

Abstract
Previous studies have shown that copy-number variants (CNVs) contribute to the risk of complex developmental phenotypes. However, the contribution of global CNV burden to the risk of sporadic congenital heart disease (CHD) remains incompletely defined. We generated genome-wide CNV data by using Illumina 660W-Quad SNP arrays in 2,256 individuals with CHD, 283 trio CHD-affected families, and 1,538 controls. We found association of rare genic deletions with CHD risk (odds ratio [OR] = 1.8, p = 0.0008). Rare deletions in study participants with CHD had higher gene content (p = 0.001) with higher haploinsufficiency scores (p = 0.03) than they did in controls, and they were enriched with Wnt-signaling genes (p = 1 × 10−5). Recurrent 15q11.2 deletions were associated with CHD risk (OR = 8.2, p = 0.02). Rare de novo CNVs were observed in ∼5% of CHD trios; 10 out of 11 occurred on the paternally transmitted chromosome (p = 0.01). Some of the rare de novo CNVs spanned genes known to be involved in heart development (e.g., HAND2 and GJA5). Rare genic deletions contribute ∼4% of the population-attributable risk of sporadic CHD. Second to previously described CNVs at 1q21.1, deletions at 15q11.2 and those implicating Wnt signaling are the most significant contributors to the risk of sporadic CHD. Rare de novo CNVs identified in CHD trios exhibit paternal origin bias.
Introduction
In the human genome, rare copy-number variants (CNVs), generally considered to be those with <1% population frequency, have recently been the focus of increasing attention as potential causative factors of complex diseases. Considered together, such individually rare CNVs are sufficiently common that case-control comparisons of their collective frequency can be conducted in large sample sets. Such comparisons have revealed association between rare CNV burden, in particular rare CNVs that overlap genes (or rare genic CNVs), and disease risk in a variety of neuropsychiatric and developmental conditions.1–4
Congenital heart disease (CHD) is the most common congenital abnormality: it has an incidence of approximately 7 in 1,000 live births.5 CHD can occur as a component of a large number of chromosomal and Mendelian malformation syndromes, but in 80% of cases, it occurs as a sporadic condition that exhibits high heritability.6 Tetralogy of Fallot (TOF [MIM 187500]) is the most common cyanotic CHD phenotype and occurs in 1 in 2,500 live births.5 TOF is considered an abnormality of the cardiac outflow tract and is characterized by anterocephalad deviation of the outlet septum; this causes overriding of the aorta, ventricular septal defects, right ventricular outflow tract obstruction, and right ventricular hypertrophy. Prior to the modern cardiac surgical era, severe CHD carried a high mortality (for example, 80% of children born with TOF died before their tenth birthday), and therefore, genetic investigation of sporadic, nonsyndromic CHD has generally focused on rare and de novo variants. Greenway et al. reported a genome-wide rare de novo CNV burden of ∼10%—it involved 10 different loci in 114 nonsyndromic TOF trios (TOF probands and their respective unaffected parents).7 However, only one locus (1q21.1) identified in that study has been replicated in an independent cohort thus far.8 Recently, Cooper et al. analyzed rare CNV burden in a large number of children, including 575 cases with CHD as a component of their phenotype, referred for genetic evaluation of intellectual disability.4 Children with CHD were shown to have a significantly increased burden of large CNVs (>400 kb) (p = 6.45 × 10−5) than were children with autism spectrum disorder. However, the population studied by these investigators included many cases with recognized deletion syndromes that typically include CHD (for example, Williams [MIM 194050] and DiGeorge [MIM 188400] syndromes); mainly large deletions were studied, and the population was not primarily ascertained for CHD.
Here, we address the disease risk associated with the global burden of CNVs > 100 kb in a case population that is nonsyndromic, non-Mendelian (i.e., sporadic), and ascertained on the basis of CHD. Our prior hypothesis was that rare genic CNVs would show association with CHD risk. We present locus-specific and functional annotation enrichments associated with CHD risk, and we propose dosage-sensitive genes for CHD. In addition, we examine the genome-wide burden of rare de novo CNVs > 30 kb in a cohort of CHD trios.
Subjects and Methods
Study Subjects and Sample Collections
CHD-affected participants (51% male and 49% female; median age = 10 years; interquartile range = 1–25 years) of European ancestry, as well as their parents and siblings (when available), were recruited from multiple centers in the UK (Newcastle, Bristol, Leeds, Liverpool, Nottingham, Leicester, and Oxford), Germany (Erlangen), Belgium (Leuven), and Australia (Sydney). Ethical approval was granted from the local institutional review boards, and informed consent was obtained from all participants (or from a parent or guardian in cases where the subjects were too young to consent themselves). All participants with CHD were screened for DiGeorge syndrome, Williams-Beuren syndrome, and other major chromosomal aberrations (e.g., trisomy 21 [MIM 190685] and trisomy 18) known to cause CHD; those found with such anomalies were excluded from further study. Case ascertainment in Bristol, Leeds, and Liverpool was principally focused on TOF, whereas case ascertainment in other centers included all CHD phenotypes. Thus, TOF was relatively overrepresented in our cohort. Case ascertainment was not focused on multiplex families; fewer than 1% of probands had an affected first-degree relative with CHD. Control subjects consisted of unrelated healthy European-ancestry individuals from a French population cohort. DNA samples from cases were extracted from blood (85%) and saliva (15%), and all DNA samples from controls were extracted from blood; quality-control (QC) assessment of CNV calls indicated no significant systematic difference between DNA derived from blood or from saliva.
Genotyping, QC Criteria, and CNV Detection on Illumina 660W SNP Arrays
A total of 2,896 CHD cases, 747 unaffected family members, and 856 unrelated controls were typed on the Illumina 660W-Quad SNP platform at the Centre National de Génotypage (Evry Cedex, France), and normalized total intensity and genotype data were obtained. For each sample, SNP QC analyses were carried out in PLINK.9 Samples with genotyping call rates < 98.5%, average heterozygosity outside the range of [0.31, 0.33], and gender mismatches and those that failed to cluster with the Phase II HapMap CEU (Utah residents with ancestry from northern and western Europe from the CEPH collection) individuals were excluded. Genome-wide identity-by-descent (IBD) sharing was calculated on all probands, and only one individual from each pair of related probands (mean proportion of alleles shared identically by descent > 0.1; n = 18) was included in the analyses. Additionally, intensity QC parameters were applied, and samples were excluded when they failed one of the following criteria: a standard deviation of autosomal log R ratio (LRR) > 3.0, a GC-wave factor of the LRR outside the range of [−0.1, 0.1],10 or a standard deviation of B-allele frequency (BAF) > 0.15 after GC correction.11 A total of 2,256 CHD cases, 697 unaffected family members, and 841 unrelated controls were included in the final analyses. The phenotype distribution of the CHD cases can be found in Table S1, available online. For case-control CNV-burden comparison, the QuantiSNP11 algorithm was used as the primary CNV detection method and PennCNV10 was used as the confirmatory method. Rare de novo CNV detection in probands and their respective unaffected parents was performed with PennCNV joint calling,10 and QuantiSNP was used for confirmation. For probands and their respective parents, both PennCNV and QuantiSNP raw data sets were further inspected manually within all the putative de novo CNV spans, as well as in the flanking regions, so that the possibility of false negatives in the parental samples could be ruled out. All CNV coordinates were mapped to NCBI build 36.1 (hg18). The coordinates for RefSeq genes and segmental duplications12 were downloaded from the UCSC Genome Browser.13 CNVs were further analyzed with custom R scripts and the “join genomic interval” tool on Galaxy14 and were visualized in the UCSC Genome Browser.
CNV Validation
Affymetrix 6.0 SNP arrays, comparative genomic hybridization (CGH) arrays, and multiplex ligation-dependent amplification (MLPA) were used for confirming CNV calls that were made on the discovery platform (Illumina 660W-Quad). A random subset of CHD cases (n = 198) that had been analyzed on the discovery platform was also typed on the Affymetrix 6.0 platform and analyzed with the Birdseye algorithm from the BirdSuite package.15 CGH was performed for all rare de novo CNVs > 30kb, CNVs in candidate loci, and recurrent CNVs that were suspected to be artifacts (because of certain properties of the genomic regions on the discovery platform) when DNA was available and when the region was adequately covered on the CGH platform. All remaining CNVs were validated with MLPA.
For the CGH experiments, 4x44K (ISCA v.2) and 2x105K Agilent (Santa Clara, CA, USA) arrays were purchased from BlueGnome (Cambridge, UK). CHD case and control DNA samples (1 μg each) were labeled, purified, hybridized, and washed with reagents according to BlueGnome protocol (Cambridge, UK; see Web Resources). Control DNA samples (catalog numbers G1471 and G1521) were purchased from Promega (Madison, WI, USA). A GenePix 4000B laser scanner (Axon Instruments, CA, USA) was used for exciting the hybridized fluorophores and for scanning the images, which were then quantified and normalized with the default settings and analyzed on hg18 (NCBI build 36.1) with BlueFuse Multi software (BlueGnome, Cambridge, UK) and visualized in the UCSC Genome Browser.
MLPA assays were performed with custom-designed synthetic probes ordered from Integrated DNA Technology (IA, USA) and with the P200 MLPA kit from MRC-Holland (Amsterdam, The Netherlands). A minimum of two probes per CNV locus were designed with the MAPD software16 in conjunction with the UCSC Extended DNA utility.13 Each MLPA assay contained a total of 11 synthetic probes with sizes ranging from 100–140 nt. MLPA reactions were carried out with the MRC-Holland protocol (see Web Resources). MLPA products were resolved on an ABI 3730xl (Applied Biosystems, CA, USA) and analyzed with GeneMarker v.1.85 software (SoftGenetics, PA, USA).
Statistical Analysis
We compared the frequency of CNVs in case and control groups by using a two-sided Fisher’s test. To make some allowance for multiple testing, we also calculated empirical p values from 1,000 random permutations of disease status by taking the minimum p value obtained over 36 tests to account for the testing of six CNV categories (see Table 1) times three CNV sizes (>100 kb, >500 kb, and >1 Mb) times two CNV types (duplications and deletions). CNV length and the number of genes spanning each CNV in cases versus controls were assessed with two-sided permutation tests, which compare the observed t statistic (normalized difference between means) with the t statistics from 10,000 random replicates of relabeling of cases and controls. Haploinsufficiency scores of the genes spanned by CNVs in cases and controls were obtained from a published source17 and compared with the use of a two-tailed Mann Whitney U test. Population attributable risk (PAR) was calculated with the following formula: 100(P(OR-1))/(1 + (P(OR−1))), in which P is the proportion of control population with the CNVs and OR is the odds ratio. The frequency of rare de novo CNVs was ascertained in 283 TOF trios. We determined the parental origin of the de novo CNV by examining the mismatches between the BAF of each SNP in the proband and both parents within each CNV region. We compared the frequency of each parental origin by using a binomial probability distribution to obtain a two-tailed p value. All statistical tests were performed with the R statistical package. Because our study included substantial numbers of CHD cases with a relatively homogeneous phenotype (TOF), we decided a priori to explore heterogeneity between the group with TOF and the group with other types of CHD. We considered that there were insufficient numbers of CHD cases with any other homogeneous phenotype to permit additional valid subgroup analyses.
Table 1.
Frequency of Deletions in Cases and Controls
| CNV Category | CHD Cases | Controls | Fold Change of CHD Cases vs. Controls | p Value |
|---|---|---|---|---|
| Rare genic | 7.8% | 4.4% | 1.8 | 0.0008 |
| Rare | 10.5% | 8.3% | 1.3 | 0.07 |
| Common genic | 6.3% | 6.5% | 1.0 | 0.80 |
| Common | 21.5% | 21.8% | 1.0 | 0.88 |
| All genic | 13.7% | 10.8% | 1.3 | 0.04 |
| All | 29.3% | 28.9% | 1.0 | 0.82 |
Statistically significant findings are shown in bold. The following abbreviations are used: CNV, copy-number variant; and CHD, congenital heart disease.
Results
CNV Validation and Inclusion Criteria
We used stringent filtering measures for case-control genome-wide CNV-burden analyses (size > 100 kb with Bayes factor11 > 100) in order to ensure comparability of detection between individuals ascertained from multiple centers. Initially, 4,551 autosomal CNV calls (1,217 deletions and 3,334 duplications) met these inclusion criteria on the Illumina 660W-Quad chip. We subjected 87 deletion calls and 216 duplication calls to validation (87% were randomly selected, and the remainder were targeted to CNVs in candidate loci and recurrent calls that were suspected to be artifacts) with one or more independent experiments (Affymetrix 6.0, array CGH, or MLPA). The resulting positive validation rates were 85% and 34% for deletions and duplications, respectively. On the basis of this validation data, we identified a number of regions that could not be genotyped reliably (see Table S2), and after these regions were excluded, 74 out of 74 (100%) deletion calls and 62 out of 62 (100%) duplication calls were successfully validated by Affymetrix 6.0, array CGH, or MLPA. In total, 1,077 out of 1,217 (88%) deletion calls and 778 out of 3,334 (23%) duplication calls that met the initial filtering criteria remained (after the unreliable regions were excluded), and they were incorporated into the final analyses.
CNV Burden in CHD Cases and Controls
The frequency of CNVs was compared between 2,256 CHD cases (808 TOF and 1,448 other CHDs) and 841 ethnically matched unrelated controls. Although our principal prior hypothesis was that rare genic CNVs would show association with CHD risk, we also examined the following CNV sets: all CNVs, genic CNVs, rare CNVs, common CNVs, and common genic CNVs. Genic CNVs were defined as those that overlap with RefSeq transcription boundaries. Rare CNVs were defined as those that occur with <1% frequency and have minimum (<20%) overlap with CNVs in the compared group—in effect, CNVs that are unique to the case or control group. We also confirmed that all CNVs deemed rare in our study had not been previously reported at >1% frequency in control populations described in the Database of Genomic Variants (DGV). Common CNVs were defined as those shared (>20% overlap) between case and control groups.
There was a highly significant (1.8-fold) difference in rare genic-deletion burden between CHD cases and controls (p = 0.0008; Table 1) and no apparent heterogeneity in risk between TOF and other CHDs. This association remained significant after permutation-test-based correction for 36 comparisons (empirical corrected p = 0.013). The strong association with rare-genic-deletion burden was responsible for the fact that borderline significant association was observed in the subgroups overlapping rare genic deletions (all rare deletions [p = 0.07]; and all genic deletions [p = 0.04]), but these became nonsignificant after correction for multiple testing. There was no difference in the frequency of common deletions, or of overall deletion burden, between case and control groups (Table 1). The excess burden of rare genic deletions corresponds to a population-attributable risk (PAR) of ∼3.5% for CHD. Given the difference in frequency of rare genic deletions > 100 kb in cases and controls, we explored association between larger rare genic deletions and CHD. We observed an apparent trend toward a greater (2.5-fold) difference in the frequency of large (>500 kb) rare deletions between cases and controls (p = 0.024); this difference was yet more marked (3.9-fold difference) when only >1 Mb deletions were considered (p = 0.017), and there was no heterogeneity between TOF and other CHDs. However, the small number of larger deletions precluded formal tests for heterogeneity between these risks. No difference was found between cases and controls in the frequency of large common deletions.
We did not detect any difference in the frequency of either rare or common duplications (see Table 2). We detected an excess of large (>500 kb) genic duplications in TOF cases compared to controls (a 1.9-fold difference; p = 0.01); this effect was solely due to a single locus (1q21.1) whose effect on TOF risk has been previously documented.8
Table 2.
Frequency of Duplications in Cases and Controls
| CNV Category | CHD Cases | Controls | Fold Change of CHD Cases vs. Controls | p Value |
|---|---|---|---|---|
| Rare genic | 8.7% | 8.1% | 1.1 | 0.61 |
| Rare | 10.5% | 10.2% | 1.0 | 0.89 |
| Common genic | 10.2% | 9.5% | 1.0 | 0.59 |
| Common | 12.1% | 12.1% | 1.0 | 1.0 |
| All genic | 18.0% | 16.3% | 1.1 | 0.29 |
| All | 21.0% | 20.7% | 1.0 | 0.88 |
The following abbreviations are used: CNV, copy-number variant; and CHD, congenital heart disease.
Properties and Functional Impact of CNVs
We compared the size of deletions and duplications in cases and controls (see Table 3) and observed larger deletions in cases than in controls (a 1.3-fold difference and p = 0.024 for TOF; a 1.6-fold difference and p = 0.022 for other CHDs) but no difference in the length of duplications. Comparing both TOF and other CHG cases to controls, we found significant differences in the numbers of genes that were spanned by both deletions and duplications (Table 4). In both case groups, these effects were driven by rare CNVs. For rare deletions, there was a 2.6-fold higher number of genes (p = 0.006) for TOF and a 3.7-fold higher number of genes (p = 0.001) for other CHDs. For rare duplications, there was a 2.8-fold higher number of genes (p = 1.0 × 10−4) for TOF and a 1.9-fold higher number of genes (p = 0.006) for other CHDs. The number of genes spanned by common CNVs did not differ between cases and controls. Furthermore, genes encompassed by deletions in CHD cases were associated with higher haploinsufficiency scores17 (p = 0.02) (see Figure S1). This effect was also due to the genes encompassed by rare deletions (p = 0.03) and not by common deletions (p = 0.40). No difference was observed in the haploinsufficiency scores of the genes encompassed by duplications in cases compared to controls (p = 0.44). The list of genes spanned by rare deletions that were associated with high haploinsufficiency scores, as well as recurrent genes overlapped by both rare deletions and rare duplications in CHD cases, can be found in Tables S3–S5.
Table 3.
CNV Size in Cases versus Controls
| Copy Number | Group | Mean Length (bp) |
Cases vs. Controls |
|
|---|---|---|---|---|
| Ratio | p Value | |||
| Deletions |
TOF cases | 285,657 | 1.3 | 0.024 |
| CHD cases | 337,288 | 1.6 | 0.022 | |
| controls | 213,262 | |||
| Duplications | TOF cases | 517,326 | 1.1 | 0.312 |
| CHD cases | 472,382 | 1.0 | 0.793 | |
| controls | 462,125 | |||
The following abbreviations are used: TOF, Tetralogy of Fallot; and CHD, congenital heart disease. p values were generated with a two-sided permutation test with 10,000 replicates.
Table 4.
Number of Genes per CNV in Cases versus Controls
| Copy Number | CNV Category | TOF Mean | CHD Mean | Control Mean |
TOF Cases vs. Controls |
CHD Cases vs. Controls |
||
|---|---|---|---|---|---|---|---|---|
| Ratio | p Value | Ratio | p Value | |||||
| Deletions |
all | 1.7 | 2.5 | 1.0 | 1.7 | 0.009 | 2.5 | 3 × 10−4 |
| rare | 3.5 | 5.1 | 1.4 | 2.6 | 0.006 | 3.7 | 0.001 | |
| common | 0.8 | 1.1 | 0.8 | 1.0 | 0.982 | 1.3 | 0.325 | |
| Duplications | all | 4.5 | 3.7 | 2.8 | 1.6 | 0.005 | 1.3 | 0.031 |
| rare | 6.1 | 4.1 | 2.2 | 2.8 | 1 × 10−4 | 1.9 | 0.006 | |
| common | 3.2 | 3.4 | 3.3 | 1.0 | 0.829 | 1.0 | 0.878 | |
The following abbreviations are used: CNV, copy-number variant; TOF, Tetralogy of Fallot; and CHD, congenital heart disease. p was generated with a two-sided permutation test with 10,000 replicates.
In order to identify pathway or ontology overrepresentation in functional regions, we performed Genomic Region Annotation Enrichment analysis (GREAT v.1.8.218) on rare deletions and rare duplications in 2,256 CHD cases. Analysis was carried out with default settings and the entire genome as background. GREAT analysis on rare deletions resulted in statistically significant enrichment genes in the Wnt-signaling pathway (2.9-fold enrichment; p = 1.2 × 10−5) and implicated 13 genes (CDH18 [MIM 603019], CDH2 [MIM 114020], CTBP1 [MIM 602618], CTNNB1 [MIM 116806], FAT1 [MIM 600976], LRP5L, NFATC1 [MIM 600489], PCDH15 [MIM 605514], PCDHB7 [MIM 606333], PCDHB8 [MIM 606334], PRKCB [MIM 176970], PRKCQ [MIM 600448], and WNT7B [MIM 601967]) in this pathway; there was involvement of Wnt genes in 28 out of 238 (12%) CHD cases with rare deletions. Phenotypes of these individuals were TOF (n = 11), atrial septal defect (n = 7), transposition of the great arteries (n = 3), atrioventricular septal defect (n = 2), coarctation of the aorta (n = 2), aortic stenosis (n = 1), congenitally corrected transposition of the great arteries (n = 1), and ventricular septal defect (n = 1). No significant enrichment was found for any other functional category. We did not find pathway or gene-ontology overrepresentation in the rare duplications.
Deletions in 15q11.2 Are Associated with CHD
Twelve 15q11.2 deletions were identified in our CHD cohort (n = 2,256), and a critical region encompassed four RefSeq genes: TUBGCP5 (MIM 608147), CYFIP1 (MIM 606322), NIPA2 (MIM 608146), and NIPA1 (MIM 608145) (see Figure 1). The phenotypes of these participants were complex left-sided malformations (n = 3), coarctation of the aorta (n = 3), atrial septal defect (n = 2), ventricular septal defect (n = 2), total anomalous pulmonary venous drainage (n = 1), and TOF (n = 1) (see Table 5 for details). We found one such deletion in 1,538 controls (841 unrelated controls and 697 unaffected family members). Therefore, 15q11.2 deletions were more frequent in CHD cases than in controls (12 out of 2,256 for cases versus 1 out of 1,538 for controls; OR = 8.2; p = 0.02).
Figure 1.
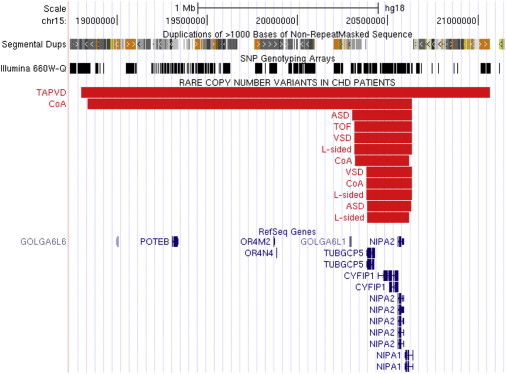
Recurrent Rare Deletions in 15q11.2
Twelve deletions (shown as red bars in the UCSC Genome Browser) were identified in three individuals with complex left-sided malformations (L-sided), three with coarctation of the aorta (CoA), two with ventricular septal defects (VSDs), two with atrial septal defects (ASDs), one with total anomalous pulmonary venous drainage (TAPVD), and one with TOF. RefSeq genes, segmental duplications, and coverage of the Illumina 660W platform in the region are shown. The smallest deletions encompass four RefSeq genes: TUBGCP5, CYFIP1, NIPA2, and NIPA1.
Table 5.
Phenotype Characteristics of CHD Individuals with 15q11.2 Deletions
| Family ID | Sex | Age (Years) | Chr | Start | Length | Cardiac Phenotype | Extracardiac Phenotype |
|---|---|---|---|---|---|---|---|
| GOCHD-2099.1 | female | N/A | 15 | 20,303,106 | 332,779 | atrial septal defect | none |
| OX-1232.1 | male | 9 | 15 | 20,388,584 | 229,594 | complex left sided | none |
| OX-941.1 | male | 8 | 15 | 20,321,135 | 297,043 | coarctation of the aorta | none |
| LEU-111.1 | male | 27 | 15 | 20,314,760 | 321,125 | complex left sided | Crohn disease |
| NOTT-301.1 | male | 6 | 15 | 18,837,563 | 1,798,322 | coarctation of the aorta | none |
| NOTT-421.1 | female | 21 | 15 | 20,384,417 | 251,468 | complex left sided | none |
| NOTT-577.1 | male | <1 | 15 | 18,802,207 | 2,263,748 | total anomalous pulmonary venous drainage | none |
| NOTT-772.1 | female | 1 | 15 | 20,314,760 | 321,125 | VSD (muscular)/ASD (secundum)/PDA | none |
| SYD-1200.1 | female | 4 | 15 | 20,388,584 | 240,866 | atrial septal defect | none |
| SYD-1366.1 | male | 3 | 15 | 20,384,417 | 251,468 | coarctation of the aorta | none |
| SYD-443.1 | female | 2 | 15 | 20,384,417 | 251,468 | ventricular septal defect | none |
| CHA-549.1 | male | N/A | 15 | 20,314,760 | 321,125 | Tetralogy of Fallot | none |
Inheritance status could not be determined because none of the parental samples were available for analysis. The following abbreviations are used: Chr, chromosome; N/A, not available; VSD, ventricular septal defect; ASD, atrial septal defect; and PDA, patent ductus arteriosus.
Recurrent CNVs at Candidate Locus 8p23.1 in CHD
We observed five rare CNVs (four deletions and one duplication) at the candidate 8p23.1 locus; the smallest deletion spanned four RefSeq genes: GATA4 (MIM 600576), NEIL2 (MIM 608933), FDFT1 (MIM 184420), and CSTB (MIM 601145) (see Figure 2). No overlapping CNV was found in our 1,538 controls or in the control populations that have been cataloged in the DGV.
Figure 2.
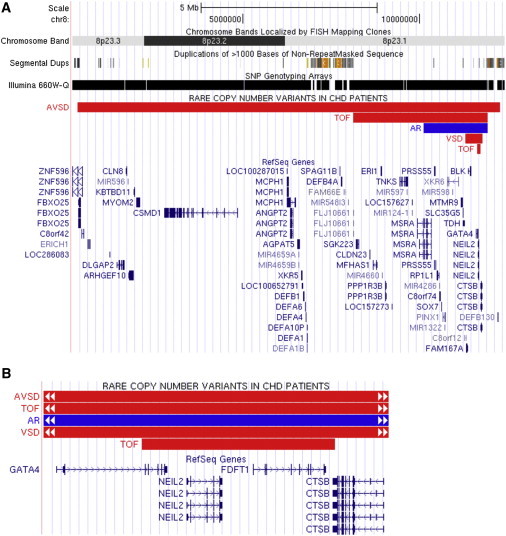
Recurrent Rare CNVs in the Candidate 8p23.1 Locus
(A and B) Four deletions were identified in one individual with an atrioventricular septal defect (AVSD), one with a ventricular septal defect (VSD), and two with TOF (all shown as red bars). The smallest deletion encompasses the last five exons of GATA4 and the whole coding regions of NEIL2 and FDFT1 (shown in B). In addition, an overlapping duplication was identified in one participant with a bicuspid aortic valve with aortic regurgitation (AR) (blue bar). The parental samples of these people are not available for analysis.
Rare De Novo CNV Burden and Paternal Origin Bias in TOF Trios
In 283 TOF probands, we used PennCNV joint calling10 to identify 13,375 putative CNV calls that did not occur in their respective unaffected parents. PennCNV calls that were previously observed in polymorphic frequency19 and found with a frequency > 0.1% in our 1,538 controls, as well as calls that were not confirmed with QuantiSNP (regardless of Bayes factor11) and those with length < 30 kb, were excluded from further analysis (see Figure S2 for details). We subjected all putative rare de novo CNVs (n = 28) to confirmation with an independent method (Affymetrix 6.0, array CGH, or MLPA), and ∼50% (13/28) were successfully validated. We thus observed rare de novo CNVs > 30 kb in ∼5% (13/283) of our TOF trios. Rare de novo CNVs were identified in loci (1q21.1, 3q29, and 4q34) that have been associated with isolated or syndromic TOF or other CHDs, as well as in regions (3q13.11, 5q14, 5q35.3, 6q27, 9p22.2, 16q11.2, 16q24.2, 19p13.3, and 22q12.3) that have not been previously described to be relevant to the risk of TOF (Table 6). We also investigated the parental origin of the de novo CNVs identified. In 10 of 11 individuals in whom this could be unequivocally determined, the CNV was on the paternal allele (see Tables 6 and S6). Thus, we identified paternal origin bias in rare de novo CNVs in TOF trios (p = 0.01). Because paternal origin bias has been previously reported in rare de novo CNV occurrences that were not mediated by segmental duplications (SDs),20,21 we additionally looked for SDs within the vicinity of the breakpoints of our de novo findings. Two out of 13 rare de novo CNV breakpoints coincided with a pair of SDs in direct orientation (see Figure S3). Thus, 85% of the rare de novo CNVs identified in this study were not mediated by SDs.
Table 6.
Rare De Novo CNVs Identified in 283 TOF Trios
| Family ID | Sex | Age (Years) | Cytoband | Hg18 Start Coordinate | Length | CN | Origin | RefSeq Genes | Cardiac and Extracardiac Phenotype |
|---|---|---|---|---|---|---|---|---|---|
| CHA-91 | male | 10 | 4q34.1-q34.3 | 173,538,773 | 6,551,325 | del | P | GALNTL6, GALNT7, HMGB2, SAP30, SCRG1, HAND2, NBLA00301, MORF4, FBXO8, KIAA1712, MIR4276, HPGD, GLRA3, ADAM29, GPM6A, MIR1267, WDR17, SPATA4, ASB5, SPCS3, VEGFC, NEIL3, AGA, LOC285501 | TOF, asthma, bilateral cryptorchidism |
| FCH-306 | male | 1 | 3q29 | 197,168,088 | 1,660,486 | del | P | SDHAP1, TFRC, LOC401109, ZDHHC19, OSTα, PCYT1A, TCTEX1D2, TM4SF19, UBXN7, RNF168, C3orf43, WDR53, FBXO45, LRRC33, C3orf34, PIGX, PAK2, SENP5, NCBP2, LOC152217, PIGZ, MFI2, DLG1, BDH1 | TOF, interrupted aortic arch, chest deformity (sternum) |
| CHA-617 | female | 10 | 16q24.2 | 85,737,889 | 177,523 | del | P | C16orf95 | TOF |
| NOTT-189b | female | 1 | 6q27 | 167,037,829 | 67,836 | del | P | RPS6KA2 | TOF |
| CHA-767 | male | 3 | 19p13.3 | 252,619 | 52,630 | del | P | MIER2 | TOF with MAPCA |
| CHA-25 | female | 54 | 16q11.2 | 45,056,281 | 40,613 | del | N/A | ANKRD26P1 | TOF |
| CHA-64a | male | 13 | 22q12.3 | 31,789,131 | 188,778 | del | M | none | TOF |
| CHA-812 | male | 8 | 3q13.11 | 105,183,599 | 97,653 | del | P | none | TOF, asthma |
| CHA-137 | female | 1 | 1q21.1 | 144,967,972 | 1,418,624 | dup | P | LOC728989, PRKAB2, PDIA3P, FMO5, CHD1L, BCL9, ACP6, GJA5, GJA8 | TOF, laryngomalacia |
| CHA-9 | female | 3 | 5q14.1-q14.3 | 80,936,354 | 4,937,263 | dup | P | SSBP2, TMEM167A, SCARNA18, XRCC4, VCAN, HAPLN1, EDIL3 | TOF |
| NOTT-389 | male | 1 | 9p22.2 | 17,735,053 | 50,117 | dup | N/A | SH3GL2 | TOF |
| CHA-750 | female | 2 | 5q35.3 | 179,681,237 | 432,453 | dup | P | GFPT2, CNOT6, SCGB3A1, FLT4, OR2Y1 | TOF |
| CHA-817 | male | 16 | 5q35.3 | 178,357,798 | 264,665 | dup | P | ZNF879, ZNF354C, ADAMTS2 | TOF |
Genes in bold are reported to be expressed in the fetal heart (Bgee database), except for SH3GL2, whose expression was detectable in the early embryo and in the myocardium of the child and adult heart. The following abbreviations are used: del, deletion; dup, duplication; P, paternal; M, maternal; N/A, not available; TOF, Tetralogy of Fallot; and MAPCA, major aortopulmonary collateral arteries.
Recurrent Rare CNVs Overlapping De Novo CNV Loci
In the remaining 1,987 CHD cases, we found additional rare CNVs that overlapped rare de novo CNVs present in our TOF trios at 1q21.1 (as previously described8), 4q34 (HAND2 [MIM 602407]), 5q14.2 (EDIL3 [MIM 606018]), and 5q35.3 (CNOT6 [MIM 608951]) (see Figure 3). In addition, we observed rare CNVs that overlapped with the rare de novo CNVs previously identified in 114 TOF trios by Greenway et al.7 at 1q21.1, 4q22.1 (PPM1K [MIM 611065]), and 7p21.3 (see Figure 4). Some of these recurrent CNVs (in 1q21.1, 4q34, and 7p21.3) were found to be inherited from an unaffected parent, whereas the inheritance status of the remaining CNVs could not be determined because of the unavailability of parental samples.
Figure 3.
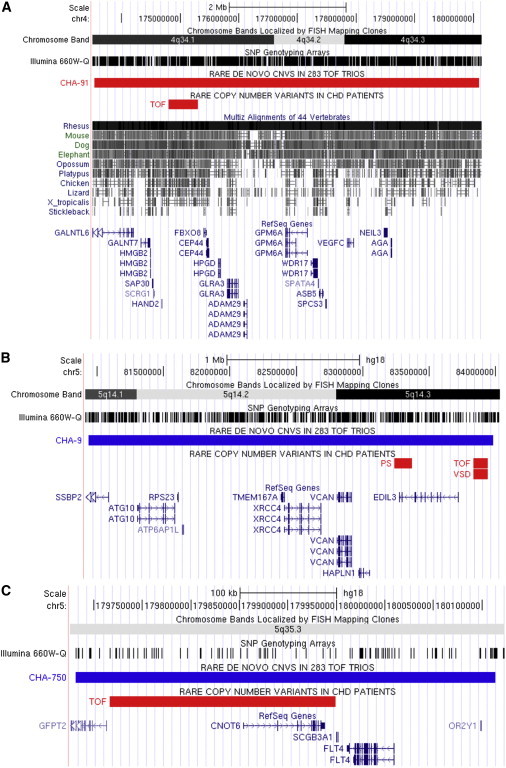
Rare CNVs Overlapping Rare De Novo CNVs Identified in TOF Trios
We examined the remaining 1,978 CHD cases for recurrent CNVs that overlap rare de novo findings in 283 TOF trios. We found overlaps in 4q34 (a rare deletion in the highly conserved region upstream of HAND2, as shown in A), 5q14.2 (one rare deletion overlapping EDIL3 and two others overlapping a conserved region ∼100 kb upstream of EDIL3, as shown in B), and 5q35 (an deletion overlapping CNOT6, as shown in C). Deletions and duplications are shown in the UCSC Genome Browser as red and blue bars, respectively. The following abbreviations are used: PS, pulmonary stenosis; and VSD, ventricular septal defect.
Figure 4.
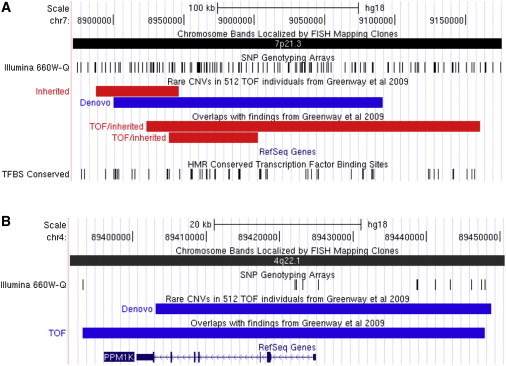
Rare CNVs Overlapping CNVs Reported by Greenway et al., 2009
We searched in 1,987 CHD cases for CNVs overlapping the rare de novo CNVs previously reported by Greenway et al. We found recurrent rare deletions in the 7p21.3 locus (A) in two TOF probands, both of whom inherited the deletions from their respective unaffected fathers. Both of these findings had been confirmed on the Affymetrix 6.0 platform and by MLPA. We did not find such variants in 841 unrelated controls or other unaffected family members (n = 695). These rare CNVs did not overlap any known RefSeq genes, although there are some overlaps with transcription-factor binding-site conservation (shown). The nearest gene is NXPH1.
(B) On the Illumina 660W platform (shown), there is insufficient coverage overlapping the 4q22.1 de novo variant reported by Greenway et al. Therefore, in addition to examining this locus in the 1,987 CHD probands who had been typed on the Illumina 660W, we also screened this locus with MLPA in 1,007 CHD cases, 866 of whom were also typed on the Illumina 660W. In a TOF proband, we detected a duplication that encompassed PPM1K (as shown). No overlapping duplication was found in 841 unrelated controls or 697 unaffected family members. The parental DNA samples of this proband were not available for analysis. Deletions and duplications are shown in the UCSC Genome Browser as red and blue bars, respectively.
Discussion
In one of the largest studies of CHD genetics thus far, we performed a genome-wide investigation of CNVs > 100 kb in sporadic, nonsyndromic CHD. Our data show that rare deletions, in particular rare genic deletions, are associated with both TOF and other CHDs and that they account for 3%–4% of the PAR of each condition. We further demonstrated significant excess of recurrent deletions at 15q11.2 and of rare deletions involving Wnt-signaling genes. Rare deletions or duplications spanning a larger number of genes confer higher risk of CHD, and rare deletions in CHD cases tend to be larger and encompass genes with higher haploinsufficiency scores.17 Additionally, de novo CNVs occurred in 5% of our trio families, implicating candidate and novel genes for CHD.
Previous large studies of CNVs in psychiatric and developmental phenotypes have demonstrated an increased burden of rare CNVs (the greatest effects were observed in single-occurrence and de novo CNVs),1,2,22 larger CNV size, and higher number of genes spanned by rare CNVs in cases compared to controls.1,3,4 The study performed by Cooper et al. (involving 15,767 cases of developmental delay and 8,329 controls) had data on 575 cases with CHD as a component of their phenotype and showed that CNVs > 400 kb with <1% frequency were present in around 25% of these cases.4 By contrast, 13.6% of our CHD cases had such CNVs; there was a highly comparable control frequency between both studies (11.5% in Cooper et al.; 10.8% in the present study). This most likely reflects the different ascertainment of the two cohorts; in the study of Cooper et al., they were chiefly ascertained through referral with a diagnosis of intellectual disability or developmental delay, and in our study, they were ascertained through pediatric and adult CHD clinics. Thus, our study is likely to provide more representative estimates of the contribution of CNVs to the population burden of CHD. Our findings complement these previous studies in providing evidence of the pathogenicity of rare CNVs and their contribution to common complex diseases, including CHD. In agreement with previous studies, our data suggest that the more genes spanned by a CNV, the greater its potential to be pathogenic. Our study also shows that cases have higher haploinsufficiency scores17 of the genes encompassed by rare deletions than do controls. Haploinsufficient genes have formerly been shown to have biased evolutionary and functional properties. They are much more highly conserved, highly expressed during early development, and highly tissue specific.
We further identified an overrepresentation of Wnt-signaling-pathway genes among those spanned by rare deletions found in CHD cases. Wnt signaling regulates diverse cellular processes, such as gene transcription and cell proliferation, migration, polarity, and division.23,24 Both the classic canonical Wnt-signaling pathway involving beta-catenin and the noncanonical branches of the pathway independent of beta-catenin are involved in a coordinated fashion at all stages of cardiac specification, differentiation, and development.23 Several model organisms with mutations in Wnt-signaling-pathway genes exhibit CHD,25,26 yet evidence to date for the involvement of the Wnt pathway in human CHD has been sparse.
We found an association between CHD and deletions of the 15q11.2 region (OR = 8.2; p = 0.02), adjacent to but not including the critical region of Prader-Willi/Angelman syndrome (MIM 176270). These deletions implicated four RefSeq genes, none of which have been previously associated with CHD. However, TUBGCP5 and NIPA1 were reportedly expressed in the fetal heart (Bgee database), thus increasing their candidacy as the causative gene for CHD. Of note, half of the cases with deletions had left-sided cardiac lesions; study of larger numbers of cases will be required for determining the significance of this apparent subphenotypic predominance. A previous study of 182 individuals with left ventricular outflow-tract obstruction identified the same 15q11.2 deletion in one person.27 Cooper et al.4 recently identified such deletions in 6 out of 575 CHD cases in their cohort (p = 0.004 when compared to controls). The penetrance of CHD associated with such deletions is clearly incomplete; only two out of nine individuals with the deletion reported in a study by Doornbos et al. had heart defects,28 and both we and Cooper et al. observed the deletion in healthy controls. Previously, 15q11.2 deletions have been associated with idiopathic generalized epilepsies,29 schizophrenia,30 and behavioral disturbances,28 and point mutations in NIPA1, one of the genes in the critical region, are known to cause autosomal-dominant spastic paraplegia.31 Our results suggest that, after the previously described 1q21.1 CNVs associated with TOF and other forms of CHD, the 15q11.2 deletion is the CNV most strongly associated with the risk of sporadic, nonsyndromic CHD. Confirmation of our findings at this region in further large studies would be of considerable interest.
Deletion of a 8p23 5 Mb region that encompasses GATA4 has previously been associated with multiple malformations that include CHD.32 GATA4 is a transcription factor essential for cardiac development.33 The first GATA4 mutations reported were in two pedigrees with multiple affected family members; in both families, all affected individuals had atrial septal defects, but in the larger pedigree, half had additional cardiac abnormalities, including pulmonary stenosis, ventricular septal defects, atrioventricular septal defects, and aortic regurgitation.34 In our CHD cases, we observed four deletions and one duplication of 8p23.1 and a critical region spanning four genes, including GATA4. The cardiac phenotypes were atrioventricular septal defects (n = 1), ventricular septal defects (n = 1), and TOF (n = 2) in the deleted individuals and a bicuspid aortic valve with aortic regurgitation in the case with the duplication. GATA4 missense changes have previously been detected at low frequencies in cases of TOF.35,36 Comparison of CNV frequency between our cases and controls did not achieve statistical significance (p = 0.08). Cooper et al. reported three deletions and one duplication that spanned GATA4 in their 575 CHD individuals, whereas they found no deletions or duplications encompassing GATA4 in their 8,329 controls (p = 1.7 × 10−5 by Fisher’s two-tailed exact test).4 Furthermore, there are no reports of CNVs overlapping GATA4 in any of the control populations that have been cataloged in the DGV. Considering these data in the context of the previously demonstrated causative nature of GATA4 missense mutations in CHD, we conclude that it is highly likely that the CHD phenotypes observed in our five individuals with 8p23.1 CNVs resulted from dosage sensitivity of GATA4.
We observed a global rare de novo CNV burden of ∼5% in our 283 TOF trios. This is broadly concordant with that previously reported in another cohort of 114 TOF trios7 given the differences in the genotyping platforms and analysis pipelines between the two studies. The rare de novo CNVs identified in this study implicate known candidate loci (1q21.1, 3q29, and 4q34), as well as other loci (3q13.11, 5q14, 5q35.3, 6q27, 9p22.2, 16q11.2, 16q24.2, 19p13.3, and 22q12.3) that have not been previously associated with CHD with recurring CNVs in 1q21.1 (GJA5), 4q34 (HAND2), 5q14.2 (EDIL3), and 5q35.3 (CNOT6).
The distal 1q21.1 locus has been previously shown to manifest a degree of phenotypic specificity in CHD, as well as in other developmental phenotypes. Duplications of 1q21.1 are associated with TOF, autism, and macrocephaly, whereas the reciprocal deletions are associated with other types of (non-TOF) CHD, schizophrenia, and microcephaly.8,37,38 Previous studies in mice and humans strongly suggest that GJA5, which encodes connexin-40, is the critical gene for the CHD phenotype in this locus.8,39 The rare de novo deletion found in one individual at the 4q34 locus spanned 24 RefSeq genes (see Table 5). One of the deleted genes was HAND2, which encodes a basic helix-loop-helix transcription factor known for its pivotal roles in cardiac development.40 The 500 kb overlapping deletion that was found in another individual with TOF, however, did not span the coding region of HAND2 but did encompass a highly conserved region that is ∼100 kb upstream of HAND2 and that overlaps previously predicted human heart-specific enhancer sequences.41 Although this deletion was inherited from an unaffected father, we did not find overlapping CNVs in our 1,577 controls or in the DGV. It is known that some CNVs in noncoding segments can profoundly affect the expression of copy-number neutral genes in the vicinity.42 Thus, taking into account the large size (>500 kb) of the deletion, its rarity, and the high degree of conservation of the involved region, we consider this deletion highly likely to have contributed to the occurrence of CHD in this individual (Figure 3 and Table S7).
We also identified recurrent CNVs at the 5q35.3 locus (Figure 3). The overlapping segment spanned a single gene: CNOT6, which encodes a subunit of the CCR4-NOT core transcriptional complex, which is known to be crucial for controlling mRNA stability during embryonic development.43,44 RNAi silencing of dNOT3, another subunit of the same complex, in Drosophila and heterozygous knockout of Cnot3 in mice both resulted in heart defects.45 We additionally found three cases with deletions overlapping the rare de novo duplication in the 5q14 locus. One of the deletions that spanned the last two exons of EDIL3 was found in a 62-year-old participant with pulmonary stenosis and a secundum atrial septal defect. The other two individuals, who had a deletion situated ∼100 kb upstream of EDIL3, were an 8-year-old with TOF and an 11-year-old with a ventricular septal defect. Glessner et al. reported the same deletion variant upstream of EDIL3 in childhood-obesity cases (6 of 2,559 cases and 0 of 4,075 lean controls); the CHD status of these cases was not reported.46 This variant was not present in our 1,578 controls or in the DGV. Both of the participants with these deletions had no notable extracardiac phenotypes. EDIL3 (epidermal growth-factor-like repeats and discoidin I-like domains 3) encodes a glycoprotein secreted by endothelial cells. It plays an important role in vessel-wall remodeling and development during angiogenesis.47,48 It is also upregulated in cardiac progenitor cells, supporting a potential role in early cardiac development; this role merits further investigation.49
With the exception of 1q21.1, we did not replicate the de novo findings that were reported by Greenway et al. in our TOF trios.7 However, in the remaining 1,987 CHD cases, we found rare CNVs overlapping those reported by Greenway et al. at 7p21.3 and 4q22.1, thus supporting the notion that they are involved in CHD risk. There is no RefSeq gene that overlaps the 7p21.3 locus, in which we found deletions of paternal origin in two TOF cases (Figure 4). The nearest gene is NXPH1 (MIM 604639), a neurexophilin family member that promotes adhesion between dendrites and axons. This region has been previously associated with autism and attention-deficit-hyperactive disorder.50 The overlapping duplication in the 4q22.1 locus, on the other hand, spanned a single gene, PPM1K (PP2C domain-containing protein phosphatase 1K or PP2C-like mitochondrial protein phosphatase [MIM 611065]), which is essential for cell survival, embryonic development, and cardiac function. Knockdown of this gene in zebrafish embryos resulted in abnormal cardiac development and heart failure from induced apoptosis.51
Interestingly, 10 out of 11 rare de novo CNVs identified in our TOF trios occurred on the paternally transmitted chromosome (p = 0.01). A recent study reported a similar magnitude of paternal bias in the origin of rare de novo CNVs in 3,443 individuals with intellectual disability (90 of 118 paternal; p = 1.14 × 10−8).20 In that study, the median paternal age of those with rare de novo CNVs that were not flanked with SDs (which represented ∼80% of deletions detected) was slightly higher (34.16 ± 4.91 years) than that of those not carrying such CNVs (32.13 ± 4.17 years; p = 0.02). A similar finding was reported in a study of 173 individuals with multisystem abnormalities:21 an excess of paternal origin in non-SD-mediated de novo CNV events (p = 0.02) was seen. No increased paternal-age bias was detected in that study, although this might have been due to a lack of power. SD-mediated chromosomal rearrangement is known to be the primary generating mechanism for CNVs,52,53 but in keeping with these two reports, most of the rare de novo CNV breakpoints in our study did not coincide with directly oriented SDs (Figure S3). A higher rate of non-SD-mediated mechanisms—e.g., nonhomologous-end joining (NHEJ) and fork stalling and template switching (FoSTeS)—as a result of the increased occurrence of double-strand breaks resulting from the greater number of germ cell divisions in spermatogenesis compared to oogenesis (particularly in older males) has been proposed as a mechanism whereby this could occur.54 Advanced paternal age has been suggested to be an independent risk factor for CHD.55 However, the number of de novo CNVs in our study was too small for any meaningful statistical analysis of paternal age to be conducted.
Considering the acknowledged limitations of the currently available CNV-detection technologies, we opted to take a conservative approach in our global CNV analyses. All of our case and control subjects were typed on the same platform at the same genotyping center, and we adopted highly stringent CNV-calling criteria in order to ensure comparability in detection between samples originating from multiple clinical centers. We further undertook extensive validation experiments in order to identify the regions that cannot be accessed reliably with our detection platform, and we excluded them from our analyses. Our approach thus effectively minimized false-positive discoveries in our data set, albeit at the expense of a higher false-negative rate. We adopted a different strategy in our search for potential highly penetrant rare de novo CNVs in our TOF trios by significantly lowering the stringency of our CNV-calling criteria in order to maximize capture. However, we subjected all putative CNVs identified by these latter criteria to confirmation with one or more independent methods. Therefore, we have high confidence in the CNV calls in both the case-control and trio arms of the study.
Our study firmly established the collective contribution of global rare CNVs to the risk of sporadic CHD. However, the bulk of our findings constitute single-occurrence CNVs, whose associations with CHD at each individual locus cannot be statistically evaluated. It is likely that large-scale meta-analyses of tens of thousands of participants will be required for accurately characterizing associations of these very rare CNVs. Moreover, the phenotypic composition of our cohort was heterogeneous and included relatively few cases of rarer malformations (e.g., hypoplastic left heart syndrome and heterotaxy). For these rare lesions, further studies involving larger numbers will be of value. We detected recurrent CNVs spanning individual genes (EDIL3 and CNOT6), which are therefore strong candidates for CHD. However, most of the recurrent rare CNVs, including the 15q11.2 deletions, for which we detected an association with CHD, identified in this study spanned multiple genes. Therefore, functional studies for characterizing the role in heart development of genes within the CNVs identified in this study are a priority for future work; among these, strongly predicted dosage-sensitive genes (those with high haploinsufficiency scores) are of particular interest. Identification of causative genes for CHD and characterization of the magnitude of their relationship with risk could potentially lead to improvement in genetic counseling in families with a high risk of CHD—perhaps particularly in CHD-affected adults contemplating reproductive choices.
Acknowledgments
We thank all the participants in the study and Linda Sneddon for assistance with sample collection. This work was funded by the British Heart Foundation (BHF), European Community’s 7th Framework Programme contract (“CHeartED”) HEALTH-F2-2008-223040, Heart Research UK, the Federated Foundation, and the Wellcome Trust. We acknowledge the support of the National Institute for Health Research through the Northumberland, Tyne, and Wear Comprehensive Local Research Network. B.D.K. and S.B. are supported by BHF personal chairs. We acknowledge the Wellcome Trust Case Control Consortium for making available data about SNP tagging of common copy-number variants.
Contributor Information
Judith A. Goodship, Email: j.a.goodship@newcastle.ac.uk.
Bernard D. Keavney, Email: b.d.keavney@newcastle.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Bgee Database, http://bgee.unil.ch/
BlueGnome Cytochip, http://www.cytochip.com
Database of Genomic Variants, http://projects.tcag.ca/variation/
Galaxy, http://main.g2.bx.psu.edu/
GREAT software, http://great.stanford.edu/
MAPD: MLPA Probe Design, http://bioinform.arcan.stonybrook.edu/mlpa2/cgi-bin/mlpa.cgi/
MRC-Holland MLPA, http://www.mrc-holland.com
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Genome Browser and extended DNA utility, http://genome.ucsc.edu/
WTCCC+ Association Study, http://www.wtccc.org.uk/wtcccplus_cnv/supplemental.shtml
References
- 1.International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu B., Roos J.L., Levy S., van Rensburg E.J., Gogos J.A., Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 3.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooper G.M., Coe B.P., Girirajan S., Rosenfeld J.A., Vu T.H., Baker C., Williams C., Stalker H., Hamid R., Hannig V. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffman J.I., Kaplan S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002;39:1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 6.Burn J., Brennan P., Little J., Holloway S., Coffey R., Somerville J., Dennis N.R., Allan L., Arnold R., Deanfield J.E. Recurrence risks in offspring of adults with major heart defects: results from first cohort of British collaborative study. Lancet. 1998;351:311–316. doi: 10.1016/s0140-6736(97)06486-6. [DOI] [PubMed] [Google Scholar]
- 7.Greenway S.C., Pereira A.C., Lin J.C., DePalma S.R., Israel S.J., Mesquita S.M., Ergul E., Conta J.H., Korn J.M., McCarroll S.A. De novo copy number variants identify new genes and loci in isolated sporadic tetralogy of Fallot. Nat. Genet. 2009;41:931–935. doi: 10.1038/ng.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soemedi R., Topf A., Wilson I.J., Darlay R., Rahman T., Glen E., Hall D., Huang N., Bentham J., Bhattacharya S. Phenotype-specific effect of chromosome 1q21.1 rearrangements and GJA5 duplications in 2436 congenital heart disease patients and 6760 controls. Hum. Mol. Genet. 2012;21:1513–1520. doi: 10.1093/hmg/ddr589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang K., Li M., Hadley D., Liu R., Glessner J., Grant S.F., Hakonarson H., Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colella S., Yau C., Taylor J.M., Mirza G., Butler H., Clouston P., Bassett A.S., Seller A., Holmes C.C., Ragoussis J. QuantiSNP: an Objective Bayes Hidden-Markov Model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007;35:2013–2025. doi: 10.1093/nar/gkm076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bailey J.A., Gu Z., Clark R.A., Reinert K., Samonte R.V., Schwartz S., Adams M.D., Myers E.W., Li P.W., Eichler E.E. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- 13.Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goecks J., Nekrutenko A., Taylor J., Galaxy Team Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Korn J.M., Kuruvilla F.G., McCarroll S.A., Wysoker A., Nemesh J., Cawley S., Hubbell E., Veitch J., Collins P.J., Darvishi K. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhi J., Hatchwell E. Human MLPA Probe Design (H-MAPD): a probe design tool for both electrophoresis-based and bead-coupled human multiplex ligation-dependent probe amplification assays. BMC Genomics. 2008;9:407. doi: 10.1186/1471-2164-9-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang N., Lee I., Marcotte E.M., Hurles M.E. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Craddock N., Hurles M.E., Cardin N., Pearson R.D., Plagnol V., Robson S., Vukcevic D., Barnes C., Conrad D.F., Giannoulatou E., Wellcome Trust Case Control Consortium Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hehir-Kwa J.Y., Rodríguez-Santiago B., Vissers L.E., de Leeuw N., Pfundt R., Buitelaar J.K., Pérez-Jurado L.A., Veltman J.A. De novo copy number variants associated with intellectual disability have a paternal origin and age bias. J. Med. Genet. 2011;48:776–778. doi: 10.1136/jmedgenet-2011-100147. [DOI] [PubMed] [Google Scholar]
- 21.Sibbons C., Morris J.K., Crolla J.A., Jacobs P.A., Thomas N.S. De novo deletions and duplications detected by array CGH: a study of parental origin in relation to mechanisms of formation and size of imbalance. Eur. J. Hum. Genet. 2012;20:155–160. doi: 10.1038/ejhg.2011.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang D., Cheng L., Qian Y., Alliey-Rodriguez N., Kelsoe J.R., Greenwood T., Nievergelt C., Barrett T.B., McKinney R., Schork N. Singleton deletions throughout the genome increase risk of bipolar disorder. Mol. Psychiatry. 2009;14:376–380. doi: 10.1038/mp.2008.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gessert S., Kühl M. The multiple phases and faces of wnt signaling during cardiac differentiation and development. Circ. Res. 2010;107:186–199. doi: 10.1161/CIRCRESAHA.110.221531. [DOI] [PubMed] [Google Scholar]
- 24.Henderson D.J., Phillips H.M., Chaudhry B. Vang-like 2 and noncanonical Wnt signaling in outflow tract development. Trends Cardiovasc. Med. 2006;16:38–45. doi: 10.1016/j.tcm.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 25.Tian Y., Yuan L., Goss A.M., Wang T., Yang J., Lepore J.J., Zhou D., Schwartz R.J., Patel V., Cohen E.D., Morrisey E.E. Characterization and in vivo pharmacological rescue of a Wnt2-Gata6 pathway required for cardiac inflow tract development. Dev. Cell. 2010;18:275–287. doi: 10.1016/j.devcel.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou W., Lin L., Majumdar A., Li X., Zhang X., Liu W., Etheridge L., Shi Y., Martin J., Van de Ven W. Modulation of morphogenesis by noncanonical Wnt signaling requires ATF/CREB family-mediated transcriptional activation of TGFbeta2. Nat. Genet. 2007;39:1225–1234. doi: 10.1038/ng2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kerstjens-Frederikse W.S., Du Marchie Sarvaas G.J., Ruiter J.S., Van Den Akker P.C., Temmerman A.M., Van Melle J.P., Hofstra R.M., Berger R.M. Left ventricular outflow tract obstruction: should cardiac screening be offered to first-degree relatives? Heart. 2011;97:1228–1232. doi: 10.1136/hrt.2010.211433. [DOI] [PubMed] [Google Scholar]
- 28.Doornbos M., Sikkema-Raddatz B., Ruijvenkamp C.A., Dijkhuizen T., Bijlsma E.K., Gijsbers A.C., Hilhorst-Hofstee Y., Hordijk R., Verbruggen K.T., Kerstjens-Frederikse W.S. Nine patients with a microdeletion 15q11.2 between breakpoints 1 and 2 of the Prader-Willi critical region, possibly associated with behavioural disturbances. Eur. J. Med. Genet. 2009;52:108–115. doi: 10.1016/j.ejmg.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 29.de Kovel C.G., Trucks H., Helbig I., Mefford H.C., Baker C., Leu C., Kluck C., Muhle H., von Spiczak S., Ostertag P. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010;133:23–32. doi: 10.1093/brain/awp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stefansson H., Rujescu D., Cichon S., Pietiläinen O.P., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E., GROUP Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rainier S., Chai J.H., Tokarz D., Nicholls R.D., Fink J.K. NIPA1 gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6) Am. J. Hum. Genet. 2003;73:967–971. doi: 10.1086/378817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pehlivan T., Pober B.R., Brueckner M., Garrett S., Slaugh R., Van Rheeden R., Wilson D.B., Watson M.S., Hing A.V. GATA4 haploinsufficiency in patients with interstitial deletion of chromosome region 8p23.1 and congenital heart disease. Am. J. Med. Genet. 1999;83:201–206. [PubMed] [Google Scholar]
- 33.Molkentin J.D., Lin Q., Duncan S.A., Olson E.N. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11:1061–1072. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]
- 34.Garg V., Kathiriya I.S., Barnes R., Schluterman M.K., King I.N., Butler C.A., Rothrock C.R., Eapen R.S., Hirayama-Yamada K., Joo K. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 35.Tomita-Mitchell A., Maslen C.L., Morris C.D., Garg V., Goldmuntz E. GATA4 sequence variants in patients with congenital heart disease. J. Med. Genet. 2007;44:779–783. doi: 10.1136/jmg.2007.052183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nemer G., Fadlalah F., Usta J., Nemer M., Dbaibo G., Obeid M., Bitar F. A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum. Mutat. 2006;27:293–294. doi: 10.1002/humu.9410. [DOI] [PubMed] [Google Scholar]
- 37.Brunetti-Pierri N., Berg J.S., Scaglia F., Belmont J., Bacino C.A., Sahoo T., Lalani S.R., Graham B., Lee B., Shinawi M. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crespi B., Stead P., Elliot M. Evolution in health and medicine Sackler colloquium: Comparative genomics of autism and schizophrenia. Proc. Natl. Acad. Sci. USA. 2010;107(Suppl 1):1736–1741. doi: 10.1073/pnas.0906080106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu H., Smith F.C., Taffet S.M., Delmar M. High incidence of cardiac malformations in connexin40-deficient mice. Circ. Res. 2003;93:201–206. doi: 10.1161/01.RES.0000084852.65396.70. [DOI] [PubMed] [Google Scholar]
- 40.Tsai C.H., Van Dyke D.L., Feldman G.L. Child with velocardiofacial syndrome and del (4)(q34.2): another critical region associated with a velocardiofacial syndrome-like phenotype. Am. J. Med. Genet. 1999;82:336–339. [PubMed] [Google Scholar]
- 41.Narlikar L., Sakabe N.J., Blanski A.A., Arimura F.E., Westlund J.M., Nobrega M.A., Ovcharenko I. Genome-wide discovery of human heart enhancers. Genome Res. 2010;20:381–392. doi: 10.1101/gr.098657.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stranger B.E., Forrest M.S., Dunning M., Ingle C.E., Beazley C., Thorne N., Redon R., Bird C.P., de Grassi A., Lee C. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Temme C., Zhang L., Kremmer E., Ihling C., Chartier A., Sinz A., Simonelig M., Wahle E. Subunits of the Drosophila CCR4-NOT complex and their roles in mRNA deadenylation. RNA. 2010;16:1356–1370. doi: 10.1261/rna.2145110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goldstrohm A.C., Wickens M. Multifunctional deadenylase complexes diversify mRNA control. Nat. Rev. Mol. Cell Biol. 2008;9:337–344. doi: 10.1038/nrm2370. [DOI] [PubMed] [Google Scholar]
- 45.Neely G.G., Kuba K., Cammarato A., Isobe K., Amann S., Zhang L., Murata M., Elmén L., Gupta V., Arora S. A global in vivo Drosophila RNAi screen identifies NOT3 as a conserved regulator of heart function. Cell. 2010;141:142–153. doi: 10.1016/j.cell.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Glessner J.T., Bradfield J.P., Wang K., Takahashi N., Zhang H., Sleiman P.M., Mentch F.D., Kim C.E., Hou C., Thomas K.A. A genome-wide study reveals copy number variants exclusive to childhood obesity cases. Am. J. Hum. Genet. 2010;87:661–666. doi: 10.1016/j.ajhg.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhong J., Eliceiri B., Stupack D., Penta K., Sakamoto G., Quertermous T., Coleman M., Boudreau N., Varner J.A. Neovascularization of ischemic tissues by gene delivery of the extracellular matrix protein Del-1. J. Clin. Invest. 2003;112:30–41. doi: 10.1172/JCI17034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fan Y., Zhu W., Yang M., Zhu Y., Shen F., Hao Q., Young W.L., Yang G.Y., Chen Y. Del-1 gene transfer induces cerebral angiogenesis in mice. Brain Res. 2008;1219:1–7. doi: 10.1016/j.brainres.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Masino A.M., Gallardo T.D., Wilcox C.A., Olson E.N., Williams R.S., Garry D.J. Transcriptional regulation of cardiac progenitor cell populations. Circ. Res. 2004;95:389–397. doi: 10.1161/01.RES.0000138302.02691.be. [DOI] [PubMed] [Google Scholar]
- 50.Salyakina D., Cukier H.N., Lee J.M., Sacharow S., Nations L.D., Ma D., Jaworski J.M., Konidari I., Whitehead P.L., Wright H.H. Copy number variants in extended autism spectrum disorder families reveal candidates potentially involved in autism risk. PLoS ONE. 2011;6:e26049. doi: 10.1371/journal.pone.0026049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu G., Ren S., Korge P., Choi J., Dong Y., Weiss J., Koehler C., Chen J.N., Wang Y. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev. 2007;21:784–796. doi: 10.1101/gad.1499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kidd J.M., Cooper G.M., Donahue W.F., Hayden H.S., Sampas N., Graves T., Hansen N., Teague B., Alkan C., Antonacci F. Mapping and sequencing of structural variation from eight human genomes. Nature. 2008;453:56–64. doi: 10.1038/nature06862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Korbel J.O., Urban A.E., Affourtit J.P., Godwin B., Grubert F., Simons J.F., Kim P.M., Palejev D., Carriero N.J., Du L. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318:420–426. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Crow J.F. The origins, patterns and implications of human spontaneous mutation. Nat. Rev. Genet. 2000;1:40–47. doi: 10.1038/35049558. [DOI] [PubMed] [Google Scholar]
- 55.Olshan A.F., Schnitzer P.G., Baird P.A. Paternal age and the risk of congenital heart defects. Teratology. 1994;50:80–84. doi: 10.1002/tera.1420500111. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.