

Abstract
Peroxynitrite (PN, ONOO−) is a potent oxidant and nitrating agent that contributes to pain through peripheral and spinal mechanisms, but its supraspinal role is unknown. We present evidence here that PN in the rostral ventromedial medulla (RVM) is essential for descending nociceptive modulation in rats during inflammatory and neuropathic pain through PN-mediated suppression of opioid signaling. Carrageenan-induced thermal hyperalgesia was associated with increased 3-nitrotyrosine (NT), a PN biomarker, in the RVM. Furthermore, intra-RVM microinjections of the PN decomposition catalyst Fe(III)-5,10,15,20-tetrakis(N-methyl-pyridinium-4-yl)porphyrin (FeTMPyP5+) dose-dependently reversed this thermal hyperalgesia. These effects of FeTMPyP5+ were abrogated by intra-RVM naloxone, implicating potential interplay between PN and opioids. In support, we identified NT colocalization with the endogenous opioid enkephalin (ENK) in the RVM during thermal hyperalgesia, suggesting potential in situ interactions. To address the functional significance of such interactions, we exposed methionine-enkephalin (MENK) to PN and identified the major metabolite, 3-nitrotyrosine-methionine-sulfoxide (NSO)-MENK, using liquid chromatography-mass spectrometry. Next, we isolated, purified, and tested NSO-MENK for opioid receptor binding affinity and analgesic effects. Compared to MENK, this NSO-MENK metabolite lacked appreciable binding affinity for δ, μ, and κ opioid receptors. Intrathecal injection of NSO-MENK in rats did not evoke antinociception, suggesting that PN-mediated chemical modifications of ENK suppress opioid signaling. When extended to chronic pain, intra-RVM FeTMPyP5+ produced naloxone-sensitive reversal of mechanical allodynia in rats following chronic constriction injury of the sciatic nerve. Collectively, our data reveal the central role of PN in RVM descending facilitation during inflammatory and neuropathic pain potentially through anti-opioid activity.
Introduction
Peroxynitrite (PN) is an oxidant and nitrating agent formed through the interaction of free radical precursor molecules, superoxide and nitric oxide (Beckman et al., 1990), that contributes to pain of several etiologies in the periphery and spinal cord (Salvemini et al., 2011). For example, preventing PN formation with systemic, intraplantar, and intrathecal administration of a superoxide dismutase mimetic or depleting PN with systemic administration of potent PN decomposition catalysts (PNDCs) such as Fe(III)-5,10,15,20-tetrakis(N-methyl-pyridinium-4-yl)porphyrin (FeTMPyP5+) (Salvemini et al., 1998a) blocks inflammation- and opioid-induced hyperalgesia, respectively (Wang et al., 2004; Muscoli et al., 2007; Ndengele et al., 2008). Despite these established peripheral and spinal effects, the role of PN at supraspinal sites during pathophysiological pain is unknown.
The rostral ventromedial medulla (RVM) functions as a biphasic output relay by which the brain modulates spinal nociception. The RVM contains physiologically defined neurons whose activities are associated with both inhibition (the so-called OFF cells) and facilitation (ON cells) of spinal nociception (Fields et al., 1983; Basbaum and Fields, 1984). During persistent and chronic pain, the net effect of this RVM output is descending facilitation (Urban et al., 1999a; Terayama et al., 2000; Porreca et al., 2001) that drives central sensitization (Porreca et al., 2002; Gardell et al., 2003; Bee and Dickenson, 2007). The underlying mechanisms within the RVM that result in descending facilitation are incompletely understood. However, enhanced glutamatergic signaling and neuroinflammation have been implicated (Urban et al., 1999b; Wei et al., 2008; Roberts et al., 2009); these are processes that PN mediates in the spinal cord during pain states (Wang et al., 2004; Muscoli et al., 2007; Chen et al., 2010). Thus, PN may have a central role in modulating RVM descending facilitation during pathophysiological pain (Little et al., 2012).
The RVM is also a key center in the endogenous opioid analgesia pathway. For example, the RVM expresses enkephalin (ENK), a potent neuropeptide with high affinity for μ- and δ-opioid receptors (Beitz, 1982; Basbaum and Fields, 1984; Raynor et al., 1996; Fields, 2004). During inflammatory pain there is enhanced ENK expression and μ- and δ-opioid receptor responsiveness in the RVM (Hurley and Hammond, 2000, 2001). If such analgesic compensation exists, why is there no pain relief? PN mediates post-translational nitroxidative modifications of leucine ENK in vitro, notably nitrating the N-terminal tyrosine residue of ENK (Yi et al., 1997; Fontana et al., 2006; Zhan and Desiderio, 2009). This tyrosine is important for the opioid ligand-receptor binding that contributes to the opioid “message” pharmacophore model (Portoghese, 1965). It is hypothesized that nitration and the subsequent lowering of the pKa of the 3-nitrotyrosyl-ENK derivate phenol from ∼10 to 7.1–7.4 (Yokoyama et al., 2010) eliminates this receptor interaction.
We therefore hypothesize that during pathophysiological pain, PN contributes to RVM descending facilitation, in part, by suppressing the endogenous opioid pathway through nitroxidative modifications of ENK and formation of functionally inert neuropeptides.
Materials and Methods
Animals and animal care.
Male Sprague Dawley rats (225–270 g) (Harlan Laboratories) were used for all studies. Rats were housed in a climate-controlled room on a 12 h light/dark cycle with food and water provided ad libitum. All experiments were performed in accordance with the International Association for the Study of Pain (Seattle, MD) and the National Institutes of Health (NIH; Bethesda, MD) guidelines on laboratory animal welfare as well as the recommendations and approval from the Saint Louis University Animal Care and Use Committee (St. Louis, MO). All observers were blinded to treatment conditions.
Intra-RVM cannulation and microinjection.
Rats received intramedullary paired guide cannulation and intra-RVM microinjections using a modification of previously described method (Roberts et al., 2009). Rats were anesthetized with an intraperitoneal (i.p.) injection of ketamine (70 mg/kg)/xylazine (9 mg/kg) solution, given a subcutaneous (s.c.) injection of an antibiotic (Baytril, 15 μg/kg), and mounted into a stereotaxic apparatus. The skull was exposed and the RVM targeted using the stereotaxic coordinates −10.8 mm bregma, −0.20 mm intra-aural, and 0.6 mm to each side of the sagittal suture (Paxinos and Watson, 1998). A paired guide cannula (1.2 mm apart; 26 gauge; Plastics One) was inserted and directed toward the RVM (7.0 mm ventral to the dura mater surface, 2.0 mm dorsal to the RVM) and then secured to the skull using stainless steel screws and dental cement. A paired dummy cannula (Plastics One) was inserted to prevent contamination and maintain patency of the guide cannula. Rats recovered on a heating pad for up to 2 h post-procedure and were then singly housed while recovering for 7 d.
For microinjections, animals were briefly anesthetized with isoflurane (3% in 100% O2 within an induction chamber and maintained on 2% isoflurane in 100% O2 at 2 L/min with a nose cone). Microinjections were performed using a 33 gauge bilateral injection cannula (Plastics One) that extended 2 mm beyond the paired guide cannula into the RVM. Drugs or their vehicles were administered using a 10 μl Hamilton syringe connected to the injection cannula through Tygon tubing (Cole-Parmer Instrument) backfilled with sterile saline. The Hamilton syringe was connected to polyethylene (PE)-10 tubing that had been heat annealed to PE-50 tubing. The PE-50 tubing was then attached to the injection cannula to provide the positive pressure required for accurate injection volumes. The total volume of the microinjections was 0.5 μl (0.25 μl/side), and drug doses are reported as the total amount administered bilaterally. The injection cannula was left in position for 1 min to prevent backflow. All cannulae were checked for patency after microinjection by slowly passing 0.1–0.2 μl of solution through each cannula.
Verification of RVM microinjection sites.
RVM microinjection sites were verified by deposits of India ink noted in 30 μm coronal sections of the medulla stained with cresyl violet. India ink microinjections (0.5 μl) were given at the conclusion of experiments. Proper placement was defined as the most distal point of the microinjection site within the rostral to caudal region of the RVM (stereotaxic coordinates: interaural, −1.30 to −2.96 mm; bregma, −10.30 to −11.96 mm), including the nucleus raphe magnus (NRM), gigantocellularis pars alpha (GiA), lateral paragigantocellularis (LPGi) nuclei, and/or adjacent ventral gigantocellularis (Gi) nucleus. In rats used for biochemical and molecular analysis the RVM was micropunched from a 2 mm coronal section, so the microinjection sites and cannula placements were confirmed by visual inspection, dissection microscopy, and light microscopy using the remaining portion of the 2 mm medullary section. Correct placement was confirmed when: (1) there were no visual signs of puncture through the ventral surface of the medulla upon tissue harvesting; (2) there was visual confirmation of a microinjection site in the proximity of the RVM upon removal of the 2 mm coronal section; and (3) the injection tracts were centered along the midsagittal plane of the micropunched tissue block in the X, Y, and Z axes. Preparations found with microinjection sites outside of the RVM were excluded.
Intra-RVM-administered drugs.
The well characterized PNDC FeTMPyP5+ (0.25–1 μg, 0.5 μl) (Salvemini et al., 1998b) and the nonselective competitive opioid receptor antagonist naloxone (5 μg; Sigma Aldrich) (Brown et al., 1986) or an equal volume of vehicle (saline) were injected in the RVM (0.5 μl). Drugs were freshly prepared by dissolving in 0.9% sterile NaCl (Hospira) before each experiment and stored on ice until used. As PN cannot be directly measured in vivo due to its rapid formation, indicators of its production are typically used for PN investigations (Szabo et al., 2007; Ferrer-Sueta and Radi, 2009). Two of the more widely used methods include detection of 3-nitrotyrosine (NT) formation to verify PN presence and PNDC pharmacological probes such as FeTMPyP5+, which prevents PN and NT formation in vivo (Salvemini et al., 1998a,b; Szabo et al., 2007).
Intrathecal catheterization.
Rats were catheterized intrathecally as previously described (Storkson et al., 1996). Following anesthesia with i.p. injection of ketamine (70 mg/kg)/xylazine (9 mg/kg), a guide cannula (20 gauge) was passed between lumbar vertebrae 5 and 6 into the intrathecal space. A PE-10 catheter was positioned 3 cm beyond the tip of the cannula near the lumbar enlargement. To prevent displacement of the catheter, the tubing was tunneled s.c. to the occipital region through a small skin incision and secured with 5-0 silk sutures. The catheter was flushed with sterile saline and heat sealed. Animals recovered for 2 h on a heating pad and 5–7 d in single-housed units before experiments were performed.
Intrathecally administered drugs.
Intrathecal injections of the α2 adrenoreceptor antagonist yohimbine (30 μg) or an equal volume of vehicle (sterile H2O) were administered following intra-RVM microinjections in carrageenan-treated rats. Methionine-enkephalin (MENK) (Sigma Aldrich) (400 μg) or 3-nitrotyrosine-methionine-sulfoxide (NSO)-MENK (400 μg) was also administered intrathecally. Injections (10 μl) were administered using a 20 μl Hamilton syringe followed by a 15 μl saline flush. Drugs were freshly prepared before each experiment and stored on ice until used.
Carrageenan-induced thermal hyperalgesia.
Lightly anesthetized (80% CO2/20% O2) rats received a s.c. injection of 1% carrageenan (50 μl; 0.9% saline) or an equal volume of saline into the plantar surface of the left hindpaw at time point 0 (baseline). Thermal hyperalgesia was defined as a significant reduction in the withdrawal response time to a noxious heat stimulus applied to the hindpaw (i.e., paw withdrawal latency in seconds). Before thermal stimulation, rats were habituated for 15 min in an enclosure (17 cm depth × 69 cm length × 14 cm height; UGO Basile) on a clear glass platform in a quiet testing room. An infrared bulb (halogen “Bellaphot,” 8 V, 50 W; UGO Basile) was positioned under the glass platform to deliver a thermal stimulus to the plantar surface of the injected hindpaw, and the animal's withdrawal latencies were electronically recorded to the nearest 0.1 s. The infrared intensity was standardized so that the baseline latencies averaged 16–20 s with a cutoff latency of 20 s. Each animal was tested three times, and its mean paw withdrawal latency was reported.
Acute nociception assay.
Antinociception following intrathecal injection of MENK or NSO-MENK was measured using the tail-flick test (UGO Basile, Italy) to determine withdrawal latencies from a noxious radiant heat source (D'Amour, 1941). Here, infrared intensity was standardized so that baseline latencies averaged 2–3 s and with a cutoff time of 10 s to prevent tissue injury. Rats were gently restrained within a Plexiglas cylinder with the distal portion of the tail (∼3 cm from the tip) resting over the infrared heat source. The tail-flick latency was recorded automatically to the nearest 0.1 s. A mean latency was determined from three such tests per animal. After measuring their baseline latencies, rats received an intrathecal injection of either MENK or NSO-MENK, and then these latencies were remeasured at 1, 2, and 3 min.
Chronic constriction injury.
Following a 7 day recovery from RVM cannulation, rats were anesthetized and received a chronic constriction injury (CCI) of the sciatic nerve using modifications of a previously described method (Bennett and Xie, 1988). The posterolateral thigh was shaved, scrubbed with Nolvasan, and a 2 cm incision was made through the skin. The left common sciatic nerve was exposed at mid-thigh by blunt dissection. Proximal to the sciatic nerve trifurcation, about 7 mm of the nerve was freed of adhering tissue and then three 4-0 silk sutures were tied loosely around the nerve, causing slight constriction with about 1 mm spacing. The incision was then closed using 5-0 silk sutures. Mechanical allodynia was determined using an automated von Frey test (dynamic plantar aesthesiometer; UGO Basile) to measure mechanical paw withdrawal thresholds (PWTs) in grams. Rats were acclimated to a Plexiglas chamber with a wire mesh floor for 15 min. Then, a pointed metallic filament was applied to the plantar surface of the hindpaw to exert a punctate pressure of up to 5 g/s and elicit a clear voluntary hindpaw withdrawal response (normally 40–45 g). The PWT was assessed three times at each time point and reported as the mean mechanical PWT (in grams) for both the ipsilateral and contralateral paws. On day 0 (D0), PWTs were assessed before surgery and subsequently on desired days post-surgery. Mechanical allodynia was defined as a significant (p < 0.05) reduction in mean PWT compared to D0. In preliminary experiments, 7 days (D7) provided the maximal PWT reduction ipsilaterally and was used as the time point for intra-RVM microinjections. At 24 h post RVM microinjection (i.e., 8 days, D8), if there were no carryover effects (i.e., changes to PWT) compared to the previous day's baseline, animals received a second microinjection of the opposite solution given on D7 followed by a microinjection of naloxone or vehicle. The experiments performed on D8 were to determine whether PN modulates opioidergic signaling during neuropathic pain.
Immunofluorescence.
Brains were harvested, fixed, and processed for immunostaining using previously described methods (Wang et al., 2004; Muscoli et al., 2010). Rats received an i.p. injection of ketamine (70 mg/kg)/xylazine (9 mg/kg) for deep anesthesia and were transcardially perfused with saline followed by 10% buffered neutral formalin, pH 7.4. The brain was harvested and post-fixed for 4 h at room temperature. Tissues were cryoprotected in a 30% sucrose solution at 4°C for 72 h or until infiltration was complete. The medulla was cryosectioned at 14 μm with every fifth section used for NT staining with double labeling for cell markers and ENK. Sections were mounted on gelatin-coated Starfrost microscopic slides (Lab Storage Systems) and stored at −20°C. For immunolabeling experiments, a rabbit polyclonal anti-NT primary antibody (4.8 μg/ml; a generous gift from Dr. Harry Ischiropoulos, University of Pennsylvania, Philadelphia, PA) was applied to sections and incubated overnight at room temperature in a sealed humid chamber. Following a series of rinses in PBS, pH 7.4, the sections were incubated with a rhodamine red-conjugated goat anti-rabbit IgG secondary antibody (1:250; Invitrogen.). After a series of rinses in PBS, slides were coverslipped with Gel Mount mounting medium (refractive index, 1.358 ± 0.002; Biomeda) containing 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI; Sigma) and stored in the dark at 4°C until imaged. Each round of immunostaining included a negative control (i.e., the primary antibody was omitted and an equal concentration of rabbit IgG was substituted) and a positive control (i.e., 200 mm PN applied directly to sections 10 μl repeated twice, 30 μl total; Cayman Chemical). Preabsorption of the NT antibody (6.4 mm) with NT (320 mm, Sigma) provided low to no fluorescent signal in NT-treated positive control, carrageenan, and saline medullary sections.
Colocalization studies were performed for NT with astrocytes (monoclonal mouse anti-GFAP; 1:100; Abcam), microglia (monoclonal mouse anti-CD11b, OX-42; 1:100; Millipore), neurons (monoclonal mouse anti-NeuN; 1:250; Millipore), as well as a general marker for ENK (mouse monoclonal anti-ENK, 1:200; Millipore) using an Alexa Fluor 488-conjugated goat anti-mouse IgG secondary antibody (1:250; Invitrogen). The location of fluorescent images within RVM nuclei (i.e., NRM, GiA, and LPGi) was confirmed by cresyl violet-stained sections from adjacent levels of the same animal corresponding to the stereotaxic coordinates: interaural, −1.30 mm to −2.96 mm; bregma, −10.30 mm to −11.96 mm (Basbaum and Fields, 1984; Paxinos and Watson, 1998).
All RVM sections were digitally imaged using an Olympus BX-41 upright epifluorescence microscope (UPlanFLN objectives: 10×, NA 0.13; 40×, NA 0.75; Plan CN 60×, NA 0.80; where NA is numerical aperture) and an Olympus DP72 camera with preset image acquisition settings. All images were taken from the same region within the NRM as well as bilateral LPGi and GiA to compare groups. Image analysis was performed using the NIH freeware program ImageJ (version 1.43) (Rasband, 2011). The mean number of NT-positive cell profiles (i.e., immunoreactivity that surrounded or extended from DAPI-stained nuclei) and processes (i.e., well demarcated processes with immunoreactivity lacking continuity with soma and/or DAPI staining) was determined using three medullary sections per animal to represent the upper, middle, and lower regions of the RVM. Images were adjusted for background using minimum brightness controls determined from the background signal of the negative control image on the same slide. Cell profiles and processes were counted using the ImageJ Cell Counter plug-in to identify and tally the NT-positive structures. The mean integrated fluorescence intensity of NT was also determined. Images were converted to 8-bit grayscale, threshold applied for background signal (2 × negative control background signal), and analyzed using the ImageJ Analyze Particle tool. NT fluorescence intensity, defined as the product of the mean gray value deviation above background correction (i) times the percentage of positive pixel area (pp2) to total pixels (p2) or i (pp2 ÷ p2), was determined using modifications of a previously described method (Marvizon et al., 2009). The NT-positive cell profiles, processes, and fluorescence intensity are reported as the group mean/area (microscopic field) normalized to the saline control group. A description of NT colocalization with cell markers was based upon observations of six merged images (two sections) per animal. Manders' overlap coefficient (M) and Pearson's colocalization correlation coefficient (Rr) were used to assess NT colocalization with ENK. Images were converted to 8 bit, threshold settings were applied, and analysis using the ImageJ Manders Coefficients plugin to determine M and Rr coefficients was performed. Manders' coefficient (Manders et al., 1993) was calculated to compare the proportion of ENK-labeled pixels that are also labeled for NT based on 10 merged images (two sections) per animal from the same region of all RVM nuclei. Pearson's colocalization coefficient (Manders et al., 1992) was used to confirm colocalization in carrageenan animals by determining whether the intensity of NT labeling per pixel positively correlated to that of ENK when they co-occur. Because the Rr is sensitive to background signal, 10 regions of interest per image (three images) where colocalization (NT and ENK overlapping) appeared to occur were used in each animal. Colocalization was also confirmed using confocal microscopy to collect Z-stacks (n = 3–4 per group; 2 Z-stacks per animal). Confocal microscopy was performed using an Olympus FV 1000 confocal microscope (multiline argon lasers with excitation at 405 nm, 488 nm, and 543 nm) with 60× oil-immersion objective (1.42 NA) and 2.4× optical zoom to provide 0.1 μm pixel dimensions in the X-Y plane and the pinhole set at 1 Airy unit. Z-stack images were obtained using sequential scanning at a 0.2 μm Z-step with a minimum of four total steps. The images were acquired within the dynamic range of the microscope (i.e., no pixel intensity values of 0 or 255 in an 8-bit image) and assessed as described above.
Formation of NSO-MENK by the reaction of MENK with peroxynitrite.
The nitro-sulfoxide (NSO-MENK) reaction product of PN and MENK was prepared from an 872 μm stock solution of MENK (5.0 mg, 8.7 × 10−6 mol; Sigma-Aldrich) in 100 mm PBS (10 ml). An aliquot (1.1 ml, 9.5 × 10−7 mol) was removed and placed in a 20 ml vial with a magnetic stir bar. To this rapidly stirred solution, PN (100 μl of a 50.0 mm stock solution in 0.1 N NaOH, 5.0 × 10−6 mol, 5.3 equivalents) was rapidly injected into the stirring vortex. The final pH was 7.2. After 1 min of vortexing so that all PN had either reacted with MENK or had decomposed (Ferrer-Sueta and Radi, 2009), the solution was analyzed immediately (without further processing) by liquid chromatography-mass spectrometry (LCMS) [electrospray ionization-positive ion mode, Waters Alliance/SQ 3100 system; Agilent Eclipse XDB-C18, 4.6 × 150 mm, 5 μm column, 15% acetonitrile to 95% gradient in 0.05% trifluoroacetic acid (TFA) over 8 min]. The major isomer is one diastereomer of NSO-MENK.
Synthesis of NSO-MENK.
NSO-MENK was synthesized using standard solution-phase peptide methods. As described in Scheme 1, 3-nitro-tyrosine was t-butyloxycarbonyl (Boc)-protected and coupled with glycine ethyl ester using ethyl-(N′,N′-dimethylamino)propylcarbodiimide hydrochloride (EDC)/1-hydroxybenzotriazole (HOBt) as the coupling reagents. The resulting dipeptide was saponified, and the dipeptide-free acid was coupled again with glycine ethyl ester. Saponification of the resulting tripeptide yielded 3-nitrotyrosyl-glycylglycine-free acid. Boc-phenylalanine was coupled with methionine methyl ester again using the EDC/HOBt method. TFA deprotection of the Boc group afforded the dipeptide phenylalanyl-methionine methyl ester. These two peptide fragments were coupled to each other using EDC/HOBt to afford the protected 3-nitro-MENK pentapeptide. Oxidation of the methionine side chain to the sulfoxide with m-CPBA followed by deprotection of the C terminus by saponification and the N terminus by TFA cleavage of the Boc group afforded NSO-MENK.
Scheme 1.

Synthesis of NSO-MENK.
Receptor binding assays.
Competitive radioligand receptor binding assays to determine the binding affinity (Ki) of MENK and NSO-MENK for μ-, δ-, and κ-opioid receptors were conducted by the National Institute of Mental Health's Psychoactive Drug Screening Program [contract no. HHSN-271-2008-00025-C (NIMH PDSP); Director Dr. Bryan L. Roth, University of North Carolina at Chapel Hill, NC, and Project Officer Jamie Driscol at NIMH, Bethesda MD) based upon a previously described method (Roth et al., 1981). Briefly, test compounds (MENK and NSO-MENK) were diluted using a standard assay buffer (50 mm Tris-HCl, 10 mm MgCl2, 0.1 mm EDTA, pH 7.4) to provide 10 pm to 10 μm assay concentrations. The radioligands for μ-opioid (0.3 nm [3H]DAMGO), δ-opioid (0.2 nm [3H]DADLE), and κ-opioid (0.3 nm [3H]U69593) receptors were diluted in the assay buffer and aliquoted into 96-well plates. Test compounds were then added to the radioligand in duplicate followed by crude membrane fractions of HEK293 cells transfected with the human recombinant opioid receptor. This solution was incubated for 1.5 h followed by harvesting using rapid filtration onto Whatman GF/B glass fiber filters pre-soaked with 0.3% polyethyleneimine using a 96-well Brandel harvester. After a series of rinses, filters were dried overnight in scintillation tubes. The next day, bound radioactivity was harvested onto 0.3% polyethyleneimine-treated, 96-well filter mats using a 96-well FilterMate harvester (PerkinElmer), and the retained radioactivity was counted in a MicroBeta scintillation counter (PerkinElmer). Raw data (dpm) representing total radioligand binding (i.e., specific plus non-specific binding) were plotted as a function of the logarithm of the molar concentration of the competitor (i.e., test or reference compound). Raw data were analyzed to determine the half-maximal inhibitory concentration (IC50) followed by the absolute inhibition constant (Ki). Reference compounds (μ-opioid, DAMGO; δ-opioid, salvinorin A; κ-opioid, naltrindole) were run simultaneously in duplicate.
Statistical analysis.
All data are expressed as mean ± SD. Behavioral data were analyzed using a two-tailed, two-way repeated-measures ANOVA with Bonferroni post hoc comparisons. To account for potential carryover and period effect on PWT in the two-day CCI experiments, time-dependent differences were analyzed by two-tailed, two-way repeated measures analysis of covariance (ANCOVA). The mean NT-positive cell profile and process counts and the normalized fluorescence intensity were compared using the Student's t test. Preliminary analysis revealed increased bilateral NT immunoreactivity in the carrageenan group. Thus, the total RVM NT immunoreactivity (i.e., the NRM and bilateral LPGi and GiA) was compared between carrageenan and saline groups. Receptor binding assays were analyzed using nonlinear regression of the normalized raw data (i.e., percent radioligand binding compared to that observed in the absence of test or reference compound) using the three parameter logistic model to obtain the IC50 followed by the Cheng–Prusoff approximation to obtain the Ki (nm). Significance was defined as p < 0.05. All statistical analyses were performed using GraphPad Prism (version 5.03; GraphPad Software) or SPSS 17.0.
Results
Carrageenan-induced thermal hyperalgesia is associated with increased 3-nitrotyrosine in the RVM
Intraplantar injection of carrageenan led to development of thermal hyperalgesia that peaked by 3 h when compared to saline control rats (Fig. 1A). The NT immunoreactivity, indicating the presence of PN, increased in cell profiles, processes, and fluorescence intensity bilaterally throughout the RVM at the time of peak hyperalgesia in carrageenan-injected rats versus saline controls (n = 4; Fig. 1C–I). We also observed that NT immunoreactivity colocalized with astrocytes (GFAP) and neurons (NeuN) in carrageenan rats (Fig. 2). Although OX-42 immunoreactivity overlapped and enveloped NT-positive processes, microglia did not colocalize with NT. All subsequent immunofluorescence and pharmacological experiments were performed at this 3 h peak thermal hyperalgesia.
Figure 1.
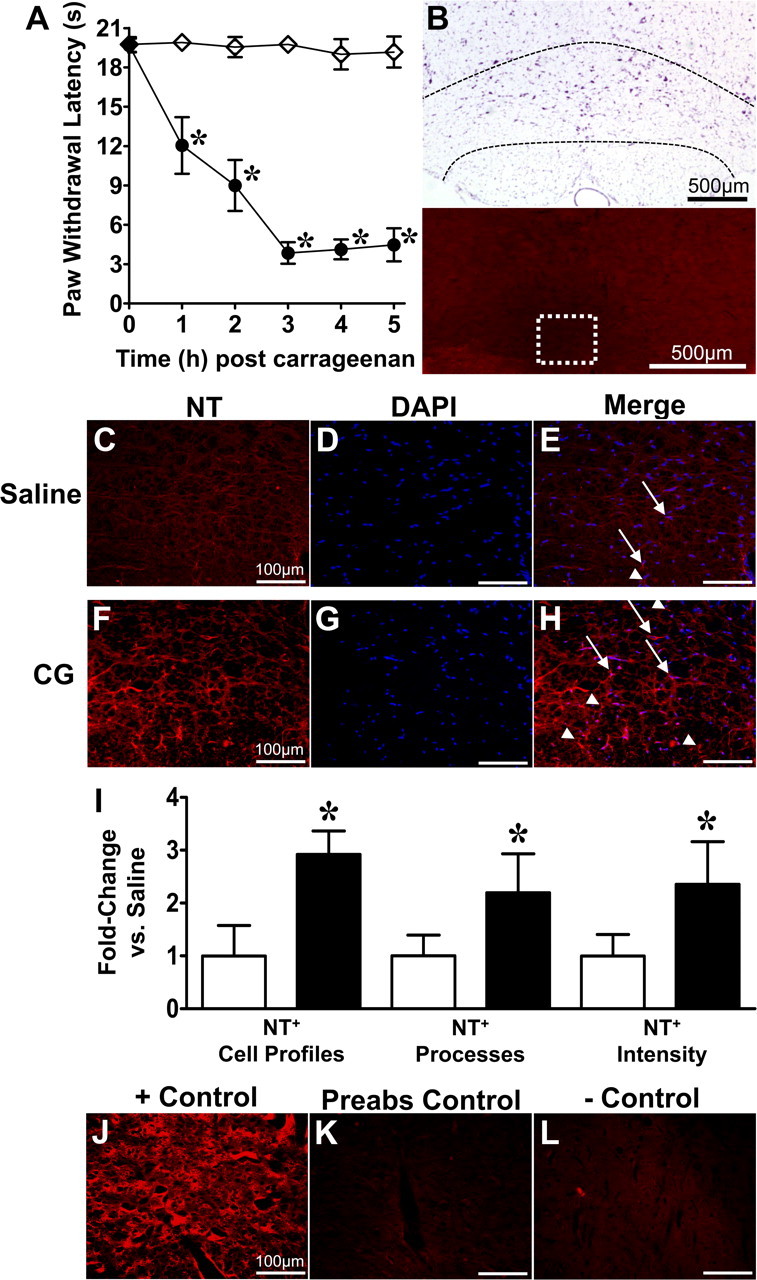
Nitrotyrosine increases in the RVM at the peak of thermal hyperalgesia. A, When compared with rats that received an intraplantar injection of vehicle (saline; s.c.; ♢), carrageenan (CG; s.c., 1%; ●) led to peak thermal hyperalgesia by 3 h. The RVM was evaluated for NT immunoreactivity at 3 h postinjection of carrageenan or saline. B, Low-magnification cresyl violet and fluorescence images demonstrate the RVM (dotted black lines) and region where subsequent images were acquired (white box). C–I, Compared to saline rats (C–E), peak thermal hyperalgesia (F–H) was associated with an increase in NT immunoreactivity (red) in cell profiles (arrows) and processes (arrowheads) (E, H, merged with DAPI, blue) as well as fluorescence intensity in the RVM [I; open bar, saline; filled bar, carrageenan (CG)]. Data in I are normalized to the mean of the saline group and represent fold change. Micrographs are from the NRM and represent three sections of the entire RVM (n = 4). J–I, The NT-positive control (+ Control) yielded intense signal (J), while preabsorption (Preabs Control) (K) and negative controls (−Control) (L) provided low levels of signal. Data are mean ± SD for n = 4 rats per group, analyzed by two-way ANOVA with Bonferroni comparisons (A) and Student's t test (I). *p < 0.05 versus saline.
Figure 2.
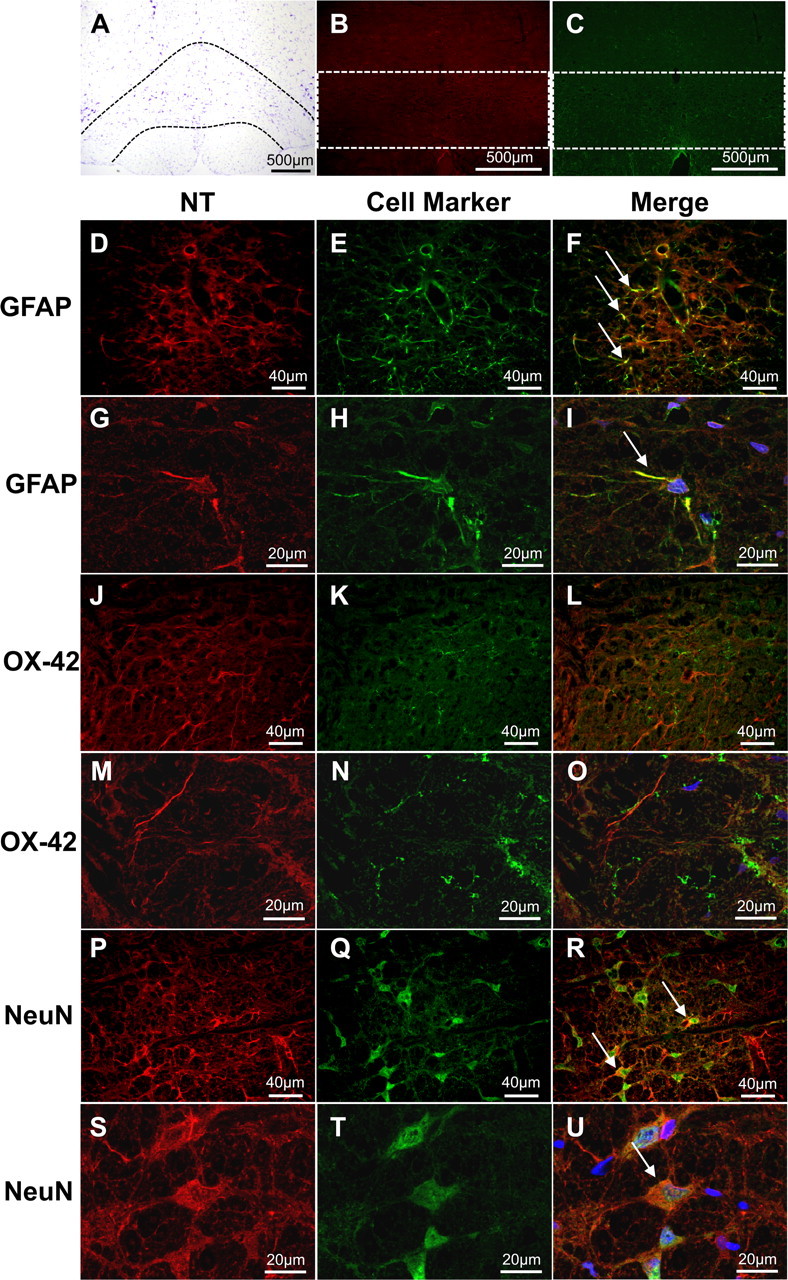
Intra-RVM NT colocalizes with astrocytes and neurons. A–C, Low-magnification cresyl violet and fluorescence images demonstrate the RVM (A, dotted black lines) and regions where subsequent colocalization images were acquired (B, C, white boxes). D–U, Fixed frozen sections from carrageenan rats at peak hyperalgesia were stained for NT (D, G, J, M, P, S, red) and astrocytes (E, H, GFAP; green), microglia (K, N OX-42; green), or neurons (Q, T, NeuN; green). These merged images (F, L, R) suggest colocalization (yellow, arrows) occurs in astrocytes and neurons. This was confirmed by observing Z-stacks from confocal microscopy at a higher magnification (I, O, U; DAPI; blue). Micrographs (F, L, R) are from the GiA and NRM nuclei and represent six merged images (n = 4).
Microinjection confirmation
Figure 3 demonstrates the microinjection sites and cannula placement in carrageenan-injected and CCI models. Thirty-micrometer-thick cresyl violet sections were evaluated with light microscopy for the microinjection sites indicated by India ink. Microscopic examination showed that these cannulae were properly placed in 76 of 82 (93%) animals.
Figure 3.

Microinjection site and cannula placement confirmation. A, Line drawings demonstrate microinjection sites in the rostral, middle, and caudal RVM identified by distance from bregma corresponding to accepted stereotaxic coordinates (Paxinos and Watson, 1998). A, B, Microinjection sites (circles, arrowheads) were confirmed in the RVM (A, gray region) using cresyl violet stained 30 μm thick coronal sections of the rostral medulla (B). C, In tissues used for micropunches of the RVM, visual inspection and dissection microscopy in the X-Y (top) and Z axes (bottom) were used for confirmation. D, Placement was also confirmed using cresyl violet stained sections from the micropunched tissues. Microinjection sites outside of the RVM were not considered for this study. Open (carrageenan) and closed (CCI) circles represent microinjection sites from two separate experiments. CN VII, Cranial nerve VII nucleus.
Reversal of thermal hyperalgesia by intra-RVM microinjection of the peroxynitrite decomposition catalyst FeTMPyP5+
Intra-RVM microinjection of FeTMPyP5+ (0.25–1 μg; n = 5) administered at peak hyperalgesia (3 h), but not its vehicle, produced a rapid (<15 min), persistent (≥2 h), and dose-dependent reversal of carrageenan-induced thermal hyperalgesia (Fig. 4A). Maximal reversal was obtained by 30 min post-FeTMPyP5+. Neither intra-RVM FeTMPyP5+ (1 μg) nor vehicle altered the withdrawal latency of saline control rats (n = 5; Fig. 4A).
Figure 4.

Intra-RVM FeTMPyP5+ reverses thermal hyperalgesia through modulation of opioid signaling. A, When compared to vehicle (○; n = 5), carrageenan-induced thermal hyperalgesia was rapidly and dose dependently reversed following intra-RVM microinjection (black arrow) of FeTMPyP5+ (■, 0.25; ♦, 0.5; ▾, 1 μg; n = 5). Paw withdrawal latencies following intra-RVM microinjection of vehicle (□; n = 5) and FeTMPyP5+ (♢; n = 5) in saline-injected rats (s.c., 50 μl) were not significantly different. B, Intrathecal injection (gray arrow) of yohimbine (▴, 30 μg; n = 5), but not vehicle (▾; n = 5), decreased paw withdrawal latencies following intra-RVM FeTMPyP5+ (1 μg; black arrow). Intrathecal injection of yohimbine in carrageenan rats receiving intra-RVM vehicle (○; n = 5) had no effect on paw withdrawal latencies. C, Intra-RVM microinjection (open arrow) of naloxone (●, 5 μg; n = 4) reversed the effects of intra-RVM FeTMPyP5+ (1 μg; black arrow) compared with vehicle (▾; n = 4). Thermal hyperalgesia was not altered following intra-RVM microinjection (open arrow) of naloxone (□, 5 μg; n = 5) or vehicle (○; n = 5). Data are mean ± SD for n rats per group and analyzed by two-way ANOVA with Bonferroni comparisons. #p < 0.001, time (t) 0 h versus t3 h; *p < 0.01 th versus 3 h; †p < 0.001 th versus 4 h.
Removal of intra-RVM peroxynitrite engages descending modulation of spinal nociception
RVM stimulation that results in inhibition of nociceptive reflexes involves multiple supraspinal descending pathways, including the recruitment of noradrenergic pathways (Hammond and Yaksh, 1984). Thus, intrathecal injections of yohimbine, an α2 adrenergic receptor antagonist, were given 1 h post-FeTMPyP5+ to determine whether intra-RVM FeTMPyP5+ reverses hyperalgesia through the activation of descending inhibitory pathways. When compared to vehicle (n = 5), intrathecal yohimbine (30 μg; n = 5) reversed the effects of intra-RVM FeTMPyP5+ in carrageenan-treated rats (Fig. 4B). In carrageenan rats receiving an intra-RVM microinjection of the vehicle for FeTMPyP5+, intrathecal yohimbine did not alter paw withdrawal latencies (Fig. 4B; n = 5). Dosages were based on previous reports (De Felice et al., 2011).
Peroxynitrite in the RVM promotes thermal hyperalgesia through altered local opioidergic signaling
An intra-RVM microinjection of naloxone, a non-subtype selective opioid receptor antagonist, was given 1 h post-FeTMPyP5+ in carrageenan-treated rats to investigate the potential interactions between PN and endogenous opioid signaling. The effects of FeTMPyP5+ (1 μg) were rapidly reversed (<15 min) with intra-RVM microinjection of naloxone (5 μg) when compared to vehicle (n = 4; Fig. 4C). Neither intra-RVM naloxone nor its vehicle altered withdrawal latencies of carrageenan-injected rats (n = 4). Naloxone doses were within the range shown by others to be effective by intra-RVM microinjection (Dickenson et al., 1979; Kalra et al., 2001).
Nitrotyrosine colocalizes with enkephalin during thermal hyperalgesia
The presence of NT coincided with ENK immunoreactivity in the RVM of carrageenan-treated rats, with an estimated 14.4% NT/ENK overlap to total ENK immunoreactivity (M = 0.144) vs 1.7% overlap in saline-treated rats (M = 0.017, n = 4; Fig. 5). Equally important, when co-occurring, the intensity of NT labeling correlated positively with ENK labeling (Rr = 0.68), consistent with the idea that ENK is a target for PN nitration.
Figure 5.
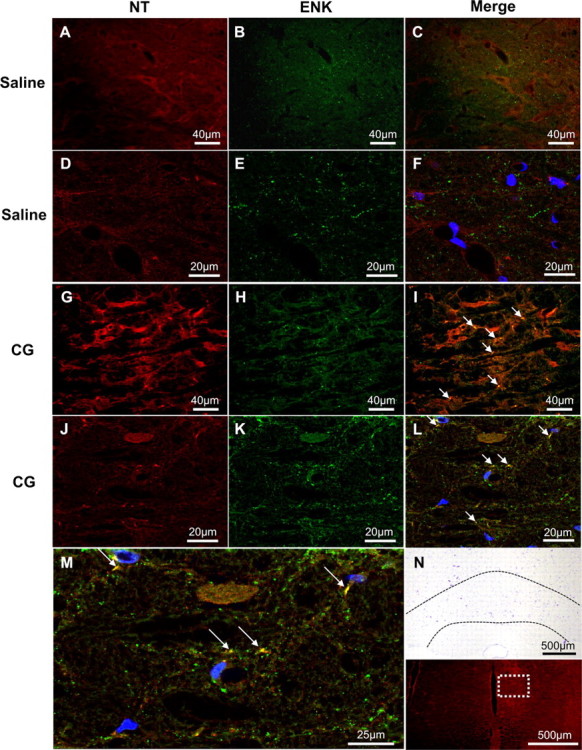
Colocalization of NT and ENK in the RVM during peak thermal hyperalgesia. A–M, When compared to vehicle (A–F), carrageenan (CG) rats at peak hyperalgesia (G–M) had increased NT (red) colocalization (yellow, arrows) with ENK (green) as determined by colocalization coefficient analysis. Lower magnification micrographs (A–C, G–I) are from the NRM nucleus and represent 10 merged images (n = 4). This was confirmed using Z-stacks from confocal microscopy at a higher magnification (D–F, J–L; DAPI in blue). M, A cropped and enlarged image of L demonstrates the colocalization (yellow, arrows) of NT and ENK in CG RVM. N, Low-magnification cresyl violet and fluorescence images demonstrate the RVM (dotted black lines) and region from where images were acquired for this figure (white box), respectively.
Nitroxidative modifications of methionine-enkephalin in vitro reduce its in vivo antinociceptive properties
To determine the effects of PN-mediated modifications of ENK, we evaluated MENK, which is the ENK with the greatest expression in the CNS (Yang et al., 1977). The major reaction product formed by exposing MENK to PN in vitro was NSO-MENK, which was nitrated at tyrosine amino acid and sulfoxidized at the methionine amino acid (Fig. 6A). As shown in Figure 6B, intrathecal injection of NSO-MENK (400 μg; n = 4) had a substantially reduced antinociceptive effect when compared to MENK (400 μg; n = 4). Dosages were within the range of previously reported concentrations for intrathecal injections of MENK (Yaksh et al., 1977). In addition, competitive binding assays of MENK and NSO-MENK to δ-, μ-, and κ-opioid receptors (Fig. 6C) demonstrated that NSO-MENK binding affinities for all opioid receptors were substantially decreased compared to those of MENK.
Figure 6.
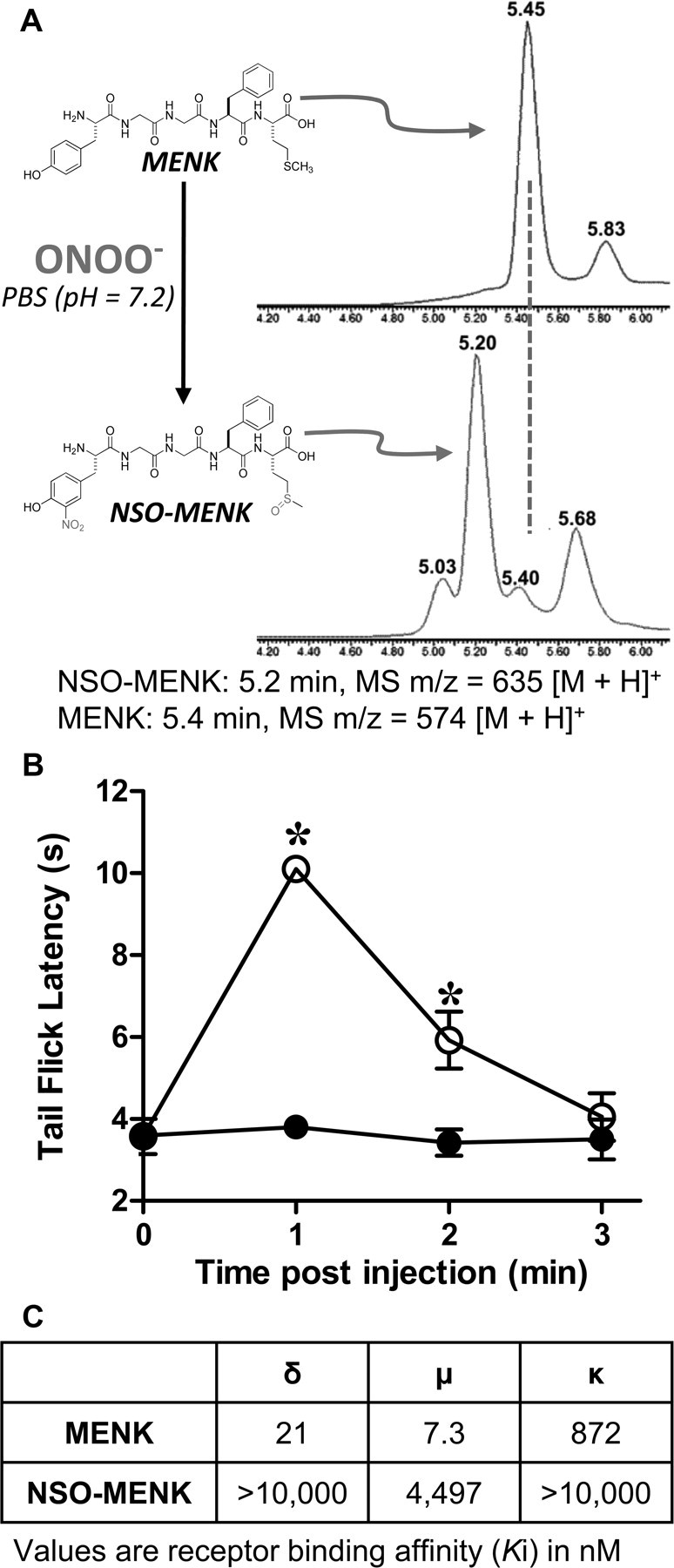
Synthesis of NSO-MENK; nitroxidative modifications of MENK suppress its antinociceptive properties in vivo. A, Treatment of a stirred solution of MENK (798 μm, 50 mm PBS, 25°C, pH 7.2) with five equivalents of PN (100 μl, 50 mm in 0.1 m NaOH) by rapid injection into the stirring vortex; final pH 7.2 afforded the nitro-sulfoxide derivative, NSO-MENK, as the major product identified with LCMS. B, Intrathecal MENK (400 μg, ○), but not NSO-MENK (400 μg, ●), led to a rapid development of antinociception. C, When compared to MENK, NSO-MENK binding affinities (Ki) for δ-, μ-, and κ-opioid receptors were substantially reduced. Behavioral data are mean ± SD for n = 4 rats and analyzed by two-way ANOVA with Bonferroni comparisons to MENK. *p < 0.001, NSO-MENK versus MENK.
Opioid-sensitive reversal of chronic constriction injury-induced mechanical allodynia by intra-RVM microinjection of FeTMPyP5+
To test our hypothesis that PN has a central role in descending modulation of chronic pain, we investigated the effects of an intra-RVM treatment with a PNDC in the CCI model of chronic neuropathic pain. Figure 7A demonstrates that intra-RVM microinjections of FeTMPyP5+ at 7 d post-CCI (D7), when allodynia is maximal, rapidly (<30 min) reversed PWT with maximal effects observed by 1 h that lasted through 4 h (n = 6). Intra-RVM microinjections of vehicle had no effect on mechanical allodynia in CCI rats (n = 6). Following the first experiment, rats were tested at 24 h (D8) for PWT, and if there were no carryover effects from the previous day's experiments (i.e., changes in baseline PWT), they received intra-RVM microinjections of the opposite solution from D7 followed by an additional microinjection of naloxone or its vehicle. As shown in Figure 7C, intra-RVM microinjection of naloxone (5 μg, n = 3) at 2 h, when FeTMPyP5+ (1 μg) effects plateaued, resulted in a rapid reversal that was significant by 30 min compared to vehicle (n = 3).
Figure 7.

Intra-RVM PN is essential to CCI-induced mechanical allodynia through the modulation of opioidergic signaling. A, When compared with day 0 (D0) PWT (pre-CCI), maximal mechanical allodynia induced by CCI occurred by D7 and was rapidly (< 30 min) reversed and maintained for 4 h following intra-RVM microinjection (black arrow) of FeTMPyP5+ (1 μg, ●; n = 6), but not vehicle (○; n = 6). B, Neither intra-RVM (black arrow) FeTMPyP5+ (●) nor vehicle (○) altered the PWT of the contralateral paw. C, Intra-RVM microinjection (open arrow) of naloxone (■, 5 μg; n = 3), but not vehicle (●; n = 3), reversed the effects of intra-RVM microinjection (black arrow) of FeTMPyP5+ (1 μg) and had no effect on CCI rats receiving vehicle (□, 5 μg; n = 3). D, Withdrawal thresholds in the contralateral paw were not altered following intra-RVM microinjections of naloxone (open arrow) in FeTMPyP5+-treated (■)- and FeTMPyP5+ vehicle-treated (□) animals. The contralateral PWT of FeTMPyP5+-treated animals were not affected by intra-RVM microinjection of the vehicle for naloxone (●). Data are mean ± SD for n rats and analyzed by two-way ANCOVA with Bonferroni comparisons. #p < 0.001 D0 versus baseline (A, D7; B, D8); *p < 0.001 th versus baseline; †p < 0.001 th versus 2 h.
Discussion
The RVM is a potential site for peroxynitrite (PN) activity during pathophysiological pain as local enhanced glutamatergic signaling and neuroimmune activation are involved in descending facilitation, processes to which PN contributes within the spinal cord to promote central sensitization (Little et al., 2012). The present study suggests that PN is essential to RVM descending facilitation by suppressing opioidergic signaling during inflammatory and neuropathic pain.
Our immunofluorescence data identified increased 3-nitrotyrosine (NT) immunoreactivity, and thus the presumptive presence of PN, within neurons and astrocytes of the RVM during thermal hyperalgesia. This agrees with previous observations that PN contributes to neuronal and glial cell-mediated spinal mechanisms that are essential to pathophysiological pain (Muscoli et al., 2004; Wang et al., 2004; Ndengele et al., 2009; Chen et al., 2010). These observations are the first to investigate the cellular sites of nitration during pain and support our recent discussions of the relative contribution of neurons and glia cells to PN activities (Little et al., 2012). That microglia did not colocalize with NT may reflect the additional protection offered by this cell's enhanced intracellular antioxidant environment compared to that of astrocytes (Chatterjee et al., 1999). Although NT is present in neurons and astrocytes, this does not implicate them as the site of PN formation. Rather, PN formation can be intracellular or extracellular, such that post-translational nitration may occur following PN diffusion into adjacent cells (Ferrer-Sueta and Radi, 2009). We hypothesize that the enhanced nociceptive signaling that stimulates RVM neurons and hyperactivates glial cells results in local PN formation. Previous studies of PN suggest that its formation depends upon N-methyl-d-aspartate receptor (NMDAR) activation as well as superoxide and nitric oxide formation in the spinal cord during central sensitization (Muscoli et al., 2004; Wang et al., 2004; Ndengele et al., 2009; Chen et al., 2010). The enhanced activities of NMDAR, nitric oxide synthase, and glial cells that occur in the RVM during pain states (Urban et al., 1999b; Wei et al., 2008; Roberts et al., 2009) may serve as potential sources for PN formation (Little et al., 2012). We are currently investigating potential sources of superoxide such as NADPH-oxidase activity as well as PN-mediated nitration and inactivation of manganese superoxide dismutase. These have been implicated as sources for superoxide during pain (Salvemini et al., 2011).
The results from our pharmacological studies using intra-RVM treatment with the PNDC FeTMPyP5+, provide novel evidence of PN′s functional contribution to descending facilitation during both inflammatory and neuropathic pain. These findings implicate PN as a key mediator of the RVM mechanisms that are essential to persistent and chronic pain. We found that intra-RVM administration of FeTMPyP5+ reversed thermal hyperalgesia, presumably by activating noradrenergic descending inhibitory pathways to modulate spinal nociception. Our findings agree with previous studies demonstrating that the RVM can recruit noradrenergic bulbospinal pathways to inhibit spinal nociception (Hammond and Yaksh, 1984). Other descending pathways, e.g., serotonergic ones, may also contribute to this effect (Hammond and Yaksh, 1984), and further investigations are required to address these possibilities.
We hypothesized that PN acts as an anti-opioid to decrease opioidergic signaling, suppress descending inhibition, and thereby lead to a net facilitation of spinal nociception. We show that the effects of intra-RVM FeTMPyP5+ are reversed by naloxone, suggesting that PN accumulation reduces opioid signaling and PN removal restores opioid signaling. Intra-RVM naloxone reversed FeTMPyP5+ effects during both inflammatory and neuropathic pain, indicating that PN may mediate similar mechanisms in either pain state to engage and maintain descending facilitation. Naloxone is a non-subtype selective opioid receptor antagonist, and we cannot conclude which receptor is responsible for our findings. However, previous studies demonstrate that intra-RVM agonism of both μ- and δ-opioid receptors provides significant antinociception during an inflammatory pain state (Hurley and Hammond, 2000, 2001).
We focused our investigation on the potential for PN to nitroxidatively modify ENK based upon previous studies (Yi et al., 1997; Fontana et al., 2006). Our colocalization experiments revealed the co-occurrence of intracellular NT and ENK in the RVM during thermal hyperalgesia, thereby providing evidence of a potential site for their in situ interactions. As PN nitration may also occur extracellularly (Ferrer-Sueta and Radi, 2009), the potential for ENK nitration likely has been underestimated here. There are numerous possibilities for the intracellular site of ENK nitration, notably within the cell body where ENK production and vesicle packaging occurs, and within presynaptic dense core vesicles (Kelly, 1993). It is unknown whether PN can traverse the vesicle membrane to nitrate ENK, although PN can pass through lysosomal and cell membranes (Ferrer-Sueta and Radi, 2009), which have the same lipid bilayer arrangement as vesicles (Kelly, 1993). Thus, PN within the cytosol or axon terminal may nitroxidatively modify ENK before its release. This may occur in local RVM neurons or in neurons that project to the RVM, such as the periaqueductal gray (PAG) neurons (Beitz, 1982). Important extensions of these findings, such as analyses to quantify and describe these nitroxidative modifications in vivo, are indicated.
As MENK is the most abundant form of ENK in the CNS (Yang et al., 1977), we investigated the interaction of PN with MENK to study the effects of nitroxidative modifications of ENK upon nociception. The major product formed by the reaction of PN with MENK was NSO-MENK, which included tyrosine nitration. Because the tyrosine in MENK is the key biologically active pharmacophore required for binding with all opioid receptors (Loew and Burt, 1978; Schwyzer, 1986), we hypothesized that NSO-MENK loses its binding properties at μ- and δ-opioid receptors, and thus its antinociceptive effects. Consistent with this hypothesis, intrathecal administration of NSO-MENK showed significantly less antinociceptive action compared to that of MENK. Indeed, we found that NSO-MENK lacks appreciable binding affinity for the δ-, μ-, and κ-opioid receptors. Since the Tyr-Gly-Gly-Phe sequence is conserved in the pentapeptide structures of leucine ENK and MENK, it is likely both will form the 3-nitrotyrosyl analog, with MENK having an additional oxidatively sensitive site in the methionine side chain. Our finding of PN-mediated MENK nitration is in agreement with previous studies of PN-mediated tyrosine nitration of leucine ENK in vitro (Yi et al., 1997; Fontana et al., 2006; Zhan and Desiderio, 2009).
In addition to interactions with ENK, PN may interact with opioid receptors and other endogenous tyrosine-containing opioid peptides. For example, oxidative stress in vitro impairs the function of opioid receptors (Raut et al., 2006), although chronic inflammatory pain is associated with enhanced RVM opioid receptor responsiveness and no change in opioid receptor number (Sykes et al., 2007). It may be that PN decreases the bioavailability of ENK by its ability to promote inflammation and activate redox-sensitive transcription factors (Matata and Galinanes, 2002) that increase the activity and/or expression of enkephalinases, which themselves can be altered during inflammation (Kondepudi and Johnson, 1993).
Although PN may interact with other endogenous opioids in the RVM, including endomorphins and dynorphin, our data suggest that restoration of RVM opioid signaling by ENK is one mechanism by which FeTMPyP5+ inhibits spinal nociceptive processing. The importance of endogenous ENK signaling in RVM descending inhibition is in agreement with previous work (al-Rodhan et al., 1990; Fields, 2004). The neuronal circuitry by which ENK exerts its effects likely differs by receptor subtype. For example, μ-opioid receptor agonism in the RVM indirectly activates OFF-cells (i.e., disinhibition following direct inhibition of gamma aminobutyric acid interneurons) and directly inactivates ON-cells (Heinricher et al., 1992, 1994). Less is known about the neuronal circuitry of δ-receptor agonism. However, the end result of both μ- and δ-receptor agonism is descending inhibition following ON-cell inactivation and OFF-cell activation (Heinricher et al., 1992, 1994; Thorat and Hammond, 1997; Harasawa et al., 2000). We anticipate that in vivo indicators of opioid receptor activation in the RVM, such as μ-receptor phosphorylation at serine 375 and/or threonine 180 (Celver et al., 2001; El Kouhen et al., 2001), are reduced during pain in response to PN activities and enhanced with FeTMPyP5+ administration. We will use such approaches in future studies to delineate additional interactions between PN and endogenous opioids.
In summary, our work suggests that PN modulates opioidergic signaling by decreasing the bioavailability of enkephalin and thus reducing the contribution of ENK to RVM descending modulation during pathophysiological pain (Fig. 8). The potential interactions between PN and opioid systems in the RVM imply that other supraspinal sites requiring opioid signaling to adequately modulate nociceptive processing (e.g., PAG and amygdala) may be susceptible to the pathophysiological environments caused by PN-mediated nitroxidative stress. It is our hypothesis that supraspinal PN formation and its subsequent activity may be essential to supraspinal changes that drive the transition of acute to chronic pain.
Figure 8.

Working hypothesis: PN decreases the bioavailability of ENK in the RVM during inflammatory and neuropathic pain. A, Normal binding of ENK (filled circles) to opioid receptors (OR) results in inhibition of neurons such as RVM ON-cells. B, During pathophysiological pain, there is an increase in PN formation (intracellular and extracellular) that results in nitroxidative modifications of ENK (circle shadow). These modifications suppress the antinociceptive properties of ENK by reducing its receptor binding affinity, potentially maintaining ON-cell activity and/or gamma aminobutyric acid interneuron inhibition of OFF-cell activity (data not shown); this contributes to RVM descending facilitation. Administration of FeTMPyP5+ prevents further modifications of ENK, which restores opioidergic signaling and contributes to the rapid reversal of hyperalgesia and allodynia, respectively.
This work was designed to elucidate the role of PN in supraspinal sites during pathophysiological pain. These data demonstrate for the first time that PN is intimately involved in the processes that culminate in RVM descending facilitation. Furthermore, PN does so by acting as an anti-descending inhibition agent potentially through nitroxidative modifications of ENK to reduce opioid signaling during inflammatory and neuropathic pain. This study provides a foundation for future investigations to characterize the roles of PN in the brain during pathophysiological pain.
Footnotes
This work was supported by NIH/National Institute on Drug Abuse Grant R01 DA024074 (D.S.), NIH/National Institute of Arthritis and Musculoskeletal and Skin Diseases Grant RC1 AR058231 (D.S. and W.L.N.), and Saint Louis University Center for Anatomical Science and Education Fellowship (J.W.L.). We express our gratitude to Dr. Andrew Lechner for his critical review of the manuscript and Dr. Jan Ryerse for his assistance with microscopy. We also thank Katie Davis, Robert Livergood, and Kevin Dietl for technical assistance and data collection.
The authors declare no competing financial interests.
References
- al-Rodhan N, Chipkin R, Yaksh TL. The antinociceptive effects of SCH-32615, a neutral endopeptidase (enkephalinase) inhibitor, microinjected into the periaqueductal, ventral medulla and amygdala. Brain Res. 1990;520:123–130. doi: 10.1016/0006-8993(90)91697-f. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Fields HL. Endogenous pain control systems: brainstem spinal pathways and endorphin circuitry. Annu Rev Neurosci. 1984;7:309–338. doi: 10.1146/annurev.ne.07.030184.001521. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bee LA, Dickenson AH. Rostral ventromedial medulla control of spinal sensory processing in normal and pathophysiological states. Neuroscience. 2007;147:786–793. doi: 10.1016/j.neuroscience.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Beitz AJ. The nuclei of origin of brain stem enkephalin and substance P projections to the rodent nucleus raphe magnus. Neuroscience. 1982;7:2753–2768. doi: 10.1016/0306-4522(82)90098-7. [DOI] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Brown RM, Clouet DH, Friedman DP. Opiate receptor subtypes and brain function. NIDA Res Monogr. 1986;71:1–188. [PubMed] [Google Scholar]
- Celver JP, Lowe J, Kovoor A, Gurevich VV, Chavkin C. Threonine 180 is required for G-protein-coupled receptor kinase 3- and beta-arrestin 2-mediated desensitization of the mu-opioid receptor in Xenopus oocytes. J Biol Chem. 2001;276:4894–4900. doi: 10.1074/jbc.M007437200. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Noack H, Possel H, Keilhoff G, Wolf G. Glutathione levels in primary glial cultures: monochlorobimane provides evidence of cell type-specific distribution. Glia. 1999;27:152–161. [PubMed] [Google Scholar]
- Chen Z, Muscoli C, Doyle T, Bryant L, Cuzzocrea S, Mollace V, Mastroianni R, Masini E, Salvemini D. NMDA-receptor activation and nitroxidative regulation of the glutamatergic pathway during nociceptive processing. Pain. 2010;149:100–106. doi: 10.1016/j.pain.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Amour FE. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- De Felice M, Sanoja R, Wang R, Vera-Portocarrero L, Oyarzo J, King T, Ossipov MH, Vanderah TW, Lai J, Dussor GO, Fields HL, Price TJ, Porreca F. Engagement of descending inhibition from the rostral ventromedial medulla protects against chronic neuropathic pain. Pain. 2011;152:2701–2709. doi: 10.1016/j.pain.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickenson AH, Oliveras JL, Besson JM. Role of the nucleus raphe magnus in opiate analgesia as studied by the microinjection technique in the rat. Brain Res. 1979;170:95–111. doi: 10.1016/0006-8993(79)90943-0. [DOI] [PubMed] [Google Scholar]
- El Kouhen R, Burd AL, Erickson-Herbrandson LJ, Chang CY, Law PY, Loh HH. Phosphorylation of Ser363, Thr370, and Ser375 residues within the carboxyl tail differentially regulates mu-opioid receptor internalization. J Biol Chem. 2001;276:12774–12780. doi: 10.1074/jbc.M009571200. [DOI] [PubMed] [Google Scholar]
- Ferrer-Sueta G, Radi R. Chemical biology of peroxynitrite: kinetics, diffusion, and radicals. ACS Chem Biol. 2009;4:161–177. doi: 10.1021/cb800279q. [DOI] [PubMed] [Google Scholar]
- Fields H. State-dependent opioid control of pain. Nat Rev Neurosci. 2004;5:565–575. doi: 10.1038/nrn1431. [DOI] [PubMed] [Google Scholar]
- Fields HL, Bry J, Hentall I, Zorman G. The activity of neurons in the rostral medulla of the rat during withdrawal from noxious heat. J Neurosci. 1983;3:2545–2552. doi: 10.1523/JNEUROSCI.03-12-02545.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana M, Pecci L, Schininá ME, Montefoschi G, Rosei MA. Oxidative and nitrative modifications of enkephalins by reactive nitrogen species. Free Radic Res. 2006;40:697–706. doi: 10.1080/10715760500467125. [DOI] [PubMed] [Google Scholar]
- Gardell LR, Vanderah TW, Gardell SE, Wang R, Ossipov MH, Lai J, Porreca F. Enhanced evoked excitatory transmitter release in experimental neuropathy requires descending facilitation. J Neurosci. 2003;23:8370–8379. doi: 10.1523/JNEUROSCI.23-23-08370.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond DL, Yaksh TL. Antagonism of stimulation-produced antinociception by intrathecal administration of methysergide or phentolamine. Brain Res. 1984;298:329–337. doi: 10.1016/0006-8993(84)91432-x. [DOI] [PubMed] [Google Scholar]
- Harasawa I, Fields HL, Meng ID. Delta opioid receptor mediated actions in the rostral ventromedial medulla on tail flick latency and nociceptive modulatory neurons. Pain. 2000;85:255–262. doi: 10.1016/s0304-3959(99)00280-8. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Morgan MM, Fields HL. Direct and indirect actions of morphine on medullary neurons that modulate nociception. Neuroscience. 1992;48:533–543. doi: 10.1016/0306-4522(92)90400-v. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Morgan MM, Tortorici V, Fields HL. Disinhibition of off-cells and antinociception produced by an opioid action within the rostral ventromedial medulla. Neuroscience. 1994;63:279–288. doi: 10.1016/0306-4522(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Hurley RW, Hammond DL. The analgesic effects of supraspinal mu and delta opioid receptor agonists are potentiated during persistent inflammation. J Neurosci. 2000;20:1249–1259. doi: 10.1523/JNEUROSCI.20-03-01249.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley RW, Hammond DL. Contribution of endogenous enkephalins to the enhanced analgesic effects of supraspinal mu opioid receptor agonists after inflammatory injury. J Neurosci. 2001;21:2536–2545. doi: 10.1523/JNEUROSCI.21-07-02536.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalra A, Urban MO, Sluka KA. Blockade of opioid receptors in rostral ventral medulla prevents antihyperalgesia produced by transcutaneous electrical nerve stimulation (TENS) J Pharmacol Exp Ther. 2001;298:257–263. [PubMed] [Google Scholar]
- Kelly RB. Storage and release of neurotransmitters. Cell [Suppl] 1993;72:43–53. doi: 10.1016/s0092-8674(05)80027-3. [DOI] [PubMed] [Google Scholar]
- Kondepudi A, Johnson A. Cytokines increase neutral endopeptidase activity in lung fibroblasts. Am J Respir Cell Mol Biol. 1993;8:43–49. doi: 10.1165/ajrcmb/8.1.43. [DOI] [PubMed] [Google Scholar]
- Little JW, Doyle T, Salvemini D. Reactive nitroxidative species and nociceptive processing: determining the roles for nitric oxide, superoxide, and peroxynitrite in pain. Amino Acids. 2012;42:75–94. doi: 10.1007/s00726-010-0633-0. [DOI] [PubMed] [Google Scholar]
- Loew GH, Burt SK. Energy conformation study of Met-enkephalin and its d-Ala2 analogue and their resemblance to rigid opiates. Proc Natl Acad Sci U S A. 1978;75:7–11. doi: 10.1073/pnas.75.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manders EMM, Verbeek FJ, Aten JA. Measurement of co-localization of objects in dual color confocal images. J Microsc. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci. 1992;103:857–862. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- Marvizón JC, Chen W, Murphy N. Enkephalins, dynorphins, and beta-endorphin in the rat dorsal horn: an immunofluorescence colocalization study. J Comp Neurol. 2009;517:51–68. doi: 10.1002/cne.22130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matata BM, Galiñanes M. Peroxynitrite is an essential component of cytokines production mechanism in human monocytes through modulation of nuclear factor-kappa B DNA binding activity. J Biol Chem. 2002;277:2330–2335. doi: 10.1074/jbc.M106393200. [DOI] [PubMed] [Google Scholar]
- Muscoli C, Mollace V, Wheatley J, Masini E, Ndengele M, Wang ZQ, Salvemini D. Superoxide-mediated nitration of spinal manganese superoxide dismutase: a novel pathway in N-methyl-d-aspartate-mediated hyperalgesia. Pain. 2004;111:96–103. doi: 10.1016/j.pain.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Muscoli C, Cuzzocrea S, Ndengele MM, Mollace V, Porreca F, Fabrizi F, Esposito E, Masini E, Matuschak GM, Salvemini D. Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. J Clin Invest. 2007;117:3530–3539. doi: 10.1172/JCI32420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscoli C, Doyle T, Dagostino C, Bryant L, Chen Z, Watkins LR, Ryerse J, Bieberich E, Neumman W, Salvemini D. Counter-regulation of opioid analgesia by glial-derived bioactive sphingolipids. J Neurosci. 2010;30:15400–15408. doi: 10.1523/JNEUROSCI.2391-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndengele MM, Cuzzocrea S, Esposito E, Mazzon E, Di Paola R, Matuschak GM, Salvemini D. Cyclooxygenases 1 and 2 contribute to peroxynitrite-mediated inflammatory pain hypersensitivity. FASEB J. 2008;22:3154–3164. doi: 10.1096/fj.08-108159. [DOI] [PubMed] [Google Scholar]
- Ndengele MM, Cuzzocrea S, Masini E, Vinci MC, Esposito E, Muscoli C, Petrusca DN, Mollace V, Mazzon E, Li D, Petrache I, Matuschak GM, Salvemini D. Spinal ceramide modulates the development of morphine antinociceptive tolerance via peroxynitrite-mediated nitroxidative stress and neuroimmune activation. J Pharmacol Exp Ther. 2009;329:64–75. doi: 10.1124/jpet.108.146290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Ed 4. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Porreca F, Burgess SE, Gardell LR, Vanderah TW, Malan TP, Jr, Ossipov MH, Lappi DA, Lai J. Inhibition of neuropathic pain by selective ablation of brainstem medullary cells expressing the mu-opioid receptor. J Neurosci. 2001;21:5281–5288. doi: 10.1523/JNEUROSCI.21-14-05281.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25:319–325. doi: 10.1016/s0166-2236(02)02157-4. [DOI] [PubMed] [Google Scholar]
- Portoghese PS. A new concept on the mode of interaction of narcotic analgesics with receptors. J Med Chem. 1965;8:609–616. doi: 10.1021/jm00329a013. [DOI] [PubMed] [Google Scholar]
- Rasband WS. ImageJ. Bethesda, MD: National Institutes of Health; 1997–2011. [Google Scholar]
- Raut A, Iglewski M, Ratka A. Differential effects of impaired mitochondrial energy production on the function of mu and delta opioid receptors in neuronal SK-N-SH cells. Neurosci Lett. 2006;404:242–246. doi: 10.1016/j.neulet.2006.05.055. [DOI] [PubMed] [Google Scholar]
- Raynor K, Kong H, Law S, Heerding J, Tallent M, Livingston F, Hines J, Reisine T. Molecular biology of opioid receptors. NIDA Res Monogr. 1996;161:83–103. [PubMed] [Google Scholar]
- Roberts J, Ossipov MH, Porreca F. Glial activation in the rostroventromedial medulla promotes descending facilitation to mediate inflammatory hypersensitivity. Eur J Neurosci. 2009;30:229–241. doi: 10.1111/j.1460-9568.2009.06813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth BL, Laskowski MB, Coscia CJ. Evidence for distinct subcellular sites of opiate receptors. Demonstration of opiate receptors in smooth microsomal fractions isolated from rat brain. J Biol Chem. 1981;256:10017–10023. [PubMed] [Google Scholar]
- Salvemini D, Wang ZQ, Stern MK, Currie MG, Misko TP. Peroxynitrite decomposition catalysts: therapeutics for peroxynitrite-mediated pathology. Proc Natl Acad Sci U S A. 1998a;95:2659–2663. doi: 10.1073/pnas.95.5.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini D, Jensen MP, Riley DP, Misko TP. Therapeutic manipulations of peroxynitrite. Drug News Perspect. 1998b;11:204–214. [PubMed] [Google Scholar]
- Salvemini D, Little JW, Doyle T, Neumann WL. Roles of reactive oxygen and nitrogen species in pain. Free Radic Biol Med. 2011;51:951–966. doi: 10.1016/j.freeradbiomed.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwyzer R. Molecular mechanism of opioid receptor selection. Biochemistry. 1986;25:6335–6342. doi: 10.1021/bi00368a075. [DOI] [PubMed] [Google Scholar]
- Størkson RV, Kjørsvik A, Tjølsen A, Hole K. Lumbar catheterization of the spinal subarachnoid space in the rat. J Neurosci Methods. 1996;65:167–172. doi: 10.1016/0165-0270(95)00164-6. [DOI] [PubMed] [Google Scholar]
- Sykes KT, White SR, Hurley RW, Mizoguchi H, Tseng LF, Hammond DL. Mechanisms responsible for the enhanced antinociceptive effects of micro-opioid receptor agonists in the rostral ventromedial medulla of male rats with persistent inflammatory pain. J Pharmacol Exp Ther. 2007;322:813–821. doi: 10.1124/jpet.107.121954. [DOI] [PubMed] [Google Scholar]
- Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- Terayama R, Guan Y, Dubner R, Ren K. Activity-induced plasticity in brain stem pain modulatory circuitry after inflammation. Neuroreport. 2000;11:1915–1919. doi: 10.1097/00001756-200006260-00022. [DOI] [PubMed] [Google Scholar]
- Thorat SN, Hammond DL. Modulation of nociception by microinjection of delta-1 and delta-2 opioid receptor ligands in the ventromedial medulla of the rat. J Pharmacol Exp Ther. 1997;283:1185–1192. [PubMed] [Google Scholar]
- Urban MO, Zahn PK, Gebhart GF. Descending facilitatory influences from the rostral medial medulla mediate secondary, but not primary hyperalgesia in the rat. Neuroscience. 1999a;90:349–352. doi: 10.1016/s0306-4522(99)00002-0. [DOI] [PubMed] [Google Scholar]
- Urban MO, Coutinho SV, Gebhart GF. Involvement of excitatory amino acid receptors and nitric oxide in the rostral ventromedial medulla in modulating secondary hyperalgesia produced by mustard oil. Pain. 1999b;81:45–55. doi: 10.1016/s0304-3959(98)00265-6. [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Porreca F, Cuzzocrea S, Galen K, Lightfoot R, Masini E, Muscoli C, Mollace V, Ndengele M, Ischiropoulos H, Salvemini D. A newly identified role for superoxide in inflammatory pain. J Pharmacol Exp Ther. 2004;309:869–878. doi: 10.1124/jpet.103.064154. [DOI] [PubMed] [Google Scholar]
- Wei F, Guo W, Zou S, Ren K, Dubner R. Supraspinal glial-neuronal interactions contribute to descending pain facilitation. J Neurosci. 2008;28:10482–10495. doi: 10.1523/JNEUROSCI.3593-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Huang SP, Rudy TA. The direct and specific opiate-like effect of met5-enkephalin and analogues on the spinal cord. Neuroscience. 1977;2:593–596. doi: 10.1016/0306-4522(77)90055-0. [DOI] [PubMed] [Google Scholar]
- Yang HY, Hong JS, Costa E. Regional distribution of LEU and MET enkephalin in rat brain. Neuropharmacology. 1977;16:303–307. doi: 10.1016/0028-3908(77)90112-5. [DOI] [PubMed] [Google Scholar]
- Yi D, Smythe GA, Blount BC, Duncan MW. Peroxynitrite-mediated nitration of peptides: characterization of the products by electrospray and combined gas chromatography-mass spectrometry. Arch Biochem Biophys. 1997;344:253–259. doi: 10.1006/abbi.1997.0218. [DOI] [PubMed] [Google Scholar]
- Yokoyama K, Uhlin U, Stubbe J. Site-specific incorporation of 3-nitrotyrosine as a probe of pKa perturbation of redox-active tyrosines in ribonucleotide reductase. J Am Chem Soc. 2010;132:8385–8397. doi: 10.1021/ja101097p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Desiderio DM. MALDI-induced fragmentation of leucine enkephalin, Nitro-Tyr leucine enkaphalin, and d(5)-Phe-Nitro-Tyr leucine enkephalin. Int J Mass Spectrom. 2009;287:77–86. doi: 10.1016/j.ijms.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]