcell.00306.2004. doi:10.1152/ajpcell.00306.2004. [DOI] [PubMed] [Google Scholar]
- 17.Mannheim D, Herrmann J, Versari D, Gossl M, Meyer FB, McConnell JP, Lerman LO, Lerman A. Enhanced expression of Lp-PLA2 and lysophosphatidylcholine in symptomatic carotid atherosclerotic plaques. Stroke. 2008;39:1448–1455. doi: 10.1161/STROKEAHA.107.503193. doi:10.1161/STROKEAHA.107.503193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herrmann J, Mannheim D, Wohlert C, Versari D, Meyer FB, McConnell JP, Gossl M, Lerman LO, Lerman A. Expression of lipoprotein-associated phospholipase A(2) in carotid artery plaques predicts long-term cardiac outcome. Eur Heart J. 2009;30:2930–2938. doi: 10.1093/eurheartj/ehp309. doi:10.1093/eurheartj/ehp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Atik B, Johnston SC, Dean D. Association of carotid plaque Lp-PLA(2) with macrophages and Chlamydia pneumoniae infection among patients at risk for stroke. PLoS One. 2010;5:e11026. doi: 10.1371/journal.pone.0011026. doi:10.1371/journal.pone.0011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaptoge S, Di Angelantonio E, Lowe G, Pepys MB, Thompson SG, Collins R, Danesh J. C-reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta-analysis. Lancet. 2010;375:132–140. doi: 10.1016/S0140-6736(09)61717-7. doi:10.1016/S0140-6736(09)61717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson A, Gao P, Orfei L, Watson S, Di Angelantonio E, Kaptoge S, Ballantyne C, Cannon CP, Criqui M, Cushman M, Hofman A, Packard C, Thompson SG, Collins R, Danesh J. Lipoprotein-associated phospholipase A(2) and risk of coronary disease, stroke, and mortality: collaborative analysis of 32 prospective studies. Lancet. 2010;375:1536–1544. doi: 10.1016/S0140-6736(10)60319-4. doi:10.1016/S0140-6736(10)60319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Colley KJ, Wolfert RL, Cobble ME. Lipoprotein associated phospholipase A(2): role in atherosclerosis and utility as a biomarker for cardiovascular risk. EPMA J. 2011;2:27–38. doi: 10.1007/s13167-011-0063-4. doi:10.1007/s13167-011-0063-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Padfield GJ, Tura O, Haeck ML, Short A, Freyer E, Barclay GR, Newby DE, Mills NL. Circulating endothelial progenitor cells are not affected by acute systemic inflammation. Am J Physiol Heart Circ Physiol. 2010;298:H2054–H2061. doi: 10.1152/ajpheart.00921.2009. doi:10.1152/ajpheart.00921.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ruzicka K, Grskovic B, Pavlovic V, Qujeq D, Karimi A, Mueller MM. Differentiation of human umbilical cord blood CD133+ stem cells towards myelo-monocytic lineage. Clin Chim Acta. 2004;343:85–92. doi: 10.1016/j.cccn.2003.11.019. doi:10.1016/j.cccn.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 25.Rabelink TJ, de Boer HC, de Koning EJ, van Zonneveld AJ. Endothelial progenitor cells: more than an inflammatory response? Arterioscler Thromb Vasc Biol. 2004;24:834–838. doi: 10.1161/01.ATV.0000124891.57581.9f. doi:10.1161/01.ATV.0000124891.57581.9f. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y, Ingram DA, Murphy MP, Saadatzadeh MR, Mead LE, Prater DN, Rehman J. Release of proinflammatory mediators and expression of proinflammatory adhesion molecules by endothelial progenitor cells. Am J Physiol Heart Circ Physiol. 2009;296:H1675–H1682. doi: 10.1152/ajpheart.00665.2008. doi:10.1152/ajpheart.00665.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gill M, Dias S, Hattori K, Rivera ML, Hicklin D, Witte L, Girardi L, Yurt R, Himel H, Rafii S. Vascular trauma induces rapid but transient mobilization of VEGFR2(+)AC133(+) endothelial precursor cells. Circ Res. 2001;88:167–174. doi: 10.1161/01.res.88.2.167. doi:10.1161/01.RES.88.2.167. [DOI] [PubMed] [Google Scholar]
- 28.Voo S, Eggermann J, Dunaeva M, Ramakers-van Oosterhoud C, Waltenberger J. Enhanced functional response of CD133+ circulating progenitor cells in patients early after acute myocardial infarction. Eur Heart J. 2008;29:241–250. doi: 10.1093/eurheartj/ehm542. doi:10.1093/eurheartj/ehm542. [DOI] [PubMed] [Google Scholar]
- 29.Pelliccia F, Cianfrocca C, Rosano G, Mercuro G, Speciale G, Pasceri V. Role of endothelial progenitor cells in restenosis and progression of coronary atherosclerosis after percutaneous coronary intervention: a prospective study. JACC Cardiovasc Interv. 2010;3:78–86. doi: 10.1016/j.jcin.2009.10.020. doi:10.1016/j.jcin.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 30.Anagnostis P, Karagiannis A, Kakafika AI, Tziomalos K, Athyros VG, Mikhailidis DP. Atherosclerosis and osteoporosis: age-dependent degenerative processes or related entities? Osteoporos Int. 2009;20:197–207. doi: 10.1007/s00198-008-0648-5. doi:10.1007/s00198-008-0648-5. [DOI] [PubMed] [Google Scholar]
- 31.Chow JT, Khosla S, Melton LJ, 3rd, Atkinson EJ, Camp JJ, Kearns AE. Abdominal aortic calcification, BMD, and bone microstructure: a population-based study. J Bone Miner Res. 2008;23:1601–1612. doi: 10.1359/JBMR.080504. doi:10.1359/jbmr.080504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hofbauer LC, Brueck CC, Shanahan CM, Schoppet M, Dobnig H. Vascular calcification and osteoporosis—from clinical observation towards molecular understanding. Osteoporos Int. 2007;18:251–259. doi: 10.1007/s00198-006-0282-z. doi:10.1007/s00198-006-0282-z. [DOI] [PubMed] [Google Scholar]
- 33.Sambrook PN, Chen CJ, March L, Cameron ID, Cumming RG, Lord SR, Simpson JM, Seibel MJ. High bone turnover is an independent predictor of mortality in the frail elderly. J Bone Miner Res. 2006;21:549–555. doi: 10.1359/jbmr.060104. doi:10.1359/jbmr.060104. [DOI] [PubMed] [Google Scholar]
- 34.Matsumoto T, Kawamoto A, Kuroda R, Ishikawa M, Mifune Y, Iwasaki H, Miwa M, Horii M, Hayashi S, Oyamada A, Nishimura H, Murasawa S, Doita M, Kurosaka M, Asahara T. Therapeutic potential of vasculogenesis and osteogenesis promoted by peripheral blood CD34-positive cells for functional bone healing. Am J Pathol. 2006;169:1440–1457. doi: 10.2353/ajpath.2006.060064. doi:10.2353/ajpath.2006.060064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tondreau T, Meuleman N, Delforge A, Dejeneffe M, Leroy R, Massy M, Mortier C, Bron D, Lagneaux L. Mesenchymal stem cells derived from CD133-positive cells in mobilized peripheral blood and cord blood: proliferation, Oct4 expression, and plasticity. Stem Cells. 2005;23:1105–1112. doi: 10.1634/stemcells.2004-0330. doi:10.1634/stemcells.2004-0330. [DOI] [PubMed] [Google Scholar]
- 36.Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the vulnerable plaque. J Am Coll Cardiol. 2006;47:C13–C18. doi: 10.1016/j.jacc.2005.10.065. doi:10.1016/j.jacc.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 37.Fadini GP, Albiero M, Menegazzo L, Boscaro E, Vigili de Kreutzenberg S, Agostini C, Cabrelle A, Binotto G, Rattazzi M, Bertacco E, Bertorelle R, Biasini L, Mion M, Plebani M, Ceolotto G, Angelini A, Castellani C, Menegolo M, Grego F, Dimmeler S, Seeger F, Zeiher A, Tiengo A, Avogaro A. Widespread increase in myeloid calcifying cells contributes to ectopic vascular calcification in type 2 diabetes. Circ Res. 2011;108:1112–1121. doi: 10.1161/CIRCRESAHA.110.234088. doi:10.1161/CIRCRESAHA.110.234088. [DOI] [PubMed] [Google Scholar]
- 38.Shintani S, Murohara T, Ikeda H, Ueno T, Honma T, Katoh A, Sasaki K, Shimada T, Oike Y, Imaizumi T. Mobilization of endothelial progenitor cells in patients with acute myocardial infarction. Circulation. 2001;103:2776–2779. doi: 10.1161/hc2301.092122. doi:10.1161/hc2301.092122. [DOI] [PubMed] [Google Scholar]
- 39.Adams V, Lenk K, Linke A, Lenz D, Erbs S, Sandri M, Tarnok A, Gielen S, Emmrich F, Schuler G, Hambrecht R. Increase of circulating endothelial progenitor cells in patients with coronary artery disease after exercise-induced ischemia. Arterioscler Thromb Vasc Biol. 2004;24:684–690. doi: 10.1161/01.ATV.0000124104.23702.a0. doi:10.1161/01.ATV.0000124104.23702.a0. [DOI] [PubMed] [Google Scholar]
- 40.Nakagawa Y, Ikeda K, Akakabe Y, Koide M, Uraoka M, Yutaka KT, Kurimoto-Nakano R, Takahashi T, Matoba S, Yamada H, Okigaki M, Matsubara H. Paracrine osteogenic signals via bone morphogenetic protein-2 accelerate the atherosclerotic intimal calcification in vivo. Arterioscler Thromb Vasc Biol. 2010;30:1908–1915. doi: 10.1161/ATVBAHA.110.206185. doi:10.1161/ATVBAHA.110.206185. [DOI] [PubMed] [Google Scholar]
- 41.Yao Y, Bennett BJ, Wang X, Rosenfeld ME, Giachelli C, Lusis AJ, Bostrom KI. Inhibition of bone morphogenetic proteins protects against atherosclerosis and vascular calcification. Circ Res. 2010;107:485–494. doi: 10.1161/CIRCRESAHA.110.219071. doi:10.1161/CIRCRESAHA.110.219071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bostrom K, Watson KE, Horn S, Wortham C, Herman IM, Demer LL. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest. 1993;91:1800–1809. doi: 10.1172/JCI116391. doi:10.1172/JCI116391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Csiszar A, Smith KE, Koller A, Kaley G, Edwards JG, Ungvari Z. Regulation of bone morphogenetic protein-2 expression in endothelial cells: role of nuclear factor-kappaB activation by tumor necrosis factor-alpha, H2O2, and high intravascular pressure. Circulation. 2005;111:2364–2372. doi: 10.1161/01.CIR.0000164201.40634.1D. doi:10.1161/01.CIR.0000164201.40634.1D. [DOI] [PubMed] [Google Scholar]
- 44.Kimura Y, Miyazaki N, Hayashi N, Otsuru S, Tamai K, Kaneda Y, Tabata Y. Controlled release of bone morphogenetic protein-2 enhances recruitment of osteogenic progenitor cells for de novo generation of bone tissue. Tissue Eng Part A. 2010;16:1263–1270. doi: 10.1089/ten.TEA.2009.0322. doi:10.1089/ten.tea.2009.0322. [DOI] [PubMed] [Google Scholar]