

Abstract
The biosynthesis of fungal bicyclo[2.2.2]diazaoctane indole alkaloids with a wide spectrum of biological activities have attracted increasing interest. Their intriguing mode of assembly has long been proposed to feature a non-ribosomal peptide synthetase, a presumed intramolecular Diels-Alderase, a variant number of prenyltransferases, and a series of oxidases responsible for the diverse tailoring modifications of their cyclodipeptide-based structural core. Until recently, the details of these biosynthetic pathways have remained largely unknown due to lack of information on the fungal derived biosynthetic gene clusters. Herein, we report a comparative analysis of four natural product metabolic systems of a select group of bicyclo[2.2.2]diazaoctane indole alkaloids including (+)/(−)-notoamide, paraherquamide and malbrancheamide, in which we propose an enzyme for each step in the biosynthetic pathway based on deep annotation and on-going biochemical studies.
Introduction
Natural products continue to be a rich source of clinical drugs for treatment of human and animal diseases.1, 2 With respect to drug development, advanced understanding of their biosynthesis is significant for rational strain improvement efforts. This includes genetic manipulation (e.g. gene knock-out, knock-in, and whole gene cluster amplification) of the key biosynthetic and regulatory genes in order to increase the yield of pharmaceuticals to a desired level.3–6 Knowledge on biosynthesis is also valuable for guiding generation of novel natural product analogs as new drug candidates by metabolic engineering, mutasynthesis and allied approaches.7–11 In addition, biochemical characterization of diverse biosynthetic enzymes continues to reveal new catalytic mechanisms that inspire inventions of novel chemical and biological catalysts in organic chemistry for production of fine-chemical and medicinal agents.12,13
Elucidation of the biosynthetic pathway of a particular natural product or a family of natural products first requires identification of the gene cluster encoding its production.14–16 Next, the combined genetic (in vivo) and biochemical characterization (in vitro) of each individual biosynthetic enzyme provides important information, including enzyme substrate specificity, cofactor requirements, and the precise order of multiple biosynthetic steps.17,18 With this information available, it becomes possible to reconstitute the entire biosynthetic pathway in a heterologous host19–21 or in a multi-component in vitro reaction.22, 23
Across all microbes, plants and animals that generate natural products, it is particularly challenging to elucidate a biosynthetic pathway completely when unprecedented steps are involved, or precedent knowledge of biosynthetic origin is limited or non-existent. Conventionally, the hunting for such enzymes catalyzing these unusual biotransformations via unexplored mechanisms depends on implementing reasonable biosynthetic principles, and the scanning of the activity of all possible candidate enzymes against all hypothetical substrates.18,24,25 Thus, the entire process can require prolonged and intensive efforts, especially for those complex natural products assembled by a large number of biosynthetic enzymes.
Due to the discovery of natural products from different microorganisms bearing the same unique structural core, but varying from one another in their tailoring groups, opportunities for facile identification of unique enzymes arise. In this scenario comparative bioinformatic analysis suggests that homologous genes can be linked to formation of a common structural core, whereas cluster-specific genes provide the basis for structural differences.26–29 Recent advances in whole genome sequencing technology have made this approach rapid and cost-effective.30–34 Thus, identification of biosynthetic gene clusters for structurally related natural products from different microorganisms has become practical for comparative analysis of these systems. Deep annotation provides adequate information to develop hypotheses regarding key gene(s) and their protein products. This in turn guides experimental strategies to explore unusual biotransformation(s) of interest using genetic and/or biochemical approaches. Although considerable information can be gleaned from biosynthetic pathway mining and annotation, putative biochemical function can only be verified by analysis of the gene product in vitro using natural or suitable model substrates.
Herein, we present an example of the comparative analysis of biosynthetic gene clusters (mined from the whole genome) and pathways for four structurally related fungal indole alkaloids bearing the unusual bicyclo[2.2.2]diazaoctane core, including the anticancer agents (−)-notoamide A ((−)-1) and (+)-notoamide A ((+)-1),35, 36 the anthelmintic paraherquamide A (2),37–39 and the calmodulin-inhibitor malbrancheamide40-42 (3) (Fig. 1A) produced by Aspergillus sp. MF297-2,43 Aspergillus versicolor NRRL35600, Penicillium fellutanum ATCC20841, and Malbranchea aurantiaca RRC1813, respectively. These fungal natural products are assembled from an L-tryptophan, a second cyclic amino acid residue, and one or two isoprene units through biosynthetic pathways that are proposed to feature an intriguing intramolecular Diels Alderase (IMDAse), and a number of unique enantiomerically selective enzymes.44–49 The diverse bioactivities of this natural product family suggests that elucidation of their biosynthesis could direct future structural diversification via biosynthetic engineering, thereby leading to enhanced biological activities.
Figure 1.
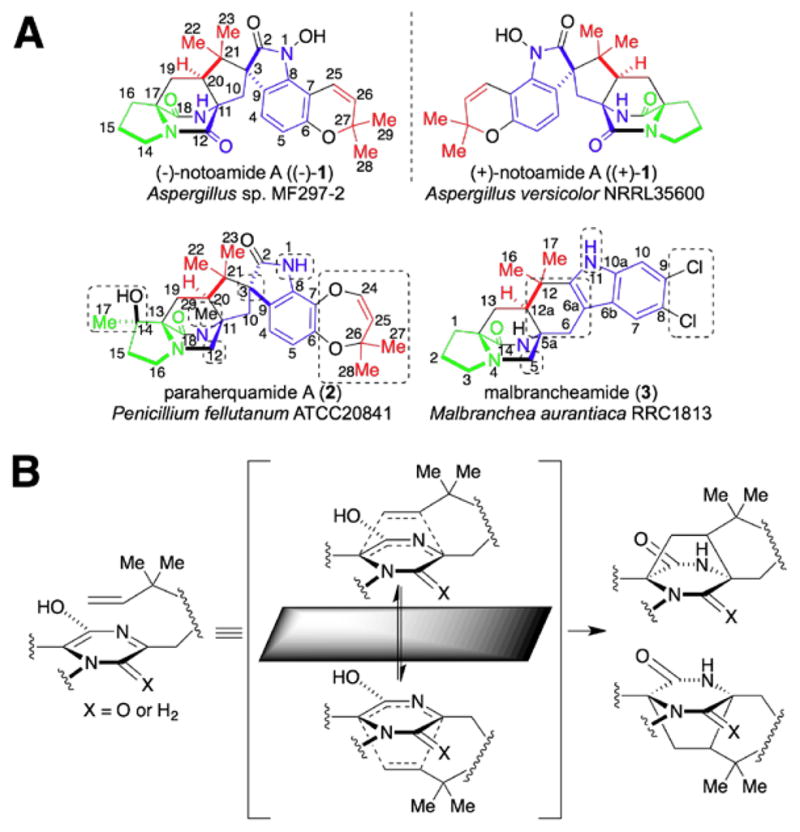
(A) Structures of (±)-notoamide A ((±)-1), paraherquamide A (2), and malbrancheamide (3). The unique structural features in 2 and 3 compared to 1 are highlighted in dashed boxes; (B) Proposed formation of the antipodal bicyclo[2.2.2]diazaoctane ring systems.
As expected, this comparative analysis provides significant insights into a number of intriguing biosynthetic questions: (1) which enzyme in each pathway is likely responsible for the formation of the bicyclo[2.2.2]diazaoctane core via the proposed intramolecular [4+2] Diels-Alder (IMDA) cyclization; (2) which enzyme in the pathway of 1 and 2 installs the spiro-oxindole functionality via a putative epoxide-initiated Pinacol-type rearrangement; and (3) what genetic difference controls formation of the dioxopiperazine in 1 versus the monooxopiperazine in 2 and 3.
Results and discussion
Structural similarity between 1–3 and its biosynthetic implications
The most significant structural similarity between 1–3 is the bicyclo[2.2.2]diazaoctane core (Fig. 1A). Biosynthetically, this unique structural moiety was proposed to arise from a [4+2] IMDA reaction (Fig. 1B).44, 46 This presumed cycloaddition reaction is also believed to catalyze the first enantiodivergent step in an otherwise common biosynthetic pathway from Aspergillus sp. MF297-2 and A. versicolor NRRL35600, leading to formation of (−)-1 and (+)-1, respectively, together with several other enantiomeric metabolites (Fig. 3).47 Currently, it remains unknown whether a specific IMDAse indeed exists in these biosynthetic pathways. However, if it does exist, one would expect its encoding gene should be present in all four gene clusters. Second, the spiro-oxindole is absent in 3, suggesting the responsible enzyme is likely absent from the pathway for 3, and present in those for 1 and 2. Third, a specific reductase responsible for reducing the tryptophan carbonyl group would be expected in the gene cluster of 2 and 3, but not 1. This genetic difference would account for the lack of the second amide carbonyl group in the piperazine ring of 2 and 3. Finally, the different hydroxylation status of the indole amide, distinct aromatic decoration among 1–3, together with other unique structural features including the tailoring of the proline moiety and N-methylation in 2, are also expected to be reflected at the genetic level.
Figure 3.

Proposed biosynthetic pathway for antipodal notoamide metabolites.
Localization and analysis of the gene clusters of (−)-notoamide (not), (+)-notoamide (not′), paraherquamide (phq), and malbrancheamide (mal) through genome mining
The genomes of A. versicolor NRRL35600, P. fellutanum ATCC20841, and M. aurantiaca RRC1813A harboring not′, phq, and mal gene clusters, respectively were sequenced to approximately 99, 84, and 181 times coverage of their estimated genome size (35 Mb), using the Illumina Solexa technology (Genome Analyzer IIx).
First, the key biosynthetic gene notE′ (Table 1) encoding a non-ribosomal peptide synthetase (NRPS) was mined from the genome sequences using the notE DNA sequence from the reported not gene cluster43 as a probe for homologous genes. NotE′, which shows 79% identity and 86% similarity to NotE at the amino acid (AA) level, was predicted to be a bimodular NRPS with the A-T-C-A-T-C (A: adenylation, T: thiolation, C: condensation) domain organization using the PKS/NRPS Analyzer (http://nrps.igs.umaryland.edu/nrps/). Genome walking from notE′ toward 5′ and 3′ ends identified another nine genes (notA′-J′, Table 1 and Fig. 2) that display high AA sequence similarity (>70%) with corresponding gene products of the not gene cluster. Notably, the overall nucleotide identity between notA′–J′ (25,440 bp) and notA–J (26,210 bp) is 71%, which is not surprising since both metabolic pathways are responsible for assembling “identical”, yet antipodal compounds. In addition to the high sequence similarity, the genetic architecture (i.e. order and direction of genes) within this region is identical in the two clusters (Fig. 2). The pattern of the exon/intron arrangement in the corresponding genes is also highly similar to each other (see Supplementary Information). In contrast, the sequence similarity is reduced drastically and the gene architecture differs after notK′/notK (Table 1, Fig. 2), strongly suggesting the previously assigned not gene cluster (notA–R) probably ends at notJ.
Table 1.
Comparative analysis* of gene clusters of not, not′, phq, and mal
| Not proteins (AA) | Function | Not′ proteins (AA) | Function (%identity to corresponding Not protein) | Phq proteins (AA) | Function (%identity to corresponding Not protein) | Mal proteins (AA) | Function (%identity to corresponding Not/Phq protein) |
|---|---|---|---|---|---|---|---|
| NotA (339) | Negative regulator | NotA′ (334) | Negative regulator (70% NotA) | PhqA (405) | Prenyltransferase (22% NotC) | MalA (667) | Halogenase (−/−) |
| NotB (456) | FAD monooxygenase | NotB′ (455) | FAD monooxygenase (88% NotB) | PhqB (2449) | NRPS [A-T-C-A-T-R] (26% NotE) | MalB (369) | Prenyltransferase (28% NotC/34% PhqA) |
| NotC (427) | Prenyltransferase | NotC′ (426) | Prenyltransferase (87% NotC) | PhqC (353) | 2OG-Fe(II)-oxygenase (−) | MalC (264) | Short chain dehydrogenase (−/52% PhqE) |
| NotD (621) | Oxidoreductase | NotD′ (612) | Oxidoreductase (80% NotD) | PhqD (322) | Pyrroline-5-carboxylate reductase (−) | MalD (336) | Negative regulator (36% NotA/55% PhqG) |
| NotE (2241) | NRPS [A-T-C-A-T-C] | NotE′ (2225) | NRPS [A-T-C-A-T-C] (79% NotE) | PhqE (265) | Short chain dehydrogenase (−) | MalE (438) | Prenyltransferase (36% NotF/34% PhqJ) |
| NotF (453) | Prenyltransferase | NotF′ (435) | Prenyltransferase (79% NotF) | PhqF (411) | Efflux pump (18% NotK) | MalF (590) | Oxidoreductase (37% NotD/39% PhqH) |
| NotG (544) | P450 monooxygenase | NotG′ (544) | P450 monooxygenase (87% NotG) | PhqG (338) | Negative regulator (34% NotA) | MalG (2345) | NRPS [A-T-C-A-T-R] (27% NotE/37% PhqB) |
| NotH (502) | P450 monooxygenase | NotH′ (499) | P450 monooxygenase (84% NotH) | PhqH (602) | Oxidoreductase (34% NotD) | ||
| NotI (434) | FAD monooxygenase | NotI′ (433) | FAD monooxygenase (85% NotI) | PhqI (462) | Prenyltransferase (−) | ||
| NotJ (371) | Unknown | NotJ′ (362) | Unknown (80% NotJ) | PhqJ (406) | Prenyltransferase (32% NotF) | ||
| NotK (564) | Efflux pump | NotK′ (577) | Efflux pump (14% NotK) | PhqK (459) | FAD monooxygenase (32% NotI) | ||
| NotL (484) | Transcriptional activator | NotL′ (620) | Transcriptional factor (15% NotL) | PhqL (563) | P450 monooxygenase (29% NotG) | ||
| NotM (402) | Unknown | NotM′ (454) | Unknown (−) | PhqM (536) | P450 monooxygenase (15% NotH) | ||
| NotN (340) | Dehydrogenase | NotN′ (416) | Unknown (−) | PhqN (326) | Methyltransferase | ||
| NotO (331) | Short-chain dehydrogenase | NotO′ (462) | Unknown (−) | PhqO (451) | P450 monooxygenase (−) | ||
| NotP (322) | Unknown | NotP′ (292) | Unknown (−) | ||||
| NotQ (152) | Unknown | NotQ′ (506) | Transcription factor | ||||
| NotR (461) | Transcriptional coactivator | NotR′ (172) | Unknown |
Genes were predicted using the FGENESH-M tool from http://www.softberry.com; Functions of gene products were predicted using BLAST search
Homology cannot be calculated due to unrelatedness
Figure 2.

The (−)-notoamide A (not), (+)-notoamide A (not′), paraherquamide (phq), and malbrancheamide (mal) biosynthetic gene clusters identified from genome sequencing and bioinformatic mining of Aspergillus sp. MF297-2, Aspergillus versicolor NRRL35600, P. fellutanum ATCC20841, and M. aurantiaca RRC1813, respectively. Homology of open reading frames across gene clusters is shown by same colored arrows. The not and not′ genes in the red box are unlikely involved in notoamide biosynthesis.
At the genetic level, it is not possible to glean the key differences that account for production of antipodal notoamide metabolites, suggesting that subtle active site sequence variation in those enantiomerically selective enzymes play a critical role in the control of absolute chirality. This requires direct biochemical analysis of the key notoamide biosynthetic enzymes, including structural biology efforts, which is currently ongoing in our laboratories.
Second, the paraherquamide (phq) gene cluster (47,884 bp) was identified from the partially assembled P. fellutanum genome by using a select group of not genes including the NRPS gene notE, the prenyltransferase genes notC and notF, and the P450 monooxygenase gene notG as in silico probes.43 Fifteen genes were identified that are likely involved in paraherquamide biosynthesis. The largest number of biosynthetic genes among the four studied metabolic pathways is consistent with 2 as the most complex structure compared to 1 and 3. Comparative bioinformatic analysis demonstrates that nine (phqA, B, F, G, H, J, K, L, and M) out of fifteen total phq genes are homologous to corresponding not genes (Table 1), although their homology is significantly lower than that between not and not′ genes. Notably, the bimodular phqB NRPS gene is different from notE in that a reductase (R) domain is located at its carboxy terminus instead of a condensation (C) domain, which is found in notE and notE′. This difference is significant because the reductase (vs condensation) domain is presumed to account for the presence of the monooxopiperazine in 2 (vs dioxopiperazine in 1) (see below).50 Among the remaining six cluster-specific genes, phqC shows high sequence similarity to 2-oxoglutarate (2OG) and Fe(II) dependent oxygenases.51,52 The phqD and phqE genes, which putatively encoding a pyrroline-5-carboxylate reductase and a short chain dehydrogenase, respectively, might be involved in the formation of the β-methyl-proline starter unit. The phqI gene that encodes the third prenyltransferase in phq is unique as it is free of introns, and therefore, distinct from the single intron-containing prenyltransferase genes phqA/notC and phqJ/notF. It is worth noting that the presence of three prenyltransferase genes is inconsistent with the two isoprene groups incorporated into the structure of 2. Thus, it is of special interest to examine whether the third prenyltransferase gene is redundant or plays an alternative, and as yet unknown function in the biosynthesis of 2. Furthermore, phqN is predicted to function as a methyltransferase, likely responsible for the N-methylation in 2. Finally, the phqO P450 gene with a unique exon/intron organization pattern is hypothesized to catalyze the C14 hydroxylation of the β-methyl-proline moiety.
Third, the seven-gene containing mal gene cluster (20179 bp) was mined from the genome of Malbranchea aurantiaca RRC1813A using phqB as an in silico probe to identify the metabolic system for 3. It has the smallest size among gene clusters of 1–3, which is consistent with the simplest structure and corresponding biosynthetic pathway. The genes malB, malD, malE, malF, and malG are common to the four gene clusters. Thus, except for the regulatory gene of malD (homologous to notA, notA′ and phqG), the remaining four biosynthetic genes (and their homologues in not, not′ and phq) are possibly responsible for installing the shared structural features of 1–3. This strongly suggests that the hypothetical Diels Alderase (if extant) should be represented by one of these four gene products (see below). Interestingly, the mal genes show greater sequence similarity to phq genes than not (or not′) genes, perhaps indicating their closer evolutionary relationship. Similar to PhqB, the NRPS MalG harbors a reductase domain at its carboxy terminus, which is consistent with the monooxopiperazine moiety in 3. Again, the apparent redundancy of the second prenyltransferase (3 only contains one isoprene group) is difficult to rationalize, but genetic disruption or RNA silencing (malB or malE) efforts are likely to shed light on the individual role of these enzymes. Finally, it is evident that the flavin-dependent halogenase MalA is likely involved in the introduction of one or both chlorine atoms in the biosynthesis of 3.
Biosynthetic pathway of notoamide A ((−)-1 and (+)-1)
Since the discovery of the biosynthetic gene cluster of (−)-1 from marine Aspergillus sp. MF297-2, in vitro biochemical characterization of the reverse prenyltransferase NotF using the NRPS (NotE) product brevianamide F53 (4) as substrate and the normal prenyltransferase NotC using 6-hydroxy-deoxybrevianamide E (6) as substrate has partially established the early steps of the notoamide pathway leading to notoamide S (7) (Fig. 3).43 The P450 monooxygenase NotG is likely catalyzing the C6 indole hydroxylation since its close homologue FtmC (59%/72% identity/similarity) in fumitremorgin biosynthesis had been characterized to hydroxylate the analogous aromatic C-H bond in the indole ring of tryprostatin B,54,55 which is structurally similar to deoxybrevianamide E (5).56
As the proposed pivotal branching point in notoamide biosynthesis,47,57,58 7 can be diverted to notoamide E (8) through an oxidative pyran ring closure putatively catalyzed by either NotH P450 monooxygenase (based on precedented examples of pyran ring formation from the epoxide intermediate generated by P450 enzymes59), or the NotD oxidoreductase. This step would be followed by an indole 2,3-epoxidation-initiated Pinacol-like rearrangement catalyzed by NotB FAD monooxygenase (FMO) leading to the formation of notoamide C (9) and notoamide D (10).58 Notably, notB (or notB′) is only observed in the not (or not′) gene cluster, consistent with the fact that this branching pathway leading to natural products 9 and 10 is only observed in notoamide biosynthesis.
On the other hand, extensive precursor feeding and incorporation studies using stable isotopically labeled intermediates have supported 7 as the substrate for the hypothetical IMDA.47 As a working hypothesis, a two-electron oxidation catalyzed by an oxidase would give rise to the achiral azadiene intermediate (11), which may immediately undergo a spontaneous stereoselective [4+2] IMDA cyclization in the active site of the same oxidase, yielding either (+)-notoamide T ((+)-12) in Aspergillus sp. MF297-2 or (−)-notoamide T ((−)-12) in A. versicolor. The opposing conformation (endo/exo) assumed by achiral 11 presumably determined by the scaffolding of each putative Diels-Alderase might account for the enantio-divergence at this key step. The five oxidases encoded by the not gene cluster, include FMO NotB and NotI, P450 enzymes NotG and NotH, and the FAD-dependent oxidoreductase NotD. NotB was recently identified as the notoamide E oxidase.58 NotI is highly similar to NotB with 42% protein sequence identity and 59% similarity, and is predicted to catalyze a similar conversion from (+)-stephacidin A60 ((+)-13) to (−)-notoamide B ((−)-14) via the 2,3-epoxidation of (+)-13 followed by a Pinacol-type rearrangement. Thus, if the putative function of NotG (see above) is correct, NotH (or NotD) is likely the bifunctional oxidase that also functions as the IMDAse responsible for generation of (+)-12. To generate antipodal (−)-12, NotH′ (or NotD′) is expected to catalyze a Diels Alder reaction leading to the opposite stereochemistry. Currently, this hypothesis is being tested in our laboratories through in vitro characterization of NotH/NotH′ (or NotD/NotD′). With comparative analysis of four gene clusters (Table 1), it appears that NotD/NotD′ is more likely to serve as the IMDAse since its homologs (PhqH and MalF) are present in all clusters. This hypothesis is based on the assumption that these four biosynthetic pathways use the same type of protein scaffolding enzyme to catayze the [4+2] cycloaddition. However, we have recently begun to challenge this assumption (see below). Presently, the possibility that NotH/NotH′ functions as the IMDAse in notoamide biosynthesis cannot be excluded. Once its identity is determined, the final oxidase NotD (or NotH) will likely be found to catalyze the oxidative pyran ring formation (Fig. 3).
Another important fact of these two related notoamide pathways is that enzymes catalyzing the biosynthetic steps after formation of 12 must also be enantiomerically and diastereochemically selective. Specifically, in previous precursor incorporation studies of racemic 13C-labeled (±)-13 with Aspergillus sp. MF297-2 and A. versicolor,61 it was ascertained that only one enantiomer of 13 can be processed (currently presumed by NotI and NotI′) to form downstream products. Understanding the subtle differences between these two enzymes will likely provide significant insights into how related enzymes have evolved to adopt opposing enantiomeric selectivity.
Finally, it remains unclear which enzyme could be responsible for the final hydroxylation steps leading to notoamide A (1) and sclerotiamide62 (15) since all five oxidative enzymes in the not(′) gene cluster has been assigned a putative function. It is possible that 1 and 15 are opportunistically produced upon the activity of unknown oxidases whose genes reside outside of the defined notoamide gene cluster. Alternatively, the possibility that a not oxidase may possess bi-functionality cannot be excluded.
Biosynthetic pathway of paraherquamide A (2)
Previous feeding studies demonstrated that L-isoleucine is the precursor to the β-methylβ-hydroxy proline moiety in 2.45, 63 Identification of the pyrroline-5-carboxylate reductase PhqD and the short chain dehydrogenase PhqE from phq cluster suggests a reasonable pathway from L-isoleucine to β-methyl proline (Fig. 4). Similar to the partially identified biosynthesis of 4-methyl proline in cyanobacterial Nostoc sp.,64 PhqE presumably oxidizes the terminally hydroxylated L-isoleucine (by an unknown enzyme) to the corresponding aldehyde. Spontaneous cyclization and dehydration would yield the 4-methyl pyrolline-5-carboxylic acid, which is then reduced by PhqD leading to the β-methyl proline precursor.
Figure 4.

Proposed biosynthetic pathway for paraherquamide A.
The presence of a C-terminal NAD(P)-dependent reductase domain in the bimodular paraherquamide NRPS (A-T-C-A-T-R) clearly indicates that the mechanism for dipeptide release by PhqB must be different from the final condensation domain of NotE (Fig. 3).50 What likely occurs is that the PhqB R domain utilizes NADPH for hydride transfer to reduce the thioester bond of the T domain-tethered linear dipeptide to a hemithioaminal intermediate, which spontaneously cleaves the C-S bond to release the aldehyde product. Subsequently, the acid-activated aldehyde is intramolecularly trapped by the nucleophilic amine from the adjacent amino acid to form a hemiaminal intermediate, which then undergoes a spontaneous dehydration and double bond rearrangement leading to formation of the monooxopiperazine intermediate 16 (likely existing as the enol form) prior to all other biosynthetic steps. This hypothesis is in good agreement with previous observations65,66 that the dioxopiperazine analog of preparaherquamide (17) cannot be incorporated into 2 by P. fellutanum since all substrates for downstream enzymes should bear the monooxopiperazine ring system. In this scheme (Fig. 4), formation of the diene in 16 is achieved by a reductive process, as opposed to the 2e− oxidation step proposed in the notoamide biosynthetic pathway (Fig. 3). If this is correct, in contrast to an oxidase (NotH/NotH′ or NotD/NotD′) proposed to be the Diels Alderase in notoamide biosynthesis, the reverse prenyltransferase (proposed to be PhqJ) might act as the scaffold for an IMDA reaction after introduction of the reverse prenyl group to 16. In this proposed route, the terminal double bond of the isoprene group would become the dienophile to react with the azadiene in the prenyltransferase active site, thus resulting in formation of the [2.2.2] diazaoctane intermediate 17.
Following formation of 17, the pyran ring formation is proposed to be installed by PhqA prenyltransferase (22% identical to NotC), PhqL (29% identical to NotG) and PhqH oxidoreductase (34% identical to NotD) (or PhqM P450 enzymes (15% identical to NotH)). The FMO PhqK (32% identical to NotI) is likely responsible for generation of the spiro-oxindole, and the N-methylation is likely mediated by the PhqN methyltransferase leading to the isolable natural product paraherquamide F38,67 (18). However, the order of these biosynthetic steps cannot be predicted without further in vivo genetic studies and/or in vitro biochemical analysis.
In late-stage paraherquamide biosynthesis, the third P450 monooxygenase PhqO is probably responsible for the C14 hydroxylation, transforming 18 to paraherquamide G38,67 (19), and paraherquamide E38,67 (20) to the final product 2. However, expansion from the 6-membered ring pyran (in 18 and 19) to the 7-membered dioxepin ring (in 2 and 20) represents a poorly understood but intriguing process. Possibly, phqC that encodes a 2OG-Fe(II)-oxygenase is involved in this ring expansion, which is consistent with previous reports showing this class of enzyme functioning as an expandase.68
Finally, the biosynthetic genes, including phqI as well as phqM (or phqH, the one uninvolved in the pyran ring formation), do not have a clearly prescribed role and appear to be redundant. This redundancy is currently being tested by gene knock-out studies in our laboratories.
Biosynthetic pathway of malbrancheamide (3)
Except for using L-proline instead of β-methyl proline as the starter unit, the biosynthetic route through premalbrancheamide (21) (Fig. 5) is proposed to parallel that of paraherquamide biosynthesis through 17 (Fig. 4). Mediated by NRPS MalG (A-T-C-A-T-R, 37% identical to PhqB) and prenyltransferase MalE (36%/34% identical to NotF/PhqJ), 21 is produced with its structure slightly different from 17 in lacking the C1 methyl group.
Figure 5.

Proposed biosynthetic pathway for malbrancheamide natural products.
Subsequently, the halogenase MalA presumably chlorinates the C9 position (malbrancheamide numbering) first to afford the isolable natural product malbrancheamide B (22), which could be further chlorinated by MalA at C8 leading to the final product malbrancheamide (3). This putative pathway is partially supported by the previous feeding study showing that the 13C labeled 21 can be incorporated into 22 by M. aurantiaca.69 Lack of observed 13C labeled 3 from the fermentation broth was interpreted to suggest that the second chlorination might be too slow to incorporate detectable levels of 13C material from 22 to 3. Notably, the order of these two chlorinations seems unexchangeable since the C8-monochloro regioisomer of 22 (C9-monochlorinated) was not detected as a natural product despite considerable effort.42 It is also possible that the dichloro species, malbrancheamide, arises from a pre-halogenated tryptophan-based assembly.
Blast (http://blast.ncbi.nlm.nih.gov/) sequence analysis revealed significant homology of MalA to the family of flavin-dependent tryptophan halogenases.70–73 This may suggest two alternative malbrancheamide biosynthetic pathways. First, MalA could chlorinate tryptophan at C4 and C5 (tryptophan numbering) sequentially prior to being loaded onto the second T domain of MalG. Then, both monochlorinated and dichlorinated tryptophan could be processed by subsequent assembly enzymes, thereby respectively leading to 22 and 3 in parallel. Second, MalA might only monochlorinate the C4 position of tryptophan, resulting in 22. Then, 22 is converted into 3 by either MalA or another unidentified halogenase that resides outside mal. To test these hypotheses, it would be the best to conduct in vitro functional analysis of purified MalA against selected substrates such as L-tryptophan and 22. Alternatively, whether or not the 13C labeled 22 can be incorporated into 3 in an in vivo precursor feeding study would also provide useful information about the timing of the two chlorination steps in malbrancheamide biosynthesis.
According to the proposed malbrancheamide biosynthetic pathway (Fig. 5), only three enzymes are required to assemble the final product 3. Inactivation of these seemingly redundant genes including malB, malC, and malF (Table 1) is currently underway. Interestingly, the MalC short chain dehydrogenase related to PhqE, which is presumed to participate in preparation of β-methyl proline starter unit in paraherquamide biosynthesis (see above), is present in the mal gene cluster although apparently unnecessary for malbrancheamide biosynthesis. This implies that malC, together with other redundant genes, might be residuals from ancestral or a horizontally transferred gene cluster (e.g. one analogous to phq). The evolving biosynthetic gene cluster not only recruits new genes, but also eliminates or retains unused genes when facing a diverse living environment and selection pressure during its evolutionary history.24
Recently, a novel malbrancheamide-type natural product named spiromalbramide (23) (Fig. 5) was isolated from a marine invertebrate-derived Malbranchea graminicola fungal strain.74 This new derivative contains the spiro-oxindole moiety that is found in notoamides and paraherquamides, but is absent from malbrancheamides. Based on the comparative analysis of not, not′, phq, and mal gene cluster, we are now capable of predicting that an FMO gene homologous to notI, notI′ or phqK should reside in the uncharacterized biosynthetic gene cluster of 23. So far, the Solexa genome sequencing of M. graminicola has been completed. This prediction will be tested in the near future as soon as the biosynthetic gene cluster is mined and annotated from genome sequences.
Conclusion
The increasing pace of whole genome sequencing projects supported by a new generation of high throughput technologies have led to exponentially increased number of natural product biosynthetic gene clusters identified in silico. The accumulated knowledge based on previously characterized biosynthetic pathways and functionally defined biosynthetic enzymes, is enabling meaningful comparative analyses of new biosynthetic gene clusters. In turn, this is enabling an efficient approach to predict new biosynthetic pathways, propose enzyme candidates for unknown biosynthetic transformations, and to prioritize targets for focused genetic and biochemical research.
In principle, the shared genes from different clusters are responsible for assembling the common structural core among similar natural products. The cluster-specific gene products are presumed to modify these structures by a series of variant tailoring steps, thereby leading to structural diversification. However, it is noteworthy that the redundant genes and multifunctional genes could complicate comparative analysis of gene clusters. Therefore, conclusions can only be unambiguously drawn after genetic and/or biochemical confirmation of enzymatic activities.
Following these simple but logical principles, we performed a comparative analysis for four related gene clusters including not, not′, phq, and mal, based on the proposed complete biosynthetic pathways for (+)/(−)-notoamides, paraherquamides, and malbrancheamides with a biosynthetic enzyme assigned for each individual step (Fig. 3–5). For example, the function of the not-specific gene notB can be readily connected to the pathway specific transformation from notoamide E (8) to notoamide C (9) and D (10). This was recently confirmed by in vitro characterization of NotB FMO enzyme.58
Furthermore, detailed comparative analysis resulted in nomination of the oxidases NotH and NotH′ (or NotD and NotD′), and the prenyltransferases PhqJ and MalE as putative Diels-Alderases to catalyze the distinctive IMDA reactions for these pathways. Next, comparative functional analysis of these enzymes in vitro will enable us to test this long standing hypothesis regarding the existence of a Diels-Alderase in the biosynthesis of fungal indole alkaloids with the bicyclo[2.2.2]diazaoctane core. It is striking that Nature has conscripted two evolutionarily related gene cluster paradigms, to construct the novel bicyclo[2.2.2]diazaoctane ring system by vastly different mechanistic protocols (Figure 6). In one instance, for the notoamides, the net transformation from the NRPS-loaded dipeptide to the bicyclo[2.2.2]diazaoctane core, a net two-electron oxidation is required to reach the key, putative azadiene species required for the proposed IMDA construction. In the other, the paraherquamide and malbrancheamide systems, the NRPS-loaded dipeptide substrate is cleaved in a net two-electron reduction, that we speculate cyclizes and dehydrates to the related (reduced) azadiene species for the homologous IMDA construction. This insight was most readily presented to us, by the analysis of the respective gene cluster annotations, and has provided a very satisfying level of corroboration with labeled precursor incorporation experiments that at first, seemed incongruous. We expect that the tremendous insights that the bioinformatics analyses have provided in these systems, will render understanding the possible biogenesis of these and related natural products more efficient, congruent and intellectually satisfying.
Figure 6.

Summary of divergent NRPS strategies that culminate in the formation of structurally related bicyclo[2.2.2]diazaoctane ring systems in distinct oxidation states.
Supplementary Material
Acknowledgments
This work was supported by NIH grant R01 CA070375 (R.M.W. and D.H.S.) and the Hans W. Vahlteich Professorship (D.H.S.). Dr. James Cavalcoli of Center for Computational Medicine and Bioinformatics, University of Michigan is gratefully acknowledged for helpful discussions on genome assembly and bioinformatic analysis. We are also thankful to Drs. Brendan Tarrier, Christine Brennan, and Robert Lyons from University of Michigan DNA Sequencing Core for genome sequencing technical support and helpful suggestions.
Contributor Information
Robert M. Williams, Email: rmw@lamar.colostate.edu.
David H. Sherman, Email: davidhs@umich.edu.
References
- 1.Li JWH, Vederas JC. Science. 2009;325:161. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- 2.Newman DJ, Cragg GM. J Nat Prod. 2007;70:461. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 3.Li R, Townsend CA. Metab Eng. 2006;8:240. doi: 10.1016/j.ymben.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Baltz RH. J Ind Microbiol Biotechnol. 1998;20:360. [Google Scholar]
- 5.Baba S, Abe Y, Suzuki T, Ono C, Iwamoto K, Nihira T, Hosobuchi M. Appl Microbiol Biotechnol. 2009;83:697. doi: 10.1007/s00253-009-1933-8. [DOI] [PubMed] [Google Scholar]
- 6.Noh J-H, Kim S-H, Lee H-N, Lee SY, Kim E-S. Appl Microbiol Biotechnol. 2010;86:1145. doi: 10.1007/s00253-009-2391-z. [DOI] [PubMed] [Google Scholar]
- 7.Strohl WR. Metab Eng. 2001;3:4. doi: 10.1006/mben.2000.0172. [DOI] [PubMed] [Google Scholar]
- 8.Cane DE, Walsh CT, Khosla C. Science. 1998;282:63. doi: 10.1126/science.282.5386.63. [DOI] [PubMed] [Google Scholar]
- 9.Walsh CT. ChemBioChem. 2002;3:125. doi: 10.1002/1439-7633(20020301)3:2/3<124::AID-CBIC124>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 10.Sanchez C, Zhu L, Brana AF, Salas AP, Rohr J, Mendez C, Salas JA. Proc Natl Acad Sci USA. 2005;102:461. doi: 10.1073/pnas.0407809102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pollier J, Moses T, Goossens A. Nat Prod Rep. 2011;28:1897. doi: 10.1039/c1np00049g. [DOI] [PubMed] [Google Scholar]
- 12.Que JL, Tolman WB. Nature. 2008;455:333. doi: 10.1038/nature07371. [DOI] [PubMed] [Google Scholar]
- 13.Goff AL, Artero V, Jousselme B, Tran PD, Guillet N, Métayé R, Fihri A, Palacin S, Fontecave M. Science. 2009;326:1384. doi: 10.1126/science.1179773. [DOI] [PubMed] [Google Scholar]
- 14.Watts KT, Mijts BN, Schmidt-Dannert C. Adv Synth Catal. 2005;347:927. [Google Scholar]
- 15.Xue Y, Zhao L, Liu H-w, Sherman DH. Proc Natl Acad Sci USA. 1998;95:12111. doi: 10.1073/pnas.95.21.12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carlson JC, Fortman JL, Anzai Y, Li S, Burr DA, Sherman DH. ChemBioChem. 2010;11:564. doi: 10.1002/cbic.200900658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kittendorf JD, Sherman DH. Bioorg Med Chem. 2009;17:2137. doi: 10.1016/j.bmc.2008.10.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carlson JC, Li S, Gunatilleke SS, Anzai Y, Burr DA, Podust LM, Sherman DH. Nat Chem. 2011;3:628. doi: 10.1038/nchem.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Boghigian BA, Armando J, Pfeifer BA. Nat Prod Rep. 2011;28:125. doi: 10.1039/c0np00037j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galm U, Shen B. Expert Opin Drug Discov. 2006;1:409. doi: 10.1517/17460441.1.5.409. [DOI] [PubMed] [Google Scholar]
- 21.Tang L, Shah S, Chung L, Carney J, Katz L, Khosla C, Julien B. Science. 2000;287:640. doi: 10.1126/science.287.5453.640. [DOI] [PubMed] [Google Scholar]
- 22.Cheng Q, Xiang L, Izumikawa M, Meluzzi D, Moore BS. Nat Chem Biol. 2007;3:557. doi: 10.1038/nchembio.2007.22. [DOI] [PubMed] [Google Scholar]
- 23.Balibar CJ, Howard-Jones AR, Walsh CT. Nat Chem Biol. 2007;3:584. doi: 10.1038/nchembio.2007.20. [DOI] [PubMed] [Google Scholar]
- 24.Gu L, Wang B, Kulkarni A, Geders TW, Grindberg RV, Gerwick L, Håkansson K, Wipf P, Smith JL, Gerwick WH, Sherman DH. Nature. 2009;459:731. doi: 10.1038/nature07870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anzai Y, Li S, Chaulagain MR, Kinoshita K, Montgomery J, Sherman DH. Chem Biol. 2008;15:950. doi: 10.1016/j.chembiol.2008.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galm U, Wendt-Pienkowski E, Wang L, Huang S-X, Unsin C, Tao M, Coughlin JM, Shen B. J Nat Prod. 2011;74:526. doi: 10.1021/np1008152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Péant B, LaPointe G, Gilbert C, Atlan D, Ward P, Roy D. Microbiology. 2005;151:1839. doi: 10.1099/mic.0.27852-0. [DOI] [PubMed] [Google Scholar]
- 28.Ryan KS. PLoS One. 2011;6:e23694. doi: 10.1371/journal.pone.0023694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buntin K, Irschik H, Weissman KJ, Luxenburger E, Blöcher H, Müller R. Chem Biol. 2010;17:342. doi: 10.1016/j.chembiol.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 30.Hawkins RD, Hon GC, Ren B. Nat Rev Genet. 2010;11:476. doi: 10.1038/nrg2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Metzker ML. Nat Rev Genet. 2010;11:31. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 32.Treangen TJ, Salzberg SL. Nat Rev Genet. 2012;13:36. doi: 10.1038/nrg3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaffer C. Nat Biotechnol. 2007;25:149. doi: 10.1038/nbt0207-149. [DOI] [PubMed] [Google Scholar]
- 34.Schuster SC. Nat Methods. 2008;5:16. doi: 10.1038/nmeth1156. [DOI] [PubMed] [Google Scholar]
- 35.Kato H, Yoshida T, Tokue T, Nojiri Y, Hirota H, Ohta T, Williams RM, Tsukamoto S. Angew Chem Intl Ed. 2007;46:2254. doi: 10.1002/anie.200604381. [DOI] [PubMed] [Google Scholar]
- 36.Greshock TJ, Grubbs AW, Jiao P, Wicklow DT, Gloer JB, Williams RM. Angew Chem Intl Ed. 2008;47:3573. doi: 10.1002/anie.200800106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamazaki M, Okuyama E, Kobayashi M, Inoue H. Tetrahedron Lett. 1981;22:135. [Google Scholar]
- 38.Ondeyka JG, Goegelman RT, Schaeffer JM, Kelemen L, Zitano L. J Antibiot. 1990;43:1375. doi: 10.7164/antibiotics.43.1375. [DOI] [PubMed] [Google Scholar]
- 39.Williams RM, Gao J, Tsujishima H, Cox RJ. J Am Chem Soc. 2003;125:12172. doi: 10.1021/ja036713+. [DOI] [PubMed] [Google Scholar]
- 40.Martinez-Luis S, Rodriguez R, Acevedo L, Gonzalez MC, Lira-Rocha A, Mata R. Tetrahedron. 2006;62:1817. [Google Scholar]
- 41.Figueroa M, Gonzalez MC, Mata R. Nat Prod Res. 2008;22:709. doi: 10.1080/14786410802012361. [DOI] [PubMed] [Google Scholar]
- 42.Miller KA, Welch TR, Greshock TJ, Ding Y, Sherman DH, Williams RM. J Org Chem. 2008;73:3116. doi: 10.1021/jo800116y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding Y, deWet JR, Cavalcoli J, Li S, Greshock TJ, Miller KA, Finefield JM, Sunderhaus JD, McAfoos TJ, Tsukamoto S, Williams RM, Sherman DH. J Am Chem Soc. 2010;132:12733. doi: 10.1021/ja1049302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams RM, Cox RJ. Acc Chem Res. 2003;36:127. doi: 10.1021/ar020229e. [DOI] [PubMed] [Google Scholar]
- 45.Stocking EM, Sanz-Cervera JF, Unkefer CJ, Williams RM. Tetrahedron. 2001;57:5303. [Google Scholar]
- 46.Stocking EM, Williams RM. Angew Chem Intl Ed. 2003;42:3078. doi: 10.1002/anie.200200534. [DOI] [PubMed] [Google Scholar]
- 47.Sunderhaus JD, Sherman DH, Williams RM. Isr J Chem. 2011;51:442. doi: 10.1002/ijch.201100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grubbs AW, Artman GDI, Tsukamoto S, Williams RM. Angew Chem Intl Ed. 2007;46:2257. doi: 10.1002/anie.200604377. [DOI] [PubMed] [Google Scholar]
- 49.Greshock TJ, Grubbs AW, Tsukamoto S, Williams RM. Angew Chem Intl Ed. 2007;46:2262. doi: 10.1002/anie.200604378. [DOI] [PubMed] [Google Scholar]
- 50.Keating TA, Ehmann DE, Kohli RM, Marshall CG, Trauger JW, Walsh CT. ChemBioChem. 2001;2:99. doi: 10.1002/1439-7633(20010202)2:2<99::AID-CBIC99>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 51.Steffan N, Grundmann A, Afiyatullov S, Ruan H, Li S-M. Org Biomol Chem. 2009;7:4082. doi: 10.1039/b908392h. [DOI] [PubMed] [Google Scholar]
- 52.Hausinger RP. Crit Rev Biochem Mol Biol. 2004;39:21. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 53.Birch AJ, Wright JJ. J Chem Soc Chem Commun. 1969:644. [Google Scholar]
- 54.Li S-M. J Antibiot. 2011;64:45. doi: 10.1038/ja.2010.128. [DOI] [PubMed] [Google Scholar]
- 55.Kato N, Suzuki H, Takagi H, Kakeya H, Uramoto M, Usui T, Takahashi S, Sugimoto Y, Osada H. Chem Bio Chem. 2009;10:920. doi: 10.1002/cbic.200800787. [DOI] [PubMed] [Google Scholar]
- 56.Steyn PS. Tetrahedron Lett. 1971;12:3331. [Google Scholar]
- 57.Tsukamoto S, Kato H, Greshock TJ, Hirota H, Ohta T, Williams RM. J Am Chem Soc. 2009;131:3834. doi: 10.1021/ja810029b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li S, Finefield JM, Sunderhaus JD, McAfoos TJ, Williams RM, Sherman DH. J Am Chem Soc. 2012;134:788. doi: 10.1021/ja2093212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oliynyk M, Stark CBW, Bhatt A, Jones MA, Hugher-Thomas ZA, Wilkinson C, Oliynyk Z, Demydchuk Y, Staunton J, Leadlay PF. Mol Microbiol. 2003;49:1179. doi: 10.1046/j.1365-2958.2003.03571.x. [DOI] [PubMed] [Google Scholar]
- 60.Qian-Cutrone J, Huang S, Shu YZ, Vyas D, Fairchild C, Menendez A, Krappitz K, Dalterio R, Klohr SE, Gao Q. J Am Chem Soc. 2002;124:14556. doi: 10.1021/ja028538n. [DOI] [PubMed] [Google Scholar]
- 61.Finefield JM, Kato H, Greshock TJ, Sherman DH, Tsukamoto S, Williams RM. Org Lett. 2011;13:3802. doi: 10.1021/ol201284y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Authrine C, Gloer JB. J Nat Prod. 1996;59:1093. doi: 10.1021/np960607m. [DOI] [PubMed] [Google Scholar]
- 63.Stocking EM, Sanz-Cervera JF, Williams RM. J Am Chem Soc. 2000;122:1675. doi: 10.1021/ja005655e. [DOI] [PubMed] [Google Scholar]
- 64.Luesch H, Hoffmann D, Hevel JM, Becker JE, Golakoti T, Moore RE. J Org Chem. 2002;68:83. doi: 10.1021/jo026479q. [DOI] [PubMed] [Google Scholar]
- 65.Ding Y, Grüschow S, Greshock TJ, Finefield JM, Sherman DH, Williams RM. J Nat Prod. 2008;71:1574. doi: 10.1021/np800292n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stocking EM, Sanz-Cervera JF, Williams RM. Angew Chem Intl Ed. 2001;40:1296. doi: 10.1002/1521-3773(20010401)40:7<1296::aid-anie1296>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 67.Liesch JM, Wichmann CF. J Antibiot. 1990;43:1380. doi: 10.7164/antibiotics.43.1380. [DOI] [PubMed] [Google Scholar]
- 68.Hewitson KS, Granatino N, Welford RWD, McDonough MA, Schofield CJ. Phil Trans R Soc A. 2005;363:807. doi: 10.1098/rsta.2004.1540. [DOI] [PubMed] [Google Scholar]
- 69.Ding Y, Greshock TJ, Miller KA, Sherman DH, Williams RM. Org Lett. 2008;10:4863. doi: 10.1021/ol8019633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.vanPée KH, Patallo EP. Appl Microbiol Biotechnol. 2006;70:631. doi: 10.1007/s00253-005-0232-2. [DOI] [PubMed] [Google Scholar]
- 71.Zeng J, Zhan J. Chem Bio Chem. 2010;11:2119. [Google Scholar]
- 72.Neumann CS, Walsh CT, Kay RR. Proc Natl Acad Sci USA. 2010;107:5798. doi: 10.1073/pnas.1001681107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dong C, Flecks S, Unversucht S, Haupt C, vanPée KH, Naismith JH. Science. 2005;309:2216. doi: 10.1126/science.1116510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Watts KR, Loveridge ST, Tenney K, Media J, Valeriote FA, Crews P. J Org Chem. 2011;76:6201. doi: 10.1021/jo2009593. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.