

Abstract
Rare sarcomere protein variants cause dominant hypertrophic and dilated cardiomyopathies. To evaluate whether allelic variants in eight sarcomere genes are associated with cardiac morphology and function in the community, we sequenced 3,600 individuals from the Framingham Heart Study (FHS) and Jackson Heart Study (JHS) cohorts. Out of the total, 11.2% of individuals had one or more rare nonsynonymous sarcomere variants. The prevalence of likely pathogenic sarcomere variants was 0.6%, twice the previous estimates; however, only four of the 22 individuals had clinical manifestations of hypertrophic cardiomyopathy. Rare sarcomere variants were associated with an increased risk for adverse cardiovascular events (hazard ratio: 2.3) in the FHS cohort, suggesting that cardiovascular risk assessment in the general population can benefit from rare variant analysis.
Main Text
More than 1,000 sarcomere protein gene variants that are thought to cause dilated cardiomyopathy (MIM 115200) or hypertrophic cardiomyopathy (MIM 192600) have been identified in familial cardiomyopathies.1 Studies of families ascertained on an index case have led to estimates that that disease penetrance of such mutations is greater than 95%.2 Next-generation-targeted resequencing enables assessment of the full allelic spectrum of disease genes in large cohorts. This study assessed the burden of structural cardiovascular disease conferred by rare variants in eight sarcomere protein genes in the general community.
Sarcomere genes ACTC1 (MIM 102540), MYBPC3 (MIM 600958), MYH7 (MIM 160760), MYL2 (MIM 160781), MYL3 (MIM 160790), TNNI3 (MIM 191044), TNNT2 (MIM 191045), and TPM1 (MIM 191010) were sequenced in 1,637 unrelated individuals in the Offspring cohort of the Framingham Heart Study3 and 1,963 unrelated individuals from the Jackson Heart Study cohort4 (Table S1 available online). These studies were performed with protocols approved by FHS, JHS, and institutional ethics committees and with informed consent. FHS is a three-generation prospective, community-based family study to identify the factors that contribute to cardiovascular disease. In 1948, researchers recruited 5,209 men and women between the ages of 30 and 62 who had not yet developed cardiovascular disease from the town of Framingham, Massachusetts. The Offspring cohort consists of 5,124 of the original participants’ adult children and their spouses enrolled in 1971. FHS participants are primarily of European ancestry. JHS is a large, community-based observational study whose participants were recruited from urban and rural areas of the Jackson, Mississippi, metropolitan statistical area (MSA). The final cohort of 5,301 participants includes 6.59% of all African American Jackson MSA residents, aged 35–84.
Targeted genes were selected with a custom designed hybrid capture array5 and then sequenced with an Illumina HiSeq. Sequence reads were first aligned to human genome assembly hg19 with the Burrows-Wheeler Aligner6 and then, in accordance with previously described procedures,7 recalibrated with the Genome Analysis Toolkit (GATK) and used for variant calling by the Unified Genotyper module of the GATK and snpEff. Samples from analysis with below 95% concordance with prior dbGAP SNP array data, with less than 50% of targeted bases covered to 20×, or with a high number of singleton variants were removed. Eighty percent of the target bases were covered with at least 20× depth of coverage (Table S1, available online).
Pathogenicity of variants was evaluated with two metrics: variant classification rules established by a Clinical Laboratory Improvement Amendments (CLIA)-approved laboratory variant calling algorithm (Partner’s Laboratory for Molecular Medicine) and the PolyPhen HCM algorithm,7 each of which classify variants as pathogenic, benign, or having unknown significance.
Statistical analysis was performed with the R software package unless otherwise noted. Significance of differences between mean values and slopes of linear regression were assessed using the t test statistic and only positive results are reported.
Across the two cohorts, 11.2% of individuals had one or more rare (MAF < 1%) nonsynonymous variants (missense, nonsense, indels, and splice variants; Tables S2 and S3). Three individuals in the FHS cohort and 19 in the JHS cohort had multiple rare nonsynonymous sarcomere variants. The frequency of rare nonsynonymous sarcomere variants was proportional to protein length, with the exception of MYBPC3 and MYL3, in which such variants were overrepresented (p < 0.0001). This might be due to underlying differences in genomic architecture. The overall frequency of rare nonsynonymous sarcomere variants was approximately the same in the FHS and JHS cohorts (Figure S1). The prevalence of individuals with rare known or likely pathogenic sarcomere variants based on the Laboratory for Molecular Medicine criteria was 14 of 1,637 sequenced FHS participants and 8 of 1,963 sequenced JHS participants (Table S4). On the basis of these findings, we estimate that 0.85% of the European Americans and 0.4% of African Americans carry a sarcomere variant defined as known or likely to be pathogenic, higher proportions than previously anticipated.8
We considered whether there was unrecognized stratification of subjects with and without rare sarcomere variants by testing for association between rare variant carrier status and measures of ancestry differences. Using common variants that were genotyped in each cohort and deposited into dbGaP, we computed the top ten principal components of genetic variation with the EIGENSTRAT software package for each distribution.9 Logistic regression of carrier status on each of the principal components revealed no evidence of statistical association between ancestry, at least informed by common variants, and the presence of a rare sarcomere gene variant (Figures S2 and S3) as association results of the 20 tests were consistent with the null distribution (one result, p < 0.05; the remainder, p > 0.1).
In addition, because rare variant stratification might not be well captured by methods to detect stratification for common variants, we selected a set of comparably sized genes with no prior evidence for involvement in cardiac morphology as negative controls and repeated our original analysis based on rare variants discovered in those genes using an empirical permutation as follows: p values were computed for 10,000 sets of 8 genes randomly selected from 30 genes included in the targeted sequencing of these cohorts. The 30 control genes used in the permutation analysis (INS, KCNQ5, ANK2, CACNA1C, CAV3, KCNE1, KCNE2, KCNH2, KCNJ2, NOS1AP, AKAP9, ATP1B1, CNOT1, GINS3, LIG3, LITAF, NDRG4, RNF207, SCN4B, ARL4D, ARL6, BBS1, BBS10, BBS12, BBS2, BBS4, BBS5, BBS7, BBS9, and CCDC28B) were selected because each is known or presumed to be pathogenic for either cardiac electrophysiological phenotypes or anthropomorphic disorders. These genes are not known to alter cardiac dimensions. All reported p values were robust to this analysis.
To assess the burden of sarcomere related pathophysiology, we assessed cardiac structure and function by using echocardiography. For our analysis, we examined maximum left ventricular diastolic wall thickness (LVWT), left ventricular diastolic diameter (LVDD), left atrial diameter (LAD), and fractional shortening (FS). FHS individuals had between one and five echocardiograms measured in M-mode over a 25 year period.10 Where multiple echocardiograms are available, the most recent observation was analyzed as cardiac dimensions increase with age. Only a single 2D echocardiography study was available for the JHS cohort, and this echocardiography study was assessed for the same parameters.11 Dimensions were scaled by each individual’s height to increase comparability across individuals as is standard practice in epidemiological echocardiography studies.12,13
The subset of traditional FHS physiologic risk factors14 previously associated with increased cardiac dimensions was considered in these analyses: hyperlipidemia (cholesterol > 240 mg/dl, HDL < 40 mg/dl, triglycerides > 150 mg/dl), hypertension (systolic blood pressure ≥ 140 mmHg or diastolic blood pressure ≥ 90 mmHg), obesity (body mass index > 30), and diabetes (fasting glucose ≥ 126 mg/dl).15–18 Smoking has not been previously associated with increased cardiac dimensions and was not considered. Phenotypes in the cohorts were stratified by genotype and presence of two or more physiologic risk factors (Table 1; Table S5 provides same data without scaling for height). Physiological risk stratification in the FHS cohort was assessed for each individual at the earliest exam between ages 35 and 50. FHS individuals who did not have a physical exam in that period were not considered for the risk-stratified analyses to control for the effects of aging. Physiological risk stratification in the JHS cohort was assessed at the first exam.
Table 1.
Stratified Summary of Mean Scaled Echocardiography Measurements
|
Framingham Heart Study |
Jackson Heart Study |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nb | Age | Female | LVWTc | LVDDc | LADc | Nb | Age | Female | LVWTc | LVDDc | LADc | |
| No risk factorsa | 855 | 58.3 (9.2) | 63% | 0.058 (0.007) | 0.284 (0.025) | 0.230 (0.028) | 972 | 55.5 (12.7) | 58% | 0.055 (0.011) | 0.285 (0.028) | 0.205 (0.027) |
| ≥1 sarcomere variant | 158 | 59.7 (9.4) | 43% | 0.060∗∗ (0.007) | 0.285 (0.025) | 0.236∗ (0.028) | 244 | 57.9 (11.8) | 62% | 0.056∗ (0.010) | 0.287 (0.028) | 0.211∗∗ (0.025) |
| MYBPC3 | 92 | 59.3 | 46% | 0.059 | 0.287 | 0.239∗∗ | 186 | 58.6 | 61% | 0.056∗ | 0.288 | 0.212∗∗∗ |
| MYH7 | 36 | 59.4 | 39% | 0.058 | 0.283 | 0.235 | 42 | 57.2 | 79% | 0.054 | 0.288 | 0.208 |
| MYL3 | 12 | 61.8 | 42% | 0.061 | 0.278 | 0.231 | 8 | 56.3 | 57% | 0.057 | 0.278 | 0.204 |
| MYL2 | 7 | 61.7 | 50% | 0.061 | 0.284 | 0.232 | 4 | 58.7 | 100% | 0.058 | 0.293 | 0.211 |
| TNNT2 | 6 | 57.0 | 50% | 0.064 | 0.279 | 0.237 | 7 | 51.8 | 25% | 0.053 | 0.294 | 0.207 |
| TPM1 | 3 | 65.7 | 0% | 0.058 | 0.284 | 0.206 | 6 | 55.0 | 40% | 0.063 | 0.298 | 0.197 |
| ACTC1 | 2 | 57.0 | 50% | 0.069 | 0.267 | 0.234 | 0 | |||||
| TNNI3 | 2 | 61.5 | 50% | 0.060 | 0.286 | 0.226 | 2 | 0.065 | 0.247 | 0.207 | ||
| LMM (likely) pathogenic | 14 | 63.0 (10.5) | 38% | 0.063∗ (0.007) | 0.286 (0.034) | 0.235 (0.028) | 8 | 54.0 (14.1) | 50% | 0.054 (0.009) | 0.287 (0.024) | 0.209 (0.029) |
| PolyPhen HCM pathogenic | 30 | 61.4 (9.2) | 42% | 0.061 (0.009) | 0.279 (0.029) | 0.226 (0.029) | 26 | 59.0 (13.3) | 59% | 0.054 (0.009) | 0.287 (0.024) | 0.206 (0.025) |
| LMM benign | 35 | 59.2 (9.4) | 32% | 0.060 (0.009) | 0.288 (0.025) | 0.246∗∗∗ (0.027) | 105 | 59.5 (11.9) | 64% | 0.056 (0.009) | 0.291 (0.029) | 0.214∗∗∗ (0.025) |
| PolyPhen HCM benign | 51 | 59.7 (9.4) | 47% | 0.059 (0.006) | 0.286 (0.024) | 0.242∗∗ (0.028) | 85 | 59.3 (11.6) | 66% | 0.057 (0.011) | 0.288 (0.023) | 0.209 (0.023) |
| ≥2 physiologic risk factorsa | 575 | 62.1 (9.1) | 35% | 0.061∗∗∗ (0.008) | 0.290∗∗∗ (0.029) | 0.246∗∗∗ (0.032) | 777 | 58.7 (11.0) | 64% | 0.058∗∗∗ (0.010) | 0.291∗∗∗ (0.028) | 0.213∗∗∗ (0.029) |
| ≥1 sarcomere variant + ≥2 physiologic risk factorsa | 62 | 61.3 (10.2) | 32% | 0.061∗∗∗ (0.007) | 0.290∗ (0.022) | 0.245∗∗∗ (0.027) | 97 | 59.5 (11.1) | 64% | 0.059∗∗∗ (0.010) | 0.288 (0.027) | 0.215∗∗∗ (0.023) |
Significance is denoted as follows: ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. The following abbreviations are used: LVDD, left ventricular diastolic diameter; and LAD, left atrial diameter.
Physiologic risk factors include hyperlipidemia, hypertension, obesity, and diabetes. Standard deviations for this group are listed parenthetically.
N is number of individuals.
Measurements (in mm) scaled by individuals’ heights (in cm) of left ventricular wall thickness (LVWT).
Significant increases in LVWT and LAD were observed among individuals carrying one or more rare sarcomere variant (Table 1). LVWT was increased by 5% and 2% for the FHS and JHS cohorts, respectively (p < 0.01, p < 0.05, Table 1) in individuals with one or more rare nonsynonymous sarcomere variants compared to individuals with no variants or physiologic risk factors. Individuals with sarcomere variants of known or likely pathogenicity had an 8.6% increased wall thickness in the FHS population (p < 0.05) but not in the JHS population. This observation might be accounted for by the fact that, historically, far more resources have been dedicated to discovery of pathogenic cardiomyopathy variants in populations of European ancestry than populations of African ancestry, underlying differences in genomic architecture19 or the availability of more extensive longitudinal data in FHS compared to JHS. Individuals with either physiologic or a combination of physiologic and genetic risk factors similarly had an increase in left ventricular dimensions in both populations.
LAD was increased by 3% in FHS and JHS participants with rare sarcomere variants compared to participants without sarcomere variants or physiologic risk factors (Table 1; p < 0.05, FHS; p < 0.01 JHS). Left atrial volume is often increased in hypertrophic cardiomyopathy because of ventricular diastolic dysfunction that is caused by sarcomere protein mutations. Increased left atrial volume is associated with an increase in adverse cardiovascular events in patients with hypertrophic cardiomyopathy or other cardiovascular conditions.20,21 Increased LAD was significantly increased in cohort participants with MYBPC3 variants and with variants classified as benign, the two largest subgroups of variants (Table 1). Whether these data imply a subtle impact of sarcomere variants on diastolic function, misclassification of some variants as benign, or an imprecise assessment of atrial size is unclear.
Out of 22 individuals with variants previously identified in the literature as likely to be pathogenic, only four had clinical criteria for HCM: two had LVWT >12 mm and two had EKG criteria for LVH22. This might reflect a lower sensitivity of population screening echocardiograms or a lower disease penetrance in the general population compared to previously studied HCM families.
We sought to further evaluate the potential pathogenicity of rare nonsynonymous sarcomere variants by comparing the two tails of the phenotype distributions with burden testing. Continuous phenotypes were transformed to dichotomous variables by comparing the top and bottom quartiles of the distribution. Because the variants were too rare to be evaluated individually, we pooled variants by gene and computed the following statistical tests with the Plink/Seq software package: burden testing of cases compared to controls, C-alpha, count of case-unique rare alleles, variable threshold test, frequency-weighted test, and sum of single-site statistics. Although some of these tests are designed to be sensitive to unusual distributions of rare variants,23,24 none produced a statistically significant result (Figure S4). This is probably due to the small number of individuals with pathogenic variants in the tails of the distribution.
We additionally considered the potential pathogenicity of nine common protein-altering variants with MAF between 1% and 10% in either of the two populations and examined their frequency and associated phenotypes individually in FHS and JHS (Table S6). Analysis was done by individual variant, rather than with a pooled approach. No common variants were significantly associated with measures of cardiac morphology when considering the top and bottom quartiles as a dichotomous variable. While considering left ventricular parameters as a continuous variable, one variant, rs3729823 (MYH7 c.4472C>G [p.Ser1491Cys]; RefSeq: NM_000257.2), was associated with increased fractional shortening in JHS (FS: 0.44, p = 0.001, n = 12) but not in FHS.
We considered the combination of 11 low frequency (0.5%–1%) or common variants (1%–10%) in addition to a rare variant (Table S6). Thirty JHS individuals and two FHS individuals shared SNP rs3729799 (MYBPC3 c.3004C>T [p.Arg1002Trp]; RefSeq: NM_000256.3). Four of these JHS participants carried a second rare sarcomere variant, all of which occurred in MYBPC3 (c.2939G>A [p.Arg980His];c.2914C>T [p.Arg972Trp]; c.646G>A [p.Ala216Thr]; and c.3682C>T [p.Arg1228Cys]; RefSeq: NM_000256.3). This combination was associated with a 14% decreased LVWT (8.1 mm, p < 10−6) compared to individuals without genetic or physiologic risk factors. These four individuals also had a 14% decreased LVWT when compared to the 26 JHS individuals who shared one of the four rare sarcomere variants but lacked rs3729799 (p < 10−4) and a 15% decreased LVWT in comparison to the 26 JHS individuals with rs3729799 but no other rare sarcomere variants (p < 10−5). This observation might imply that multiple variants can have more effect on cardiac morphology than each alone.
Given that the FHS cohort had multiple echocardiographic evaluations over time, we had the opportunity to evaluate the burden of sarcomere variants longitudinally. Rare variants were associated with increased risk for greater left ventricular dimensions with advancing age (Figures 1A and 1B). A linear one way within groups ANOVA model was constructed that predicted LVWT scaled by an individual’s height as a function of age and covariates genetic risk (≥1 sarcomere variant) and physiologic risk (≥2 physiologic risk factors).
Figure 1.
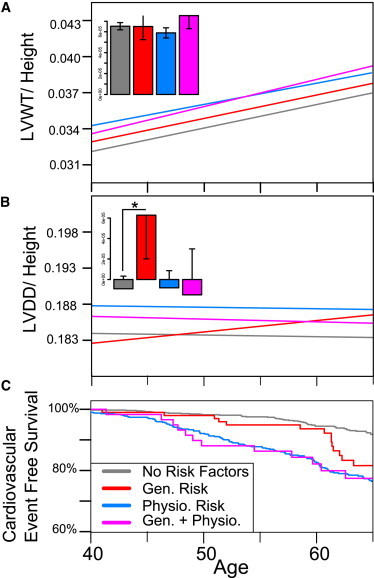
Stratified Framingham Cardiovascular Phenotypes
(A) Longitudinal regression of LVWT scaled to height over time stratified by genetic risk only (defined by one or more sarcomere variants in the absence of physiological risk factors), presence of two or more physiological risk factors (hyperlipidemia, hypertension, obesity, or diabetes), or the combination of genetic and physiologic risk factors. The inset shows a bar graph of regression line slopes, with standard deviations.
(B) Longitudinal regression of left ventricular diastolic diameter (LVDD) scaled to height, stratified as above. A statistically significant decline (p < 0.05) is observed between the slope of individuals with and without genetic risk factors in the absence of physiologic risk factors.
(C) Kaplan-Meier curves showing age of first adverse cardiovascular event, stratified as described above. Increased risk for first adverse cardiovascular event conferred by genetic risk factor is significant (p < .01).
In this model, the sarcomere genetic risk covariate was statistically significant (p < 0.05) and age and physiologic risk were statistically significant (p < 0.001). The result remained significant when evaluated by empirical permutation as described above.
A significant increase in rate of LV diastolic diameter change over time was observed in individuals with genetic risk in the absence of physiologic risk (p < 0.05) but not in the other groups analyzed (Figure 1B). This might occur because a subset of the genetic variants predisposes an individual to a dilated cardiomyopathy phenotype and/or reflect hemodynamic responses in individuals with only genetic risk factors. Alternatively, treatments might have diminished the LV diastolic diameter in individuals with physiologic risks.
We next sought to evaluate the association between rare sarcomere variants and adverse cardiovascular-event free survival. To perform this analysis, we stratified individuals by physiologic and genetic risk between ages 35 and 50, excluding individuals who did not have a physical exam in that time period (n = 168) and those individuals who had a prior cardiovascular event (n = 32). The distribution of cardiac dimensions in individuals excluded from the analysis was no different than the overall population. Significance was determined with a multivariable Cox-proportional hazards model with physiologic risk (≥2 physiologic risk factors) and genetic risk (≥1 sarcomere variant) modeled as dichotomous covariate terms as well as a term for the interaction of the two variables. All three terms were statistically significant (genetic risk, p < 0.01; physiologic risk, p < 0.001; genetic/physiologic risk interaction term p < 0.05). We evaluated whether the fitted Cox-proportional hazards regression model adequately describes the data by testing proportionality of hazards, influential observations, and nonlinearity assumptions. Analysis could not be extended beyond the age of 65 because the assumption of proportionality of hazards for our physiologic risk covariate did not hold beyond 30 years, which is comparable to the longest duration Framingham Risk Score.25 Significance of the Cox- proportional hazards model result was confirmed through empirical permutation as described above.
Rare sarcomere variants were found to increase risk for adverse cardiovascular events in the FHS cohort (Figure 1C). Comparison of Kaplan-Meier survival curves and a multivariable Cox proportional hazard model accounting for presence of genetic and physiologic risk factors with mean follow up of 25 years indicated that individuals with one or more rare nonsynonymous variants either with or without physiologic risk had significantly earlier onset of first cardiovascular events (hazard ratio: 2.3; 95% confidence interval: [1.2, 4.1]; p < .01; Table S7). Remarkably, the significance of this association is even stronger (hazard ratio: 2.8; 95% confidence interval: [1.5, 5.1]; p < 0.001) when adverse cardiovascular events are restricted to those affecting the myocardium (e.g., angina pectoris, myocardial infarction, coronary insufficiency, and congestive heart failure). Although 12 out of the 14 first observed cardiovascular disease events in individuals with genetic risk and no physiologic risk factors were in diseases of the myocardium as compared to 32 out of 48 disease events in individuals with no genetic or physiologic risk factors, a chi-square test failed to identify significant differences in event distribution between groups (Table S8). Previous work has linked cardiac hypertrophy to earlier adverse cardiovascular event onset of all manifestations of cardiovascular disease,26,27 so it is possible that the increased cardiovascular dimensions in individuals with genetic risk explains this survival curve. Testing this hypothesis proved inconclusive because of the small number of individuals in our study with both genetic risk and observed cardiovascular events.
Our analysis of rare variants in eight sarcomere genes suggests that rare genetic variants might contribute to cardiovascular risk assessment in the community. We were surprised to see that sarcomere protein gene variants previously described as pathogenic were present in 0.6% of the general population, twice the estimated prevalence of HCM.12 Moreover, this fraction of pathogenic variants is likely an underestimate given our findings that >10% of the population carries a rare nonsynonymous sarcomere variant and the possibility that some of these variants are also pathogenic. On the other hand, we find that the estimated effect sizes of these variants are smaller when ascertained in the general population than was previously predicted from clinically identified families. Going forward, functional analyses in combination with computational and additional clinical data (e.g., assessment of diastolic function) might better discern pathogenic from benign rare variants found in the general population. Such information will be essential for accurate interpretation of personal genomes. Nevertheless, the association of sarcomere genetic risk with differences in left ventricular dimensions and with earlier onset of adverse cardiovascular events (hazard ratio: 2.3) suggests that analysis of rare variants might potentially be an informative tool in assessing cardiovascular risk in the general population in the future.
Acknowledgments
We gratefully acknowledge the contribution of Framingham and Jackson Heart Study cohort participants. This work was supported by grants from the National Human Genome Research Institute (Medical Sequencing Program grant U54 HG003067, to the Broad Institute PI, Lander), the National Heart, Lung and Blood Institute (HL080494-05 to C.E.S. and J.G.S.), and the Howard Hughes Medical Institute (to C.E.S). A.G.B. and D.S.H. are supported by NIH Medical Scientist Training Program fellowship 5T32GM007753-33. S.C. is supported in part by grant K99HL107642 and the Ellison Foundation. The Jackson Heart Study is supported by contracts N01-HC-95170, N01-HC-95171, and N01-HC-95172 from the National Heart, Lung, and Blood Institute, the National Institute for Minority Health and Health Disparities, and additional support from the National Institute of Biomedical Imaging and Bioengineering. The Framingham Heart Study was supported by contracts N01-HC-25195 and 6R01-NS 17950 from the National Heart, Lung and Blood Institute and genotyping services from Affymetrix, Inc. (contract No. N02-HL-6-4278 for the SNP Health Association Resource [SHARe] project).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Genome Analysis Toolkit (GATK), http://gatk.sourceforge.net/
Online Mendelian Inheritance in Man, http://www.omim.org
Partners Healthcare Laboratory for Molecular Medicine Variant Classification Rules, http://pcpgm.partners.org/LMM
PolyPhen HCM, http://genetics.bwh.harvard.edu/hcm/
Plink/Seq, http://atgu.mgh.harvard.edu/plinkseq
snpEff, http://snpeff.sourceforge.net/
Accession Numbers
The dbGaP accession numbers for the 3,600 sequences and cardiovascular phenotype data reported in this paper are NHLBI Framingham Cohort, phs000007.v16.p6 and NHLBI Jackson Heart Study Candidate Gene Association Resource, phs000286.v2.p1.
References
- 1.Konno T., Chang S., Seidman J.G., Seidman C.E. Genetics of hypertrophic cardiomyopathy. Curr. Opin. Cardiol. 2010;25:205–209. doi: 10.1097/HCO.0b013e3283375698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Georgakopoulos D., Christe M.E., Giewat M., Seidman C.M., Seidman J.G., Kass D.A. The pathogenesis of familial hypertrophic cardiomyopathy: early and evolving effects from an alpha-cardiac myosin heavy chain missense mutation. Nat. Med. 1999;5:327–330. doi: 10.1038/6549. [DOI] [PubMed] [Google Scholar]
- 3.Govindaraju D.R., Cupples L.A., Kannel W.B., O’Donnell C.J., Atwood L.D., D’Agostino R.B., Sr., Fox C.S., Larson M., Levy D., Murabito J. Genetics of the Framingham Heart Study population. Adv. Genet. 2008;62:33–65. doi: 10.1016/S0065-2660(08)00602-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson J.G., Rotimi C.N., Ekunwe L., Royal C.D., Crump M.E., Wyatt S.B., Steffes M.W., Adeyemo A., Zhou J., Taylor H.A., Jr., Jaquish C. Study design for genetic analysis in the Jackson Heart Study. Ethn. Dis. 2005;15(4, Suppl 6):S6–S30. 37. [PubMed] [Google Scholar]
- 5.Gnirke A., Melnikov A., Maguire J., Rogov P., LeProust E.M., Brockman W., Fennell T., Giannoukos G., Fisher S., Russ C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat. Biotechnol. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morita H., Larson M.G., Barr S.C., Vasan R.S., O’Donnell C.J., Hirschhorn J.N., Levy D., Corey D., Seidman C.E., Seidman J.G., Benjamin E.J. Single-gene mutations and increased left ventricular wall thickness in the community: the Framingham Heart Study. Circulation. 2006;113:2697–2705. doi: 10.1161/CIRCULATIONAHA.105.593558. [DOI] [PubMed] [Google Scholar]
- 9.Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 10.Lieb W., Xanthakis V., Sullivan L.M., Aragam J., Pencina M.J., Larson M.G., Benjamin E.J., Vasan R.S. Longitudinal tracking of left ventricular mass over the adult life course: clinical correlates of short- and long-term change in the framingham offspring study. Circulation. 2009;119:3085–3092. doi: 10.1161/CIRCULATIONAHA.108.824243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Samdarshi T.E., Taylor H.A., Edwards D.Q., Liebson P.R., Sarpong D.F., Shreenivas S.S., Howard G., Garrison R.J., Fox E.R. Distribution and determinants of Doppler-derived diastolic flow indices in African Americans: the Jackson Heart Study (JHS) Am. Heart J. 2009;158:209–216. doi: 10.1016/j.ahj.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maron B.J., Mathenge R., Casey S.A., Poliac L.C., Longe T.F. Clinical profile of hypertrophic cardiomyopathy identified de novo in rural communities. J. Am. Coll. Cardiol. 1999;33:1590–1595. doi: 10.1016/s0735-1097(99)00039-x. [DOI] [PubMed] [Google Scholar]
- 13.Vasan R.S., Larson M.G., Levy D., Evans J.C., Benjamin E.J. Distribution and categorization of echocardiographic measurements in relation to reference limits: the Framingham Heart Study: formulation of a height- and sex-specific classification and its prospective validation. Circulation. 1997;96:1863–1873. doi: 10.1161/01.cir.96.6.1863. [DOI] [PubMed] [Google Scholar]
- 14.D’Agostino R.B., Sr., Vasan R.S., Pencina M.J., Wolf P.A., Cobain M., Massaro J.M., Kannel W.B. General cardiovascular risk profile for use in primary care: the Framingham Heart Study. Circulation. 2008;117:743–753. doi: 10.1161/CIRCULATIONAHA.107.699579. [DOI] [PubMed] [Google Scholar]
- 15.de Simone G., Palmieri V., Bella J.N., Celentano A., Hong Y., Oberman A., Kitzman D.W., Hopkins P.N., Arnett D.K., Devereux R.B. Association of left ventricular hypertrophy with metabolic risk factors: the HyperGEN study. J. Hypertens. 2002;20:323–331. doi: 10.1097/00004872-200202000-00024. [DOI] [PubMed] [Google Scholar]
- 16.Lin T.H., Chiu H.C., Su H.M., Voon W.C., Liu H.W., Lai W.T., Sheu S.H. Association between fasting plasma glucose and left ventricular mass and left ventricular hypertrophy over 4 years in a healthy population aged 60 and older. J. Am. Geriatr. Soc. 2007;55:717–724. doi: 10.1111/j.1532-5415.2007.01134.x. [DOI] [PubMed] [Google Scholar]
- 17.Devereux R.B., Pickering T.G., Alderman M.H., Chien S., Borer J.S., Laragh J.H. Left ventricular hypertrophy in hypertension. Prevalence and relationship to pathophysiologic variables. Hypertension. 1987;9:II53–II60. doi: 10.1161/01.hyp.9.2_pt_2.ii53. [DOI] [PubMed] [Google Scholar]
- 18.Lauer M.S., Anderson K.M., Kannel W.B., Levy D. The impact of obesity on left ventricular mass and geometry. The Framingham Heart Study. JAMA. 1991;266:231–236. [PubMed] [Google Scholar]
- 19.Lohmueller K.E., Indap A.R., Schmidt S., Boyko A.R., Hernandez R.D., Hubisz M.J., Sninsky J.J., White T.J., Sunyaev S.R., Nielsen R. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsang T.S., Abhayaratna W.P., Barnes M.E., Miyasaka Y., Gersh B.J., Bailey K.R., Cha S.S., Seward J.B. Prediction of cardiovascular outcomes with left atrial size: is volume superior to area or diameter? J. Am. Coll. Cardiol. 2006;47:1018–1023. doi: 10.1016/j.jacc.2005.08.077. [DOI] [PubMed] [Google Scholar]
- 21.Tani T., Yagi T., Kitai T., Kim K., Nakamura H., Konda T., Fujii Y., Kawai J., Kobori A., Ehara N. Left atrial volume predicts adverse cardiac and cerebrovascular events in patients with hypertrophic cardiomyopathy. Cardiovasc. Ultrasound. 2011;9:34. doi: 10.1186/1476-7120-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Casale P.N., Devereux R.B., Alonso D.R., Campo E., Kligfield P. Improved sex-specific criteria of left ventricular hypertrophy for clinical and computer interpretation of electrocardiograms: validation with autopsy findings. Circulation. 1987;75:565–572. doi: 10.1161/01.cir.75.3.565. [DOI] [PubMed] [Google Scholar]
- 23.Price A.L., Kryukov G.V., de Bakker P.I., Purcell S.M., Staples J., Wei L.J., Sunyaev S.R. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 2010;86:832–838. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neale B.M., Rivas M.A., Voight B.F., Altshuler D., Devlin B., Orho-Melander M., Kathiresan S., Purcell S.M., Roeder K., Daly M.J. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7:e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pencina M.J., D’Agostino R.B., Sr., Larson M.G., Massaro J.M., Vasan R.S. Predicting the 30-year risk of cardiovascular disease: the framingham heart study. Circulation. 2009;119:3078–3084. doi: 10.1161/CIRCULATIONAHA.108.816694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kannel W.B., Abbott R.D. A prognostic comparison of asymptomatic left ventricular hypertrophy and unrecognized myocardial infarction: the Framingham Study. Am. Heart J. 1986;111:391–397. doi: 10.1016/0002-8703(86)90156-0. [DOI] [PubMed] [Google Scholar]
- 27.Brown D.W., Giles W.H., Croft J.B. Left ventricular hypertrophy as a predictor of coronary heart disease mortality and the effect of hypertension. Am. Heart J. 2000;140:848–856. doi: 10.1067/mhj.2000.111112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.