

Abstract
The basis for impaired differentiation in TP63 mutant ankyloblepharon-ectodermal dysplasia-clefting (AEC) syndrome is unknown. Human epidermis harboring AEC TP63 mutants recapitulated this impairment, along with downregulation of differentiation activators, including HOPX, GRHL3, KLF4, PRDM1, and ZNF750. Gene-set enrichment analysis indicated that disrupted expression of epidermal differentiation programs under the control of ZNF750 and KLF4 accounted for the majority of disrupted epidermal differentiation resulting from AEC mutant TP63. Chromatin immunoprecipitation (ChIP) analysis and ChIP-sequencing of TP63 binding in differentiated keratinocytes revealed ZNF750 as a direct target of wild-type and AEC mutant TP63. Restoring ZNF750 to AEC model tissue rescued activator expression and differentiation, indicating that AEC TP63-mediated ZNF750 inhibition contributes to differentiation defects in AEC. Incorporating disease-causing mutants into regenerated human tissue can thus dissect pathomechanisms and identify targets that reverse disease features.
Introduction
TP63 (MIM 603273) is essential for the development and homeostasis of stratified epithelia.1–5 TP63 exists in multiple isoforms; however, in developmentally mature epidermis, the predominant isoform is ΔNp63α.6 Consistent with this, genetic ablation of TA isoforms does not markedly impact epidermal homeostasis or differentiation.7 Importantly, previous studies have demonstrated that TP63 is required for both the maintenance of epidermal progenitor proliferation as well as differentiation.4,8 How TP63 executes both functions remains incompletely understood.
Heterozygous, dominant mutations in TP63 cause a number of monogenic human diseases that share the common feature of abnormal epithelia, including ectodermal dysplasia. Among these disorders are ankyloblepharon-ectodermal dysplasia-clefting syndrome (AEC [MIM 106260])9, ectrodactyly-ectodermal dysplasia-clefting syndrome (EEC [MIM 604292]),10 limb-mammary syndrome (LMS [MIM 603543]),10 acro-dermato-ungual-lacrimal-tooth syndrome (ADULT [MIM 103285]),11 Rapp-Hodgkin syndrome (RHS [MIM 129400]),12,13 and split hand/foot malformation (SHFM [MIM605289]).14 Intriguingly, despite the fact that all these disorders result from heterozygous TP63 mutations, impaired epidermal differentiation with epidermal erosions is characteristic in individuals with AEC but is rarely seen in other syndromes,15 whereas severe limb abnormalities common to EEC are not seen in individuals with AEC.16 These findings suggest that the mutations causing each syndrome impair specific and distinct activities of TP63.
TP63 syndromes arise from mutations that affect discrete domains of TP63. AEC itself is characterized by mutations in the C-terminal portion of the sterile alpha motif (SAM) domain, which is specific to TP63α isoforms.17 EEC mutations, in contrast, predominantly localize in the C-terminal portion of the TP63 DNA binding domain. Previous work has shown that TP63-EEC mutants are defective in the induction of DLX5 and DLX6, which are implicated in the pathogenesis of EEC phenotypes (MIMs 600028 and 600030).18–20 Remarkably, AEC mutants are capable of DLX5 and DLX6 transactivation, demonstrating that AEC mutants retain important TP63 functions.20 The mechanism by which AEC mutation impairs epidermal differentiation is unknown.
Here, we introduce AEC TP63 mutants in regenerated human epidermal tissue, both to model epidermal features of AEC and to gain insight into the mechanisms whereby these mutants disrupt epidermal differentiation. Multiple independent AEC TP63 mutants confirmed that introducing these mutants into postnatal regenerated epidermis is sufficient to disrupt epidermal differentiation. Profiling AEC model tissue showed repression of established transcriptional activators of epidermal differentiation, including HOPX (MIM 607275), GRHL3 (MIM 608317), KLF4 (MIM 602253), and PRDM1 (MIM 6034230). Also repressed was ZNF750 (MIM 610226), a recently characterized effector of epidermal differentiation that is also associated with impaired epidermal differentiation in a single extended family (MIM 610227).21,22 ZNF750 was found to be both TP63-dependent and bound by TP63 via chromatin immunoprecipitation sequencing (ChIP-Seq) studies in keratinocyte (KC) differentiation. ChIP analysis of AEC mutants in differentiating KCs showed that AEC mutants, like wild-type TP63, retained the ability to bind the ZNF750 promoter in spite of their repression of ZNF750 induction. Remarkably, enforcing ZNF750 expression in AEC model tissue significantly rescued HOPX, GRHL3, KLF4, and PRDM1 differentiation-activator expression and impaired epidermal differentiation. These findings indicate that ZNF750 repression by AEC TP63 mutants contributes to the impaired epidermal differentiation seen in AEC and suggest that introducing mutant genes responsible for monogenic disorders within regenerated human tissue may represent a useful approach to understanding disease pathogenesis.
Material and Methods
Human Subjects
Specific consent was obtained for use of the AEC tissue biopsy. All experiments adhered to institutional review board-approved protocols of Stanford University and the Barnes-Jewish Hospital of St. Louis, Washington University School of Medicine.
Cell Culture and Organotypic Culture of Human Skin
Primary human neonatal KCs were isolated from freshly discarded foreskin specimens. KCs were grown in KC serum-free medium (KSFM) (Gibco BRL) supplemented with epidermal growth factor and bovine pituitary extract. KC differentiation was induced via the addition of 1.2 mM calcium for 3 days at full confluence. For organotypic skin cultures, 5 × 105 cells were seeded onto devitalized human dermis and raised to the air/liquid interface for inducing KC stratification and differentiation, as described previously.23,24 Organotypic tissue experiments spanned a duration of 4 days. Puromycin drug selection (1 μg/ml) was maintained for sustaining delivered vectors throughout this time course; sustained stable wild-type TP63 and AEC TP63 mutant expression was confirmed throughout the organotypic tissue time course for all experiments via immunoblotting and quantitative PCR (qPCR).
Retroviral Constructs
cDNA of wild-type murine TP63-ΔNα and human ZNF750 was subcloned into the pBABE-puro retroviral vector via the BamHI and SalI sites. We used the murine cDNA sequence of TP63-ΔNα (RefSeq accession number AF075439.1) to prevent human-specific small interfering RNA (siRNA) (see Figure S1 available online) from targeting exogenously expressed TP63. The amino acid sequence of murine TP63-ΔNα was humanized via sequential QuikChange (Stratagene)-mediated mutagenesis. Substituted nucleotides were c.G820>A, c.A1085>C, c.C1558_1559delinsAG, c.G1582>A, and c.T1717>G, for generation of p.Ala274Thr, p.Gln362Pro, p.Pro520Ser, p.Gly528Ser, and p.Ser537Ala, respectively. AEC point mutants were then generated via QuikChange, with the humanized pBABE-puro-TP63-ΔNα used as a template. Substituted nucleotides were c.A1377>C, c.C1401>G, c.T1445>C, and c.G1499>C, for generation of AEC mutants p.Leu459Phe, p.Cys467Trp, p.Ile482Thr, and p.Arg500Pro, respectively. AEC amino acid residues are numbered according to the TP63-ΔNα sequence. Retrovirus was produced via Lipofectamine 2000 (Invitrogen)-mediated transfection of helper-free recombinant 293T-based retroviral packaging cells. KCs were infected via 1 hr spin at 32°C in the presence of 5 μg/ml of polybrene. Immediately after spin infection, cells were rinsed with PBS and returned to culture in standard KSFM. Two days post infection, KC media was supplemented with 1 μg/ml puromycin, and cells were allowed to grow for 1 week prior to experimental use.
RNAi
siRNA oligomers against the DNA binding domain (which targets all isoforms) of TP63 were designed and synthesized by Dharmacon: 5′-CGACAGTCTTGTACAATTT. siRNA oligomers were mixed with 5 × 106 primary human KCs, and the cells were then electroporated with a nucleofection kit (Amaxa, Gaithersburg, MD, USA) according to the manufacturer’s protocol. Dharmacon-scrambled siRNA oligomer, which is complementary to no known cellular RNA, was used as a control. In knockdown experiments, KCs were electroporated with the use of the T-007 high-viability neonatal-KCs setting with 1,500 picomoles of the indicated oligomer. For protein analysis, electroporated cells were cultured in KSFM for 3 days and then harvested for western blot analysis.
mRNA Expression Analysis
Total RNA from organotypic cultures or cultured cells was extracted with the RNeasy mini kit (QIAGEN) and quantified with NanoDrop. One μg of total RNA was reverse transcribed with the iScript RT-PCR kit from Bio-Rad. qPCR was performed with 2X Maxima SYBR (Fermentas) on the Mx3000P (Stratagene) thermocycler. Samples were run in triplicate and normalized to the ribosomal protein L32. See Supplemental Data for primer sequences used for qPCR (Table S1).
Protein Levels
For immunoblotting, protein was harvested from cells in lysis buffer, which consisted of 20 mM Tris (pH 7.5), 250 mM NaCL, 1 mM EDTA, 1% NP40, and 0.2% SDS supplemented with protease inhibitor (Complete Mini EDTA-free; Roche) and phosphatase inhibitor mixture II (Sigma-Aldrich, St. Louis). For each sample, 30 μg of protein was boiled in SDS for 5 min and then run in a 4%–12% SDS-PAGE gel. The protein was subsequently transferred to polyvinylidene fluoride membranes. Antibodies used for immunoblotting included mouse anti-TP63 (4A4) (Santa Cruz Biotechnology) and mouse anti-beta-actin (Sigma-Aldrich), and sheep anti-mouse immunoglobulin G (IgG) conjugated to horseradish peroxidase was purchased from Amersham Biosciences (Piscataway, NJ, USA). For immunofluorescence experiments, 7-μm-thick epidermal sections were fixed in ice-cold methanol for 15 min, followed by blocking in PBS with 10% horse serum for 30 min. Sections were incubated in primary antibodies for 1 hr. Primary antibodies included keratin 1 (Covance: PRB-149P, 1:2,000) and filaggrin (FLG) (PRB-417P, 1:1,000), with Alexa 555-conjugated goat anti-rabbit IgG (Molecular Probes, 1:1,000) used as secondary. Nuclear dye, Hoechst 33342, was used at 1:1,000 (Molecular Probes).
ChIP and ChIP-Seq Assay
The formaldehyde crosslinking and immunoprecipitation (IP) protocol was adapted from Wells and Farnham.25 Formaldehyde (16% solution methanol-free; Pierce) was added to KCs resuspended in 10 ml PBS at a final concentration of 0.5%. Pellets from approximately 1 × 106 cells were resuspended in 1 ml of swelling buffer (50mM Tris [pH 7.6], 1% NP-40, 10 mM KAc, 15 mM MgAc, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1X Protease Inhibitor Cocktail [PIC] without EDTA [Roche]). After 20 min incubation on ice, cells were Dounce homogenized in a B Dounce homogenizer. Nuclei were collected through centrifugation at 5,000 rpm, resuspended in 400 μl/1 × 106 cells of nuclei lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl [pH 8.0], 0.5 mM PMSF, and PIC), and incubated on ice for 30 min. Samples were sonicated with a Bioruptor-200 (Diagenode) on the “high” setting for eight cycles of 30-s-on and 30-s-off pulses, for an average length of 300 to 500 bp, and then microcentrifuged at 14,000 rpm. The chromatin solution was precleared with the addition of Staph A cells (prepared as described previously25 for 15 min at 4°C). Precleared chromatin from 1.5 × 106 cells was incubated with 2 μg of nonspecific rabbit IgG or anti-TP63 (Santa Cruz 4A4) at 4°C for 12–18 hr with rotation. IP, washing, and elution of immune complexes were carried out as described previously. Prior to the first wash, 10% of the supernatant from the rabbit IgG primary-antibody reaction for each sample was saved as total input chromatin and was processed with the eluted immunoprecipitates, beginning at the crosslink-reversal step. After addition of NaCl to 200 mM and 10 μg of RNase A, samples were incubated at 65°C overnight to reverse the crosslinks. Prior to DNA purification with QIAGEN PCR-purification columns, 500 μl of Buffer PB and 7.5 μl of 2M NaAc (pH 5.1) were added to the samples. Columns were eluted twice with 50 μl of Buffer EB and 2 μl used for all qPCRs. For ChIP-Seq experiments, the experimental conditions were modified as follows: Chromatin was sonicated for 24 cycles of 30-s-on and 30-s-off pulses for an average length of 150 to 300 bp. Approximately 7.5 × 106 cells were used per IP. Twenty μg of p63 (4A4) antibody was preconjugated onto Protein G Dynabeads (Invitrogen) per IP. The libraries were prepared per Illumina’s instructions. Libraries were sequenced on the Genome Analyzer IIx (Illumina), with read lengths of 36 bp. Raw reads were mapped to hg18 with Bowtie. ChIP signals were normalized to 10 million mapped reads for each condition. Peaks of each sample were called with MACS26 against input with a p value cutoff of 1 × 10−9, false-discovery-rate cutoff of 0.05, tags cutoff of 10, and fold-enrichment cutoff of 10. Normalized signal within 2 kb upstream and downstream of the summits of predicted peaks was extracted with a smooth window size of 50 bp. The control library was prepared identically, with the use of ∼50 ng of input DNA.
Gene Expression Profiling
Amplification and labeling of cDNA probes and hybridization to the Human Genome U133 Plus 2.0 microarray chip (Affymetrix) were performed by the Stanford Protein and Nucleic Acid Facility. Data analysis was performed with R. Each data set for an experiment was filtered for probes that had an expression value ≥ 100 in at least one of the samples along with a p value ≤ 0.05, based on SAM analysis. Pairwise comparisons between the RNAi-treated samples and the control samples were performed for discovery of probes that showed ≥ 1.8-fold expression change. Hierarchical clustering and heat-map generation were performed with Cluster and TreeView. p values indicating the significance of the overlap between various gene sets were calculated with Fisher’s exact test. Gene ontology term enrichment was performed with DAVID with the total set of genes on the appropriate microarray as the background. p values represent a Bonferroni-corrected modified Fisher’s exact test. Gene sets for epidermal regulators significantly associated with the AEC and TP63i signature were acquired from the Gene Expression Omnibus (GEO) or the European Bioinformatics Institute (accession numbers: ZNF750 and KLF4, GSE32685; IRF6, GSE5800; GRHL3, GSE7381; CEBPa and CEBPb, E-MEXP-1719; EFNA, GSE26521; Notch1, GSE23782) or from a supplemental table for ETS1. Gene expression data was processed in R with the use of SAM with a p value ≤ 0.05 cutoff for differentially expressed genes with ≥ 2-fold expression change. Gene-set enrichment analysis (GSEA) was conducted with Genomica software, with p value ≤ 0.05 for significant enrichment.
Results
The impaired epidermal differentiation observed in AEC syndrome might result both from the persistence of a deficit programmed during development and from TP63-mutant-mediated disruption of differentiation in developmentally mature tissue. For examination of the latter role of AEC mutant TP63, cDNA encoding TP63-ΔNα-WT or AEC mutants found in humans, including TP63-ΔNα-p.Cys467Trp, TP63-ΔNα-p.Leu459Phe, TP63-ΔNα-p.Ile482Thr, and TP63-ΔNα-p.Arg500Pro, was introduced within regenerated organotypic human epidermis. Importantly, for accurate modeling of the heterozygous TP63 state of AEC human tissue, rigorous controls were used for verifying stable physiological levels of TP63 mutants in the context of normal endogenous TP63 for all studies (see Material and Methods; Figures S1A–S1C). Resulting cells were then used to regenerate epidermal tissue on intact human dermis in organotypic culture via an approach demonstrated to recapitulate much of the morphology and gene expression of normal human skin.21,23,24,27 In contrast to controls, each of the AEC TP63 mutants profoundly impaired the levels of canonical epidermal differentiation markers, including KRT1 (MIM 139350) and FLG (MIM 135940) (Figures 1A and B), as well as SPRR1A (MIM 182265), SPRR3 (MIM 182271), TGM1 (MIM 606776), DSC1 (MIM 125643), LOR (MIM 152445), LCE3D (MIM 612616), and LCE1E (MIM 612607) (Figures S2A and S2B). TUNEL staining indicated that this disrupted differentiation-gene expression was not accompanied by apoptosis. Diminished levels of differentiation proteins, such as KRT1 and FLG, in AEC organotypic tissue agree with a previous study of AEC skin,28 which we also independently reverified using AEC skin biopsy specimens (Figures 1C and 1D). Whereas control tissue demonstrated proliferation that was appropriately localized to the basal layer, as previously noted in organotypic epidermis, wherein proliferation and differentiation faithfully recapitulate their normal spatial localization by 1 week time points of full regeneration,23 AEC tissue exhibited abnormal suprabasal Ki67 staining, consistent with observations seen in AEC skin28 (Figure S3). Clonogenic assays did not reveal defective proliferation of AEC mutant KCs, as recently observed in a knockin mouse model of AEC syndrome,29 indicating that this phenotype may arise from a developmental defect caused by mutant TP63 (Figure S4). The disrupted epidermal differentiation seen in regenerated human tissue upon introduction of AEC mutants suggests that these mutants can also impair this process in developmentally mature postnatal cells and also generate a human epidermal tissue model in which to study the differentiation defects observed in AEC.
Figure 1.
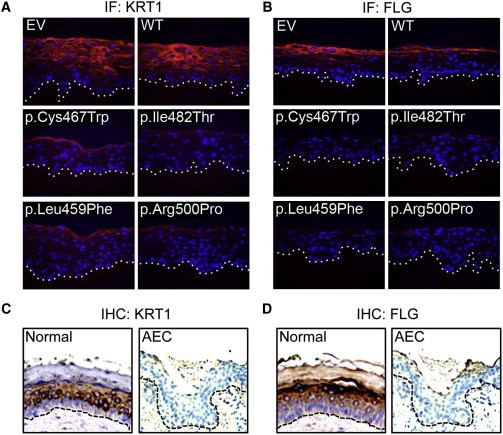
Disrupted Epidermal Differentiation by AEC TP63 Mutants in Regenerated Organotypic Human Epidermal Tissue
Differentiation markers (A) KRT1 and (B) FLG in control (EV), TP63-ΔNp63α-WT (WT), TP63-ΔNp63α-Cys467Trp, TP63-ΔNp63α-Leu459Phe, TP63-ΔNp63α-Ile482Thr, and TP63-ΔNp63α-Arg500Pro AEC mutant-expressing regenerated organotypic human epidermal tissue (orange = differentiation protein, blue = Hoechst 33342 nuclei, dotted line denotes basement membrane). Differentiation markers (C) KRT1 and (D) FLG in normal skin and in AEC lesional skin from a 1-day-old AEC-affected individual with a confirmed p.Phe458Ser alteration.
The dominant effects on epidermal differentiation of AEC-associated mutations in the TP63 SAM domain raised the question as to whether these effects are due to lost function of the entire TP63α C-terminal region, or whether they require retention of mutated C-terminal TP63 residues. For testing of this, cDNA encoding TP63-ΔNα-WT, TP63-ΔNα-p.Arg500Pro, or TP63-ΔNδ, which lacks the entire C-terminal SAM domain mutated in AEC and is normally not present at significant levels in epidermis,30 was introduced within regenerated organotypic human epidermis. Physiologic levels of TP63 constructs were confirmed by specific silencing of endogenous TP63 (Figure 2A). In contrast to AEC p.Arg500Pro mutant tissue, regenerated epidermal tissue harboring TP63-ΔNδ in the context of normal endogenous TP63 engaged differentiation-protein production normally (Figure 2B), suggesting that retention of mutated TP63 SAM-domain-containing C-terminal residues are important for the differentiation-suppressive effects of AEC mutant TP63.
Figure 2.
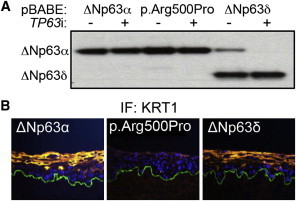
Retention of Mutated SAM Domain in TP63 AEC Mutants Is Required for Inhibition of Epidermal Differentiation
(A) Immunoblot analysis of TP63-ΔNp63α levels in human KCs transduced with TP63-ΔNp63α-WT, TP63-ΔNp63α-Arg500Pro, and TP63-ΔNp63δ. KCs were treated with control or TP63 targeting siRNA, as indicated.
(B) KRT1 in TP63-ΔNp63α-WT, TP63-ΔNp63α-Arg500Pro, and TP63-ΔNp63δ expressing regenerated organotypic human epidermal tissue (orange = differentiation protein, blue = Hoechst 33342, green = basement membrane).
To characterize the impaired epidermal differentiation observed in AEC model tissue further, we performed transcriptional profiling on AEC mutant regenerated human organotypic epidermis. This revealed 1,185 (533 repressed; 652 induced) and 864 (499 repressed; 365 induced) differentially expressed genes in the AEC-p.Ile482Thr and AEC-p.Arg500Pro mutant organotypic tissues, respectively (Tables S2 and S3). Comparison of these gene sets showed a significant (p < 10−250) overlap of 651 genes between these mutants (Figure 3A and Table S4). Repressed genes in the shared gene set were significantly enriched for gene ontology terms associated with epidermal development and differentiation, whereas the induced genes showed no significant enrichment for any gene ontology terms (Figure 3B). AEC TP63 mutants thus broadly block induction of genes involved in epidermal differentiation.
Figure 3.
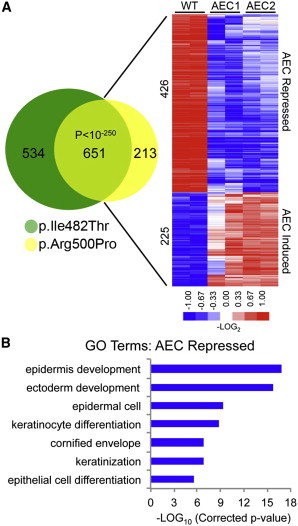
Transcriptional Profiling of AEC Model Tissue
(A) Venn diagram illustrating overlap between changes identified in p.Ile482Thr (AEC1) and p.Arg500Pro (AEC2) AEC mutant versus wild-type (WT) control organotypic tissue. Heat map of the 651 genes shared between the two profiles (p value < 10−250, Fisher’s exact test); blue (repressed) and red (induced), log2-based scale.
(B) Gene ontology (GO) analysis of the p.Ile482Thr and p.Arg500Pro gene-profile overlap (p values represent a Bonferroni-corrected EASE score).
TP63 is required for normal epidermal differentiation.3,4,8,28 To assess the degree to which AEC TP63 mutants may prevent normal induction of TP63-dependent differentiation genes, we compared AEC TP63 mutant impacts on global mRNA expression during differentiation with those of TP63 depletion (TP63i). Comparison of the TP63i (Figures S5A and S5B and Table S5) and AEC gene sets revealed a highly significant (p < 10−148) overlap of 206 genes (Figure 4A and Table S6), with the repressed genes in this narrowed set being enriched for the same gene ontology terms identified in the p.Ile482Thr and p.Arg500Pro AEC set (Figure 3B). AEC TP63 mutants therefore block induction of a number of genes that are dependent on wild-type TP63 for their normal induction during differentiation.
Figure 4.
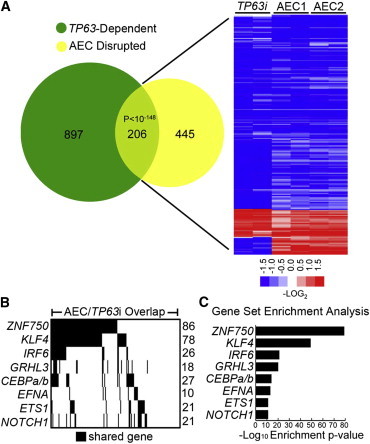
Identification of TP63-Dependent Candidate Regulators Responsible for the AEC Phenotype
(A) Venn diagram illustrating overlap between changes identified in AEC cultures and KCs treated with siRNA targeting total TP63. Heat map of the 206 genes shared between the two profiles (p value < 10−148, Fisher’s exact test); blue (repressed) and red (induced), log2-based scale.
(B) Barcode plot showing AEC and TP63i gene-number overlap with indicated epidermal gene set.
(C) GSEA enrichment values for top overlapping gene sets.
To gain insight into how AEC TP63 blocks differentiation, we performed GSEA on the set of genes that are both TP63-dependent during normal differentiation and inhibited by AEC TP63 mutants, on the basis of the possibility that AEC TP63 mutants might act by inhibiting the normal TP63-dependent activation of specific differentiation-enabling factors. GSEA comparison of published gene sets for regulators of epidermal homeostasis and differentiation identified the most significant overlap with the epidermal differentiation regulators ZNF750,31 KLF4,32 and GRHL333 (Figures 4B and 4C; Tables S7 and S8), each of which was repressed by AEC mutant TP63 (Table S4). Interestingly, IRF634 (MIM 607199) and CEBPα and CEBPβ35 (MIMs 116897 and 189965) transcriptional targets also showed significant overlap with the AEC and TP63i gene set. Neither IRF6 nor CEBPα/β levels were repressed by AEC mutant TP63 (Table S4), suggesting that their full transcriptional programs may require coordination with the TP63-regulated transcriptional network.
To investigate the relationship between mutant and wild-type TP63 and ZNF750, we performed TP63 ChIP-Seq in proliferating versus differentiating human KCs. This identified 6,050 total TP63-bound regions (Table S9). Analysis of the read-signal intensities within these regions between proliferating and differentiating KCs revealed stable, unchanged TP63 binding in undifferentiated and differentiated cells at > 94.4% of these binding sites (Figure 5A), including two binding peaks identified near the transcription start site (TSS) of ZNF750 (Figure 5B), both of which contain canonical TP63 binding sequences.36 For confirmation of these results and examination of whether AEC TP63 mutants retain the capacity to bind TP63 DNA motifs at the ZNF750 locus, TP63 ChIP was performed on human KCs depleted of endogenous TP63 by RNAi and with either wild-type TP63 or AEC TP63 mutants introduced at near-endogenous levels (Figure 5C). In agreement with ChIP-Seq data, TP63 ChIP in control cells showed similar TP63 binding near the ZNF750 TSS in both progenitor and differentiated KCs. Additionally, AEC TP63 mutants bound ZNF750 target sequences at similar levels to wild-type TP63 (Figure 5D). ZNF750 is thus a TP63-bound and -regulated gene whose induction during epidermal differentiation is inhibited by AEC TP63 mutants, which can bind the ZNF750 locus comparably to wild-type TP63.
Figure 5.
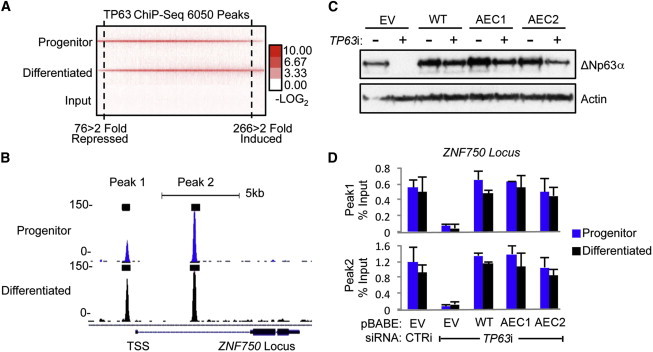
TP63 ChIP-Seq Identifies TP63 Binding to the ZNF750 Locus during Epidermal Differentiation
(A) Heat map showing stable p63 genomic binding in undifferentiated progenitor-containing KC populations and in differentiated KCs.
(B) TP63 binding near the TSS of the ZNF750 locus in undifferentiated progenitor-containing KC populations and in differentiated KCs.
(C) Immunoblot analysis of TP63-ΔNp63α in KCs infected with pBABE-Empty Vector (EV), pBABE-TP63-ΔNp63α-WT (WT), pBABE-p.Ile482Thr (AEC1), and pBABE-p.Arg500Pro (AEC2) AEC TP63 mutants. Each population of KCs was treated with control or TP63 targeting siRNA, as indicated.
(D) ChIP analysis comparing binding capacity of wild-type TP63 versus AEC TP63 mutants at the ZNF750 locus in undifferentiated and differentiated KCs treated with TP63i (specific for endogenous TP63 mRNA). Error bars are ± SD between qPCR triplicate samples.
The tremendous overlap of the AEC-repressed and ZNF750-dependent gene sets raised the possibility that ZNF750 may be an important pathologic target of AEC TP63 mutants that resides upstream of TP63-dependent transcriptional targets. Consistent with this, restoring ZNF750 expression to AEC TP63 mutant epidermal tissue (Figure 6A) rescued expression of the majority of TP63 transcriptional targets, including GRHL3, KLF4, and PRDM1, along with epidermal differentiation-gene expression (Figures 6B–6D). ZNF750 is therefore bound and repressed by AEC TP63 mutants, and restoring ZNF750 expression in human AEC model tissue can rescue defects in differentiation caused by AEC mutant TP63.
Figure 6.

Rescue of AEC Differentiation Defects by ZNF750
Human epidermal tissue was regenerated from KCs infected with either pBABE-Empty Vector (EV) or pBABE-p.Arg500Pro AEC mutant TP63, then with either pBABE-EV or pBABE-ZNF750. Quantitative mRNA levels of (A) ZNF750 and (B) epidermal differentiation markers and regulators in the indicated day 4 organotypic tissues. Error bars are ± SD between triplicate samples. Differentiation proteins KRT1 (C) and FLG (D) in the indicated organotypic tissues.
Discussion
Here, we generated a human epidermal tissue model of AEC and then used this model to identify ZNF750 as an AEC TP63 mutant target capable of rescuing differentiation defects in this setting. The modeled recapitulation by AEC mutant TP63 of the impaired differentiation seen in AEC human skin demonstrates that repressed epidermal differentiation seen in AEC does not solely arise from defects occurring during epidermal-progenitor development, but also from dysfunction within developmentally mature cells that can be directly induced by AEC mutant TP63. At the same time, introduction of AEC mutant TP63 into developmentally mature KCs did not result in diminished clonogenicity (Figure S4), as seen in a recent mouse knockin model of AEC,29 indicating that additional AEC phenotypes arise from AEC mutant TP63 disruption of epidermal development. Differentiation-suppressive effects of AEC mutant TP63 appeared to require retention of mutant C-terminal TP63-ΔNα residues, because enforced expression of the normally nonabundant TP63-ΔNδ isoform, which entirely lacks the C-terminal sequences encompassing the SAM domain, had no such effects. Genome-wide ChIP-Seq analysis of TP63 led to the observation that the vast majority of wild-type TP63 DNA binding peaks are stable between proliferating and differentiating KCs. This is consistent with the retained levels of TP63 in both undifferentiated basal as well as differentiating suprabasal KCs within human epidermis.37 These data also suggest that genomic mobilization of TP63 may not significantly contribute to TP63 gene regulatory control of epidermal differentiation. The retention of AEC mutant TP63 DNA binding at canonical TP63 DNA binding sites located near the ZNF750 TSS suggests that dominant-negative AEC TP63 mutant impacts on differentiation do not require disruption of TP63 DNA binding capacity.
Prior work studying AEC skin9,38 demonstrated normal suprabasal KRT10 levels in several AEC individuals, in contrast to other published work,28 which showed diminished-to-absent KRT1 in lesional AEC tissue, which is in agreement with the present study. Several factors may account for this discrepancy. First is the issue of lesional versus nonlesional AEC skin. The skin lesions in AEC generally do not affect the majority of the skin surface and have been observed to affect neonates most severely and then improve over time, indicating that AEC mutant TP63-associated skin defects undergo eventual compensation. The findings of Koster et al.28 are consistent with this, in that impaired differentiation was only observed in the lesional, but not the nonlesional, AEC skin of the same individual. In spite of this, however, it is important to note that the study by McGrath et al.9 did carefully demonstrate abnormal distribution of another differentiation marker, namely FLG, providing further support for abnormal epidermal differentiation in AEC. Moreover, the work by Browne et al.38 demonstrated that AEC mutant TP63 failed to normally transactivate epidermal differentiation promoters, further consistent with an adverse differentiation impact of AEC mutant TP63. In the present study, lesional skin from an AEC-affected newborn recapitulated Koster et al.’s28 findings of impaired KRT1 levels, as well as also showing diminished FLG. It is also possible that the differences in these bodies of work could be due to characteristics of individual AEC mutations. Differentiation-protein levels were studied in only three individuals total in the two papers by McGrath et al.9 and Browne et al.38 noted above, with alterations p.Gly534Trp, p.Thr533Pro, and p.Gly561Asp, which were not analyzed in the current work. Thus, this possibility cannot be excluded; however, the noted strikingly different differentiation picture seen between lesional and nonlesional skin in the same AEC individual by Koster et al.28 and the FLG abnormalities observed even in what appears to have been nonlesional skin by McGrath et al.9 support a primary differentiation impact that undergoes compensation over time, consistent with the well-observed clinical skin improvements in individuals with AEC postinfancy.
The finding that restored expression of ZNF750 rescued defective differentiation-gene expression in AEC model tissue supports a model in which AEC mutants block proximal differentiation mediators required for the activation of the epidermal differentiation program. In this regard, transcriptional profiling of model AEC tissue led to the identification of a set of repressed transcriptional activators of epidermal differentiation regulators, which included HOPX, GRHL3, KLF4, PRDM1, and ZNF750. These regulators were confirmed to be both normally TP63-dependent and also suppressed by AEC TP63 mutants (Table S6). Remarkably, GSEA revealed that the recently characterized ZNF750-KLF4 differentiation axis31 accounted for a major portion of the epidermal differentiation repressed by AEC mutant TP63. In AEC model tissue, expression of ZNF750 rescued both the expression of these transcription factors as well as epidermal differentiation-gene expression. This suggests that ZNF750 repression by AEC TP63 mutants may be a proximal event in the pathogenesis of disrupted differentiation in AEC and supports the rationale for potential genetic and other therapeutic efforts to upregulate epidermal ZNF750 in AEC.
Acknowledgments
We thank L. Morcom for expert administrative assistance. This work is supported by the U.S. Department of Veterans Affairs Office of Research and Development and by the National Institutes of Health/National Institute of Arthritis and Musculoskeletal and Skin Diseases R01 AR45192 to P.A.K.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Human Protein Atlas, http://www.proteinatlas.org
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Accession Numbers
Microarray and ChIP-Seq data have been deposited with the accession numbers GSE33495 and GSE33572.
References
- 1.Mills A.A., Zheng B., Wang X.J., Vogel H., Roop D.R., Bradley A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature. 1999;398:708–713. doi: 10.1038/19531. [DOI] [PubMed] [Google Scholar]
- 2.Yang A., Schweitzer R., Sun D., Kaghad M., Walker N., Bronson R.T., Tabin C., Sharpe A., Caput D., Crum C., McKeon F. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature. 1999;398:714–718. doi: 10.1038/19539. [DOI] [PubMed] [Google Scholar]
- 3.Koster M.I., Kim S., Mills A.A., DeMayo F.J., Roop D.R. p63 is the molecular switch for initiation of an epithelial stratification program. Genes Dev. 2004;18:126–131. doi: 10.1101/gad.1165104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Truong A.B., Kretz M., Ridky T.W., Kimmel R., Khavari P.A. p63 regulates proliferation and differentiation of developmentally mature keratinocytes. Genes Dev. 2006;20:3185–3197. doi: 10.1101/gad.1463206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Senoo M., Pinto F., Crum C.P., McKeon F. p63 Is essential for the proliferative potential of stem cells in stratified epithelia. Cell. 2007;129:523–536. doi: 10.1016/j.cell.2007.02.045. [DOI] [PubMed] [Google Scholar]
- 6.Yang A., Kaghad M., Wang Y., Gillett E., Fleming M.D., Dötsch V., Andrews N.C., Caput D., McKeon F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell. 1998;2:305–316. doi: 10.1016/s1097-2765(00)80275-0. [DOI] [PubMed] [Google Scholar]
- 7.Su X., Paris M., Gi Y.J., Tsai K.Y., Cho M.S., Lin Y.L., Biernaskie J.A., Sinha S., Prives C., Pevny L.H. TAp63 prevents premature aging by promoting adult stem cell maintenance. Cell Stem Cell. 2009;5:64–75. doi: 10.1016/j.stem.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su X., Cho M.S., Gi Y.J., Ayanga B.A., Sherr C.J., Flores E.R. Rescue of key features of the p63-null epithelial phenotype by inactivation of Ink4a and Arf. EMBO J. 2009;28:1904–1915. doi: 10.1038/emboj.2009.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McGrath J.A., Duijf P.H., Doetsch V., Irvine A.D., de Waal R., Vanmolkot K.R., Wessagowit V., Kelly A., Atherton D.J., Griffiths W.A. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum. Mol. Genet. 2001;10:221–229. doi: 10.1093/hmg/10.3.221. [DOI] [PubMed] [Google Scholar]
- 10.Celli J., Duijf P., Hamel B.C., Bamshad M., Kramer B., Smits A.P., Newbury-Ecob R., Hennekam R.C., Van Buggenhout G., van Haeringen A. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell. 1999;99:143–153. doi: 10.1016/s0092-8674(00)81646-3. [DOI] [PubMed] [Google Scholar]
- 11.Duijf P.H., Vanmolkot K.R., Propping P., Friedl W., Krieger E., McKeon F., Dötsch V., Brunner H.G., van Bokhoven H. Gain-of-function mutation in ADULT syndrome reveals the presence of a second transactivation domain in p63. Hum. Mol. Genet. 2002;11:799–804. doi: 10.1093/hmg/11.7.799. [DOI] [PubMed] [Google Scholar]
- 12.Bougeard G., Hadj-Rabia S., Faivre L., Sarafan-Vasseur N., Frébourg T. The Rapp-Hodgkin syndrome results from mutations of the TP63 gene. Eur. J. Hum. Genet. 2003;11:700–704. doi: 10.1038/sj.ejhg.5201004. [DOI] [PubMed] [Google Scholar]
- 13.Kantaputra P.N., Hamada T., Kumchai T., McGrath J.A. Heterozygous mutation in the SAM domain of p63 underlies Rapp-Hodgkin ectodermal dysplasia. J. Dent. Res. 2003;82:433–437. doi: 10.1177/154405910308200606. [DOI] [PubMed] [Google Scholar]
- 14.Ianakiev P., Kilpatrick M.W., Toudjarska I., Basel D., Beighton P., Tsipouras P. Split-hand/split-foot malformation is caused by mutations in the p63 gene on 3q27. Am. J. Hum. Genet. 2000;67:59–66. doi: 10.1086/302972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brunner H.G., Hamel B.C., Van Bokhoven H. The p63 gene in EEC and other syndromes. J. Med. Genet. 2002;39:377–381. doi: 10.1136/jmg.39.6.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Julapalli M.R., Scher R.K., Sybert V.P., Siegfried E.C., Bree A.F. Dermatologic findings of ankyloblepharon-ectodermal defects-cleft lip/palate (AEC) syndrome. Am. J. Med. Genet. A. 2009;149A:1900–1906. doi: 10.1002/ajmg.a.32797. [DOI] [PubMed] [Google Scholar]
- 17.Chi S.W., Ayed A., Arrowsmith C.H. Solution structure of a conserved C-terminal domain of p73 with structural homology to the SAM domain. EMBO J. 1999;18:4438–4445. doi: 10.1093/emboj/18.16.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marinoni J.C., Stevenson R.E., Evans J.P., Geshuri D., Phelan M.C., Schwartz C.E. Split foot and developmental retardation associated with a deletion of three microsatellite markers in 7q21.2-q22.1. Clin. Genet. 1995;47:90–95. doi: 10.1111/j.1399-0004.1995.tb03930.x. [DOI] [PubMed] [Google Scholar]
- 19.Crackower M.A., Scherer S.W., Rommens J.M., Hui C.C., Poorkaj P., Soder S., Cobben J.M., Hudgins L., Evans J.P., Tsui L.C. Characterization of the split hand/split foot malformation locus SHFM1 at 7q21.3-q22.1 and analysis of a candidate gene for its expression during limb development. Hum. Mol. Genet. 1996;5:571–579. doi: 10.1093/hmg/5.5.571. [DOI] [PubMed] [Google Scholar]
- 20.Lo Iacono N., Mantero S., Chiarelli A., Garcia E., Mills A.A., Morasso M.I., Costanzo A., Levi G., Guerrini L., Merlo G.R. Regulation of Dlx5 and Dlx6 gene expression by p63 is involved in EEC and SHFM congenital limb defects. Development. 2008;135:1377–1388. doi: 10.1242/dev.011759. [DOI] [PubMed] [Google Scholar]
- 21.Birnbaum R.Y., Zvulunov A., Hallel-Halevy D., Cagnano E., Finer G., Ofir R., Geiger D., Silberstein E., Feferman Y., Birk O.S. Seborrhea-like dermatitis with psoriasiform elements caused by a mutation in ZNF750, encoding a putative C2H2 zinc finger protein. Nat. Genet. 2006;38:749–751. doi: 10.1038/ng1813. [DOI] [PubMed] [Google Scholar]
- 22.Yang C.F., Hwu W.L., Yang L.C., Chung W.H., Chien Y.H., Hung C.F., Chen H.C., Tsai P.J., Fann C.S., Liao F., Chen Y.T. A promoter sequence variant of ZNF750 is linked with familial psoriasis. J. Invest. Dermatol. 2008;128:1662–1668. doi: 10.1038/jid.2008.1. [DOI] [PubMed] [Google Scholar]
- 23.Ridky T.W., Chow J.M., Wong D.J., Khavari P.A. Invasive three-dimensional organotypic neoplasia from multiple normal human epithelia. Nat. Med. 2010;16:1450–1455. doi: 10.1038/nm.2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sen G.L., Reuter J.A., Webster D.E., Zhu L., Khavari P.A. DNMT1 maintains progenitor function in self-renewing somatic tissue. Nature. 2010;463:563–567. doi: 10.1038/nature08683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wells J., Farnham P.J. Characterizing transcription factor binding sites using formaldehyde crosslinking and immunoprecipitation. Methods. 2002;26:48–56. doi: 10.1016/S1046-2023(02)00007-5. [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sen G.L., Webster D.E., Barragan D.I., Chang H.Y., Khavari P.A. Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes Dev. 2008;22:1865–1870. doi: 10.1101/gad.1673508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koster M.I., Marinari B., Payne A.S., Kantaputra P.N., Costanzo A., Roop D.R. DeltaNp63 knockdown mice: A mouse model for AEC syndrome. Am. J. Med. Genet. A. 2009;149A:1942–1947. doi: 10.1002/ajmg.a.32794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferone G., Thomason H.A., Antonini D., De Rosa L., Hu B., Gemei M., Zhou H., Ambrosio R., Rice D.P., Acampora D. Mutant p63 causes defective expansion of ectodermal progenitor cells and impaired FGF signalling in AEC syndrome. EMBO Mol. Med. 2012;4:192–205. doi: 10.1002/emmm.201100199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mangiulli M., Valletti A., Caratozzolo M.F., Tullo A., Sbisà E., Pesole G., D’Erchia A.M. Identification and functional characterization of two new transcriptional variants of the human p63 gene. Nucleic Acids Res. 2009;37:6092–6104. doi: 10.1093/nar/gkp674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sen G.L., Boxer L.D., Webster D.E., Bussat R.T., Qu K., Zarnegar B.J., Johnston D., Siprashvili Z., Khavari P.A. ZNF750 is a p63 target gene that induces KLF4 to drive terminal epidermal differentiation. Dev. Cell. 2012;22:669–677. doi: 10.1016/j.devcel.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Segre J.A., Bauer C., Fuchs E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat. Genet. 1999;22:356–360. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 33.Ting S.B., Caddy J., Hislop N., Wilanowski T., Auden A., Zhao L.L., Ellis S., Kaur P., Uchida Y., Holleran W.M. A homolog of Drosophila grainy head is essential for epidermal integrity in mice. Science. 2005;308:411–413. doi: 10.1126/science.1107511. [DOI] [PubMed] [Google Scholar]
- 34.Ingraham C.R., Kinoshita A., Kondo S., Yang B., Sajan S., Trout K.J., Malik M.I., Dunnwald M., Goudy S.L., Lovett M. Abnormal skin, limb and craniofacial morphogenesis in mice deficient for interferon regulatory factor 6 (Irf6) Nat. Genet. 2006;38:1335–1340. doi: 10.1083/ng1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lopez R.G., Garcia-Silva S., Moore S.J., Bereshchenko O., Martinez-Cruz A.B., Ermakova O., Kurz E., Paramio J.M., Nerlov C. C/EBPalpha and beta couple interfollicular keratinocyte proliferation arrest to commitment and terminal differentiation. Nat. Cell Biol. 2009;11:1181–1190. doi: 10.1038/ncb1960. [DOI] [PubMed] [Google Scholar]
- 36.Yang A., Zhu Z., Kapranov P., McKeon F., Church G.M., Gingeras T.R., Struhl K. Relationships between p63 binding, DNA sequence, transcription activity, and biological function in human cells. Mol. Cell. 2006;24:593–602. doi: 10.1016/j.molcel.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 37.Uhlen M., Oksvold P., Fagerberg L., Lundberg E., Jonasson K., Forsberg M., Zwahlen M., Kampf C., Wester K., Hober S. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 2010;28:1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 38.Browne G., Cipollone R., Lena A.M., Serra V., Zhou H., van Bokhoven H., Dötsch V., Merico D., Mantovani R., Terrinoni A. Differential altered stability and transcriptional activity of ΔNp63 mutants in distinct ectodermal dysplasias. J. Cell Sci. 2011;124:2200–2207. doi: 10.1242/jcs.079327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.