

Abstract
Heme-copper oxidase (HCO) performs efficient four-electron reduction of oxygen to water without releasing toxic, reactive oxygen species (ROS). Essential for this function is a post-translationally modified histidine–tyrosine cross-link (Tyr-His) in its heme a3/CuB oxygen reduction center. Through the genetic incorporation of the Tyr-His ligand and CuB site into myoglobin, we recapitulated important features of HCO into this small soluble protein, which exhibits selective O2 reduction activity while generating less than 6% ROS, at more than 1000 turnovers. These results support that Tyr-His crosslink is indeed important for HCO function, and creates the exciting opportunity to rapidly evolve better HCO model proteins to achieve higher activity and selectivity, which may be suitable as alternatives to precious metal catalyst in fuel cells.
Keywords: post-translational modification, tyrosine-histidine crosslink, protein design, heme copper oxidase, oxygen reduction
In aerobic respiration, heme-copper oxidase[1] (HCO) performs efficient four-electron reduction of oxygen to water without releasing toxic, reactive oxygen species (ROS) such as superoxide and peroxide.[2] Due to its importance in biogenetics, fascinating chemistry and potential applications in fuel cells, HCO has been intensively investigated by structural biologists,[3] enzymologists,[1, 4] and synthetic chemists.[5] While tremendous progress has been made from the above studies, several important questions about the HCO structural features and mechanism still remain. Of particular interests is the unique post-translationally modified covalent cross-link between C6 of tyrosine and Nε2 of a nearby histidine (Tyr-His crosslink, Figure 1A), revealed first by high-resolution x-ray structure[3a] and then confirmed by biochemical and biophysical studies.[6] Importantly, the Tyr-His crosslink is conserved in HCO from bacterial to mammal,[5b] implicating a strong evolutionary connection and its functional significance.
Figure 1.

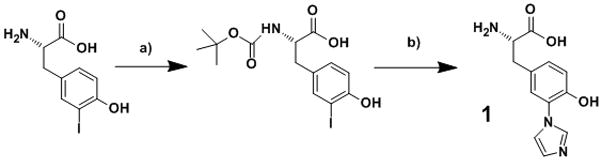
(A) CuB site of cytochrome c oxidase (bovine numbering). (B) Structure model overlay of imiTyrCuBMb (cyan) and F33YCuBMb (yellow). (C) Synthesis of imiTyr 1. a) Di-tert-butyl dicarbonate, THF, 1N NaOH rt, 4 h. b) imidazole, Cs2CO3, CuI, DMF, reflux, 16 h.
While the presence of covalent cross-linked Tyr-His is firmly established in HCOs, its exact role in HCO function is still not fully understood. A major limitation of our understanding is that such an post-translational modification cannot be directly probed through traditional site-directed mutagenesis methods. To overcome this limitation, synthetic model compounds have been made, whose studies have suggested that that the crosslink between Tyr and His significantly decreased pKa of phenol and imidazole sidechains. The decreased pKa of phenol is believed to facilitate proton delivery and tyrosyl radical formation.[5b, 7] Despite these advances, questions about the roles of Tyr-His crosslink still remain, as no study has been able to compare the functions of His and Tyr residues at the same positions in the same protein with and without cross-linking.
To answer the above question and to gain new insights into the HCO catalytic process,[7c] we report herein the genetic incorporation of an unnatural amino acid (UAA) imiTyr 1 (Figure 1C) that mimics the cross-linked Tyr-His ligand into a functional model of HCO (Figure 1B) in sperm whale myoglobin[8](termed imiTyrCuBMb). We then compared its activity with a mutant bearing His and Tyr at the same position in the same protein but lack such a crosslink (F33YCuBMb). Our results show that imiTyrCuBMb recapitulated important features of HCO, exhibiting selective O2 reduction activity while generating less than 6% ROS, at more than 1000 turnovers. Importantly, incorporation of imiTyr in HCO model resulted in eight fold increase in selectivity and threefold increase in catalytic turnover, supporting that Tyr-His crosslink is indeed essential for HCO function,[5b] and provides direct evidence for the first time that the covalent cross-linked Tyr-His moiety can dramatically enhance the oxidase activity.
We choose myoglobin for genetic incorporation of the unnatural amino acids and to carry out functional studies because, in comparison the large (~200 kDa) membrane HCO proteins consisting of multiple subunits that are difficult to produce homogeneously in large amounts, myoglobin is a much smaller (18 kD) water-soluble protein that can be expressed and purified easily in gram quantities.[9] Previous biochemical and spectroscopic studies have indicated that the designed heme-copper center in myoglobin can be a functional model of HCOs.[8]
We first synthesized (S)-2-amino-3-(4-hydroxy-3-(1H-imidazol-1-yl)phenyl)propanoic acid 1 (hereafter termed imiTyr), which contains the ortho-imidazole–phenol linkage (Tyr-His ligand), and then genetically encoded it into myoglobin. The unnatural amino acid imiTyr 1 was synthesized by first protecting the amine group of 3-iodotyrosine using Di-tert-butyl dicarbonate,[10] affording Boc-L-3-iodotyrosine, which was then allowed to react with imidazole in the precence of Cs2CO3 and CuI in DMF under refluxing conditions to afford 1 in 50% yield[7c] (Figure 1C).
To selectively incorporate imiTyr at defined sites in proteins in E. coli, a mutant Methanococcus jannaschii tyrosyl amber suppressor tRNA (MjtRNATyrCUA)/tyrosyl-tRNA synthetase (MjTyrRS) pair was evolved that uniquely specifies 1 in response to the TAG codon. An MjTyrRS library, pBK-lib-jw1, in which six residues (Tyr32, Leu65, Phe108, Gln109, Asp158, and Leu162) were randomized, and any one of the six residues (Ile63, Ala67, His70, Tyr114, Ile159, Val164) was either mutated to Gly or kept unchanged, was used to screen for active and selective mutants for 1, as previously reported.[11] Nine MjTyrRS clones emerged after three rounds of positive selections and two rounds of negative selections (Table S1). The best clone grew at 120 μg/mL of chloramphenicol in the presence of 1 mM 1, but only at 20 μg/mL chloramphenicol in its absence, and was named imiTyrRS. Sequencing of this clone revealed five mutations: Tyr32Glu, Leu65Ser, His67Gly, Asp158Tyr and Leu162Asn. Among the five mutations, the His67Gly mutation was required to create additional space to accommodate the imidazole group. The other four mutations may make specific hydrogen bonding interactions to stablize the imiTyr sidechain. Work is underway to obtain a structure of the imiTyrRS in order to gain insight into these interactions.
To determine the efficiency and fidelity for the incorporation of 1 into proteins, an amber stop codon was substituted for Ser4 in sperm whale myoglobin containing a C-terminal His6 tag. Protein expression was carried out in E. coli in the presence of the selected synthetase (imiTyrRS), MjtRNATyrCUA and 1 mM 1, or in the absence of 1 as a negative control. Analysis of the purified protein by SDS-PAGE showed that full-length myoglobin was expressed only in the presence of 1 (Figure 2A), indicating that imiTyrRS is specifically active for 1 but does not utilize any natural amino acid. The yield of the mutant myoglobin was 10 mg/L. For comparison, the yield of wild-type sperm whale myoglobin (wtMb) under similar conditions was 50 mg/L. ESI-MS spectrometric analysis of the mutant myoglobin gave an observed average mass of 18497 Da (H+), in close agreement with the calculated mass of 18496 Da for the Ser4→1 myoglobin mutant.
Figure 2.

(A) Coomassie-stained SDS-PAGE of TAG4 mutant (left) and mutant (right) myoglobin (indicated by black arrow) expression in the presence and absence of 1 mM UAA 1. (B) ESI-MS spectra of the TAG4 mutant. The insert shows the deconvoluted spectrum; expected mass: 18496 Da, found: 18497 Da.
Having demonstrated the efficient genetic incorporation of imiTyr 1 into wtMb, we then expressed myoglobin double mutant Leu29His/Phe33→1 (called imiTyrCuBMb) as a functional HCO model,[8] with a yield of 5 mg/L. The selective incorporation of imiTyr at position 33 was again confirmed by SDS-PAGE (Figure 2A) and ESI-MS (Figure S2). Together with distal His64 in sperm whale Mb, the newly introduced His29 and imiTyr33 constitute a CuB binding site according to our model, placing the copper ion in close proximity to heme iron (Figure 1B). The UV-vis spectrum of imiTyrCuBMb at pH 7.4 is similar to that observed for wtMb and is typical of a six-coordinate, H2O-bound, high-spin heme with a Soret band absorption maximum at 408 nm (Figure S3). The deoxy-imiTyrCuBMb was obtained by reducing the purified met-imiTyrCuBMb with excess dithionite. The UV-vis spectrum (Figure S3) of deoxy-imiTyrCuBMb exhibits a Soret band at 430 nm and a visible absorption band at 562 nm. This spectrum is nearly identical to that of deoxy-wtMb, indicating that mutation in imiTyrCuBMb did not cause major structural changes of the protein. Addition of Cu2+ at pH 7.4 to imiTyrCuBMb resulted in spectrum difference in the Soret region (Figure S4), which was fitted to a double reciprocal plot and a Hill plot.[8b] These results indicate that a single Cu2+ binding site were present in imiTyrCuBMb, with a KD of 1.6 μM.
Since the hallmark of native HCOs is the clean catalytic reduction of O2 to water without releasing ROS, we then measured the rate imiTyrCuBMb catalyzes oxygen reduction by using an O2 electrode in the presence of 1000 equivalents of ascorbic acid as the reductant and 100 equivalents of TMPD (tetramethyl-p-phenylenediamine dihydrochloride) as a redox mediator. Similar method was previous used to measure enzymatic activity of native HCO.[12] To differentiate between ROS and water products, we employed catalase which selectively reacts with H2O2 to produce O2, and superoxide dismutase (SOD) which selectively reacts with superoxide to produce H2O2 and O2. If O2 consumption results in release of ROS but not formation of water, the apparent O2 reduction rate will slow down in the presence of catalase and SOD, because they will convert ROS back to O2. By comparing the rates of reduction in the absence and presence of catalase and SOD, the portion of O2 reduction due to water formation (in blue) and due to ROS formation (in red) can be calculated (Figure 3A). Not surprisingly, most of the O2 consumed by wtMb lead to ROS, both in the presence and absence of Cu2+. By contrast, imiTyrCuBMb catalyzed O2 reduction without generating more than 6% H2O2 in the presence of 1 eq Cu2+, with oxygen consumption rate of 12 μM/min (enzyme activity = 2 oxygen/min). In the absence of Cu2+, however, more than 30% of O2 was converted to ROS, indicating that CuB is important for the catalytic turnover of O2 into H2O.
Figure 3.


(A) Rates of oxygen reduction to form either water (blue) or ROS (red) catalyzed by 6 μM wtMb, F33YCuBMb, or imiTyrCuBMb, in the presence or absence of 6 μM of Cu2+, with 0.6 mM TMPD and 6 mM ascorbic acid as reductants (B) O2 reduction turnover number catalyzed by imiTyrCuBMb or F33YCuBMb.
The myoglobin system described here provides a unique capability to compare directly the effect of His and Tyr with and without cross-linking. To take advantage this unique feature, we also measured O2 consumption for the myoglobin mutant Leu29His/Phe33Tyr/Phe43His (F33YCuBMb). This mutant harbors a CuB site consisting of residues His29, His43 and His64, as we have shown previously,[8b] and a tyrosine residue in position 33. While His43 and Tyr33 sidechains are in similar positions as the imidazole group and phenol group of imiTyr33, respectively (Figure 1B), no crosslink is present between them. Not only the overall activity of the F33YCuBMb was less (~40%) than imiTyrCuBMb, but also more than 50% of O2 was converted to ROS with or without Cu2+ added, confirming that the cross-linked Tyr-His ligand plays a critical role for the selective reduction of O2 into H2O.
To test if imiTyrCuBMb is a robust O2 reduction catalyst, capable of performing many turnovers without self-destruction, we then measured O2 consumption under multiple turnover conditions. To a solution containing 6 μM imiTyrCuBMb, 6 μM CuSO4 and 800 μM O2, 0.6 mM TMPD and 6 mM ascorbic acid were added. O2 reduction was then monitored by using an oxygen electrode until all the oxygen was consumed. Subsequently, a further addition of 800 μM O2 was carried out by purging the solution with pure oxygen, and then oxygen reduction was again measured until all the oxygen was reduced. These stepwise additions resulted in the multiple plateaus observed in the traces (Figure 3B), and these processes were repeated until imiTyrCuBMb achieved more than 1000 turnovers (Figure 3B). The turnover number reported here is an underestimate as the extra turnovers occurring during oxygen addition were not included in the calculation. Notably, little decrease in O2 consumption rate was observed after the catalyst reached 1000 turnovers, indicating that at this point the imiTyrCuBMb catalyst has remained intact. By contrast, F33YCuBMb underwent less than 400 turnovers under similar conditions. These results again support that the cross-linked Tyr-His ligand is required for optimal O2 reduction to H2O. Here, the different catalytic activity between imiTyrCuBMb and F33YCuBMb can also be attributed to the different geometry of the copper center. To further investigate the mechanism, we are currently working to solve the crystal structure of imiTyrCuBMb, and examine the enzymatic property of another mutant having a Phe-His type crosslinkage at the 33 position.
In conclusion, by directly incorporating the unnatural amino acid imiTyr 1 into myoglobins in E. coli in response to the amber codon TAG, we have successfully designed a functional HCO model imiTyrCuBMb which catalyzes selective and efficient oxygen reduction to water. The imiTyrCuBMb HCO model bearing Tyr-His crosslink is eight fold more selective with three fold more turnovers than F33YCuBMb, which does not contain the crosslink but harbor His and Tyr at the same positions in the same protein. Since the synthesis of imiTyr is only two steps with 50% overall yield, and mutant proteins bearing imiTyr 1 at any site can be easily obtained and purified in milligram quantities through site-directed mutageneis and recombinant protein expression, further systematic investigation of Tyr-His crosslink function is now possible. While imiTyrCuBMb exhibits lower enzymatic activity (2 oxygen/min)[12] in comparison to native heme copper oxidase (~ 300 oxygen/sec), it is possible to rapidly improve our HCO model to achieve higher activity through directed evolution and unnatural amino acid incoporation. Our designed enzyme harbors unnatural amino acid imiTyr which is highly analogous to the post-translationally modified tyrosine-histidine ligand found in CuB site of HCO, serves as an ideal model for a more detailed understanding of HCOs, and allows for potential applications in synthetic biology and alternative energy.[5b, 9, 13]
Experimental Section
To express mutant myoglobin protein imiTyrCuBMb, plasmids pBAD-JYAMB-His29TAG33 was cotransformed with pBK-imiTyrRS into GeneHog®-Fis E. coli cells. Cells were amplified in LB media (5 mL) supplemented with kanamycin (50 μg/mL) and tetracycline (15 μg/mL). Starter culture (1 mL) was used to inoculate 100 mL of liquid LB supplemented with appropriate antibiotics and 1 mM 1. Cells were then grown at 37°C to OD600 of 0.5, and protein expression was induced by the addition of 0.2% arabinose. After 12 hours of growth at 37°C, cells were harvested by centrifugation. Wild type or mutant myoglobin was then purified by Ni-NTA affinity chromatography under denaturing conditions, and reconstituted with heme as previously described.[9]
Oxygen consumption was measured by using an Oxygraph Clark-type oxygen electrode (Hansatech Instruments) at 25°C in 20 mM Tris buffer, pH 7.4. All experiments were performed three times to obtain standard deviation. The electrode was calibrated against air-saturated buffer and O2-depleted buffer prior to use. Protein was exchanged to 20 mM Tris, pH7.4, which was treated with chelex beads overnight. The final concentration of protein was adjusted to 6 μM. For experiment with Cu2+, CuSO4 was added to final concentration of 6 μM and stirred for 10 min. For experiment with catalase and SOD, catalase (Sigma-Aldrich) was added to final concentration of 7.3 U/L, SOD (Sigma-Aldrich) was added to final concentration of 0.5 U/μL. The reaction is initiated by adding TMPD and ascorbic acid to a final concentration of 0.6 mM and 6 mM, respectively. Oxygen concentration was read immediately after adding reductant. The turn-over experiment was performed similarly except after O2 in the reaction chamber being exhausted, the chamber was vented with pure O2 to boost O2 concentration to 800 μM. Ascorbic acid was added to keep concentration at 6 mM.
Supplementary Material
Acknowledgments
We gratefully acknowledge the Major State Basic Research Program of China (2010CB912301, 2009CB825505), National Science Foundation of China (30870592, 90913022, Y2JY141001) to J.W. and X. L, and National Institutes of Health (GM062211) to Y. L. We thank Z. Xie, X. Peng for help in protein mass spectroscopy.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author
Contributor Information
Yi Lu, Email: yi-lu@illinois.edu.
Jiangyun Wang, Email: jwang@ibp.ac.cn.
References
- 1.Trumpower BL, Gennis RB. Annu Rev Biochem. 1994;63:675–716. doi: 10.1146/annurev.bi.63.070194.003331. [DOI] [PubMed] [Google Scholar]
- 2.a) Babcock GT, Wikstrom M. Nature. 1992;356:301–309. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]; b) FergusonMiller S, Babcock GT. Chem Rev. 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 3.a) Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]; b) Buschmann S, Warkentin E, Xie H, Langer JD, Ermler U, Michel H. Science. 2010;329:327–330. doi: 10.1126/science.1187303. [DOI] [PubMed] [Google Scholar]
- 4.a) Kaila VRI, Verkhovsky MI, Wikstrom M. Chem Rev. 2010;110:7062–7081. doi: 10.1021/cr1002003. [DOI] [PubMed] [Google Scholar]; b) Yoshikawa S, Muramoto K, Shinzawa-Itoh K. Annu Rev Biophys. 2011;40:205–223. doi: 10.1146/annurev-biophys-042910-155341. [DOI] [PubMed] [Google Scholar]; c) Fann YC, Ahmed I, Blackburn NJ. Biochemistry. 1995;34:10245–10255. doi: 10.1021/bi00032a019. [DOI] [PubMed] [Google Scholar]; d) Jiang JJ, Bank JF, Zhao WW, Scholes CP. Biochemistry. 1992;31:1331–1339. doi: 10.1021/bi00120a008. [DOI] [PubMed] [Google Scholar]
- 5.a) Collman JP, Devaraj NK, Decreau RA, Yang Y, Yan YL, Ebina W, Eberspacher TA, Chidsey CED. Science. 2007;315:1565–1568. doi: 10.1126/science.1135844. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kim E, Kamaraj EEK, Karlin KD. Chem Rev. 2004;104:1077–1133. doi: 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- 6.a) Brzezinski P, Gennis RB. J Bioenerg Biomembr. 2008;40:521–531. doi: 10.1007/s10863-008-9181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Buse G, Soulimane T, Dewor M, Meyer HE, Bluggel M. Protein Sci. 1999;8:985–990. doi: 10.1110/ps.8.5.985. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hemp J, Robinson DE, Ganesan KB, Martinez TJ, Kelleher NL, Gennis RB. Biochemistry. 2006;45:15405–15410. doi: 10.1021/bi062026u. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Proshlyakov DA, Pressler MA, DeMaso C, Leykam JF, DeWitt DL, Babcock GT. Science. 2000;290:1588–1591. doi: 10.1126/science.290.5496.1588. [DOI] [PubMed] [Google Scholar]; e) Tomson F, Bailey JA, Gennis RB, Unkefer CJ, Li ZH, Silks LA, Martinez RA, Donohoe RJ, Dyer RB, Woodruff WH. Biochemistry. 2002;41:14383–14390. doi: 10.1021/bi026370c. [DOI] [PubMed] [Google Scholar]
- 7.a) Pesavento RP, Pratt DA, Jeffers J, van der Donk WA. Dalton Trans. 2006:3326–3337. doi: 10.1039/b516090a. [DOI] [PubMed] [Google Scholar]; b) van der Donk WA. J Org Chem. 2006;71:9561–9571. doi: 10.1021/jo0614240. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McCauley KM, Vrtis JM, Dupont J, van der Donk WA. J Am Chem Soc. 2000;122:2403–2404. [Google Scholar]; d) Pratt DA, Pesavento RP, van der Donk WA. Org Lett. 2005;7:2735–2738. doi: 10.1021/ol050916g. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) White KN, Sen I, Szundi I, Landaverry YR, Bria LE, Konopelski JP, Olmstead MM, Einarsdottir O. Chem Comm. 2007:3252–3254. doi: 10.1039/b703835f. [DOI] [PubMed] [Google Scholar]; f) Liu JG, Naruta Y, Tani F, Chishiro T, Tachi Y. Chem Comm. 2004:120–121. doi: 10.1039/b311538k. [DOI] [PubMed] [Google Scholar]; g) Collman JP, Ghosh S, Dey A, Decreau RA, Yang Y. J Am Chem Soc. 2009;131:5034–5035. doi: 10.1021/ja9001579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Sigman JA, Kim HK, Zhao XA, Carey JR, Lu Y. Proc Nat Acad Sci. 2003;100:3629–3634. doi: 10.1073/pnas.0737308100. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sigman JA, Kwok BC, Lu Y. J Am Chem Soc. 2000;122:8192–8196. [Google Scholar]
- 9.Lu Y, Yeung N, Sieracki N, Marshall NM. Nature. 2009;460:855–862. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmidt EW, Nelson JT, Fillmore JP. Tetrahedron Lett. 2004;45:3921–3924. [Google Scholar]
- 11.Wang JY, Zhang W, Song WJ, Wang YZ, Yu ZP, Li JS, Wu MH, Wang L, Zang JY, Lin Q. J Am Chem Soc. 2010;132:14812–14818. doi: 10.1021/ja104350y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pawate AS, Morgan J, Namslauer A, Mills D, Brzezinski P, FergusonMiller S, Gennis RB. Biochemistry. 2002;41:13417–13423. doi: 10.1021/bi026582+. [DOI] [PubMed] [Google Scholar]
- 13.a) Gewirth AA, Thorum MS. Inorg Chem. 2010;49:3557–3566. doi: 10.1021/ic9022486. [DOI] [PubMed] [Google Scholar]; b) Xiong P, Nocek JM, Vura-Weis J, Lockard JV, Wasielewski MR, Hoffman BM. Science. 2010;330:1075–1078. doi: 10.1126/science.1197054. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chen H, Ikeda-Saito M, Shaik S. J Am Chem Soc. 2008;130:14778–14790. doi: 10.1021/ja805434m. [DOI] [PubMed] [Google Scholar]; d) Matsui T, Unno M, Ikeda-Saito M. Acc Chem Res. 2010;43:240–247. doi: 10.1021/ar9001685. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.