

Abstract
To date, the widely used genome-wide association studies (GWASs) of the human genome have reported thousands of variants that are significantly associated with various human traits. However, in the vast majority of these cases, the causal variants responsible for the observed associations remain unknown. In order to facilitate the identification of causal variants, we designed a simple computational method called the “preferential linkage disequilibrium (LD)” approach, which follows the variants discovered by GWASs to pinpoint the causal variants, even if they are rare compared with the discovery variants. The approach is based on the hypothesis that the GWAS-discovered variant is better at tagging the causal variants than are most other variants evaluated in the original GWAS. Applying the preferential LD approach to the GWAS signals of five human traits for which the causal variants are already known, we successfully placed the known causal variants among the top ten candidates in the majority of these cases. Application of this method to additional GWASs, including those of hepatitis C virus treatment response, plasma levels of clotting factors, and late-onset Alzheimer disease, has led to the identification of a number of promising candidate causal variants. This method represents a useful tool for delineating causal variants by bringing together GWAS signals and the rapidly accumulating variant data from next-generation sequencing.
Introduction
After the first wave of genome-wide association studies (GWASs), thousands of common variants associated with hundreds of human traits have been identified.1 Although these findings shed light on the genetic architectures of human traits, the variants reaching genome-wide significance for any particular phenotype cumulatively explain only a small portion of the phenotypic variation.2 Moreover, in most cases the variants identified by GWASs are proxies of the causal variants that still remain to be discovered.3 Under the “common disease, common variant” hypothesis, one frequently used approach for causal-variant discovery is to look for the variants in the genomic region showing strong linkage disequilibrium (LD) (e.g., r2 > 0.8) with the variant discovered by the GWAS. However, this approach will not work well when the allelic frequencies of the causal variants are below the frequencies of the common variants used for describing “blocks” of LD. For example, the well-documented causal variants of Crohn disease (MIM 266600) in NOD2 (MIM 605956)4 and the causal variants of anemia in individuals treated for chronic hepatitis C in ITPA (MIM 147520)5 are in both cases considerably more rare than the disease-associated variants identified by GWASs. As a result, their LD with the GWAS-discovered variants is, at best, moderate (r2 < 0.5). In fact, there are a number of examples where the signals identified in GWASs are caused or contributed by variants with substantially lower allele frequencies than the GWAS-discovered variants4–8 (Table 1). These experiences suggest that it would be valuable to establish efficient algorithms for systematically searching for variants that contribute to GWAS signals but are substantially rarer than the discovery variants from GWASs. This effort should be viewed as complementary to widely performed discovery efforts that assume that the causal variant is similar in frequency to the GWAS-discovered variant12–18 and as an addition to the recently increasing screens for rare causal variants.6–8,19–23 We also note that the effort to make effective use of GWAS signals to look for causal variants makes no assumptions or claims about the collective importance of synthetic associations to GWAS signals.24,25 Rather, we recognize that some GWAS signals will be synthetic, and for this reason, discovery strategies well suited to this possibility are needed.26 It is also worth noting that even in cases where a given genomic region carries both common and rare causal variants, the approach described here might be especially helpful for identifying any rare causal variants that contribute to the original GWAS signal. In theory, our approach can also be applied to the situation where the causal variant has a similar frequency to the GWAS-discovered variant. In this case, however, we anticipate little benefit from our approach because the fact that variants in the same frequency range and their LD properties with the discovery variant will mostly already be known makes it possible to directly consider all variants in high LD with the discovery variant.
Table 1.
Variants Responsible for the Associations Detected by GWASs
| Phenotype | Gene |
Causal Variants |
GWAS Variants |
References | ||
|---|---|---|---|---|---|---|
| Name | MAF | Name | MAF | |||
| Crohn disease | NOD2 | rs2066844 | 4.1% | rs17221417 | 28.7% | Wang et al.4 |
| rs2066845 | 1.5% | |||||
| rs2066847 | 1.9% | |||||
| Ribavirin-induced hemolytic anemia | ITPA | rs1127354 | 7.6% | rs6051702 | 19.4% | Fellay et al.5 |
| rs7270101 | 12.3% | |||||
| Therapeutic warfarin dose | CYP2C9 | rs1799853 | 11% | rs4917639 | 18% | Takeuchi et al.9 and Wadelius et al.10 |
| rs1057910 | 7% | |||||
| Bladder cancer | UGT1A6 | rs17863783 | 2.5% | rs11892031 | 8.5% | Tang et al.11 |
| Low-density lipoprotein cholesterol | PCSK9 | rs11591147 | 3.7% | rs11206510 | 24.3% | Sanna et al.8 |
| Sick sinus syndrome | MYH6 | c.2161C>Ta | 0.38% | rs28730774 | 1% | Holm et al.6 |
| Ovarian cancer | BRIP1 | c.2040_2041insTTb | 0.41% | rs34289250 | 0.89% | Rafnar et al.7 |
The following abbreviations are used: GWAS, genome-wide association study; and MAF, minor allele frequency.
RefSeq NM_002471.3; reference genome build 36.
RefSeq NM_032043.2; reference genome build 36.
To prioritize candidate causal variants in genomic regions surrounding GWAS signals, especially when the LD between the causal variants and the GWAS-discovered variant is relatively weak, we designed a computational method called the “preferential LD” approach. This approach is based upon the idea that when the allelic frequency of a causal variant is lower than that of the GWAS-discovered variant, the LD between them, although relatively weak, will be larger than the LD values between the causal variant and most other variants interrogated in the GWAS. Thus, instead of simply looking for any candidate causal variants in high LD with the GWAS-discovered variant, as is typical, we focus on those variants that show the strongest preferential LD with the GWAS-discovered variant, regardless of the absolute magnitude of the LD. We calculated the LD values by using genotypes from 479 individuals of European ancestry; these genotypes included a combination of those that were from whole-genome sequencing, whole-exome sequencing, chip genotyping, and imputation. Starting with a variant discovered by the GWAS (this variant is called here the “discovery” variant), we first applied our approach to several examples where the causal variants are known. All the cases we considered had the property that the identified causal variants were rarer than the discovery variant. Our approach successfully identified the known causal variants of Crohn disease, hemolytic anemia following treatment for hepatitis C virus (HCV), therapeutic warfarin dose (MIM 122700), bladder cancer (MIM 109800), and hearing loss. This proof of concept strongly suggests that our approach will succeed in identifying unknown cases in which the causal variants are rarer than discovery ones, regardless of how often they might occur overall in GWAS signals. We applied this approach to 33 independent GWAS discovery variants across three different traits and report the leading candidate causal variants from the analysis. Included in this list of possible causal variants are a number of suggestive candidates that might contribute to the identified GWAS signals. The results and scripts of the preferential LD approach are available at our website (Web Resources).
Material and Methods
Study Subjects
All samples collected at Duke were approved by the local institutional review board (IRB) so they could be used as controls. All samples from outside institutions were received in a deidentified state. All deidentified samples were received under a Duke IRB exemption and were therefore classified as nonhuman subjects.
Sequencing
The genomic DNA of 75 individuals was directly sequenced with the Illumina Genome Analyzer IIx or the HiSeq 2000, whereas the genomic DNA of the other 282 and 122 individuals was captured on the Agilent SureSelect Human All Exon 37 Mb Kit and 50 Mb Kit, respectively, before Illumina sequencing. The average coverage of whole-genome- and whole-exome-sequenced samples was 34× and 78×, respectively. The sequence reads were aligned to the reference genome (NCBI build 36, release 50) with the Burrows-Wheeler Aligner.27 SAMtools28 was used for calling genotypes and identifying single-nucleotide variants (SNVs). For quality control, SNVs were required to pass four filters: a consensus quality score of no less than 20, a SNP quality score of no less than 20, no fewer than three reads supporting the variant allele, and a maximum read depth of 500. For exome-sequencing samples, we further required at least 90% of the capture regions in each sample to be sequenced with ≥5× coverage. When no variant call was made at a particular position of a sample, we assumed that the genotype was a homozygous reference if the position was covered by no fewer than eight reads; otherwise, we considered the genotype to be missing at that position. For each of the three sequencing platforms, SNVs with a missing rate > 10% were removed.
Genotyping and Imputation
In order to expand the number of variants in exome-sequencing samples, we performed microarray genotyping and computational imputation in these samples. Specifically, a total of 298 and 106 exome-sequencing samples were genotyped with the Illumina Human610-Quad BeadChip and Illumina Human1M-Duo BeadChip, respectively. For each genotyping platform, SNPs with a missing rate > 10% were removed and the set of the genotypes satisfying a minor allele frequency (MAF) ≥ 1%, a Hardy-Weinberg equilibrium (HWE) p value > 1 × 10−6, and a SNP missing rate ≤ 5% were used for imputation with MACH29,30 (50 rounds; all other parameters were kept at their default values). We used two reference panels in the imputation: HapMap II phased CEU (Utah residents with ancestry from northern and western Europe from the CEPH collection) chromosomes and HapMap III phased European-ancestry chromosomes (CEU + TSI [Toscans in Italy]). After filtering out SNVs that were not reliably imputed (MACH-estimated r2 < 0.3), we merged the imputed genotypes from the two reference panels by using PLINK.31 If a SNV was imputed from both panels, the genotypes from the HapMap III haplotypes overwrote the genotypes from the HapMap II haplotypes. The imputed genotypes were then integrated with chip genotypes from genotyping arrays. The chip genotypes were overwritten by imputed genotypes only when they were missing. Next, the integrated genotypes of the 298 samples genotyped with the Illumina Human610-Quad BeadChip were combined with the integrated genotypes of the 106 samples genotyped with the Illumina Human1M-Duo BeadChip. Platform-specific SNPs were removed. Finally, we merged the genotypes obtained from sequencing in all 479 samples with the genotypes integrated from chip genotyping and imputation in the 404 exome-sequencing samples. The sequencing genotypes were overwritten only when they were missing. After removing SNVs with a MAF equal to zero, we obtained a total of 13,418,055 autosomal SNVs in the 479 samples.
The Preferential LD Approach
Step 1. Collecting Candidate SNVs around the Discovery SNP
From our SNV collection described above, we first extracted the genotypes of SNVs that had been evaluated in the original GWAS, which included all SNVs in the genotyping platform(s) of the GWAS and SNVs in the imputation reference panel if the GWAS was a meta-analysis. The extracted SNVs that were not on the same chromosome as the discovery SNP were removed. The remaining SNVs were also filtered by MAF and a HWE test. For meta-GWASs and GWASs for which the cutoff values were not reported in their publications, we applied the commonly used cutoffs (MAF ≥ 1% and HWE p value > 1 × 10−6); otherwise, the cutoff values reported in the publications were used. We called this group of SNVs “GWAS SNVs.” We then collected a group of “candidate SNVs” from our SNV collection by using the following criteria: 500 kb upstream and downstream of the discovery SNP, MAF ≤ the MAF of the discovery SNP, MAF ≤ 15%, HWE p value > 1 × 10−6, r2 with the discovery SNP > 0.005, and absence from the GWAS SNV group. The pairwise LD was calculated with Haploview.32 Similar results were obtained when we extended the distance to 1 Mb around the discovery SNP (data not shown). We observed little effect on the approach’s ability to identify causal variants when we changed the MAF cutoff of the candidate SNVs, even when the candidate SNVs were not required to be as common as the discovery SNP (Table S1, available online).
Step 2. Identifying Candidate SNVs in Preferential LD with the Discovery SNP
For each candidate SNV, we calculated the statistic “preferential LD” (PLD) and filtered out the candidate SNVs that could not be specifically tagged by the discovery SNP by requiring the value of PLD to be ≤0.05. Specifically, for the ith candidate SNV, , where I is an indicator function, N is the number of SNVs in the GWAS SNV group, rij2 is the r2 between the ith candidate SNV and the jth GWAS SNV, and ri2 is the r2 between the ith candidate SNV and the discovery SNP. PLD,i assesses the proportion of GWAS SNVs that have equal or higher r2 with the ith SNV than the r2 between the ith SNV and the discovery SNP.
Step 3. Identifying Candidate SNVs Whose LD with the Discovery SNP Is Not Random
For each of the remaining candidate SNVs from step 2, we performed a permutation test to determine whether its r2 with the discovery SNP was due to chance. Specifically, we permuted the genotypes of the candidate SNV and the discovery SNP 2,000 times and calculated the empirical p value as the fraction of permutations for which the r2 calculated from the permuted genotypes was equal to or greater than the observed r2. The empirical p value estimates the probability of observing the same or better r2 value for two random variants with the same frequencies as the two particular variants. Only the candidate SNVs with an empirical p value ≤ 0.1 were kept. We used a less stringent cutoff here to compensate for the smaller sample size of SNVs observed only in whole-genome samples.
Step 4. Prioritizing Candidate SNVs
Finally, we prioritized the candidate SNVs that were preferentially tagged by the discovery SNP and functionally important by using a sorting score (S). For the ith candidate SNV, , where PhastConsi is the PhastCons score33 for primates at the corresponding genomic position, w is the weight for the PhastCons score (by default, w = 0.3), and PLD,i is the corresponding preferential LD value described above. We noticed that as long as the PhastCons score is incorporated into the sorting score, changing the weight of the PhastCons score only marginally affects the ranking of the known causal variants, except for one case in which the causal variant is very rare (Table S2). The PhastCons scores were downloaded from the UCSC Genome Browser (Web Resources), and a larger value corresponds to greater selective constraint. Using PhyloP34 score instead of PhastCons score was found to generate overall similar results in ranking the known causal variants (Table S3). Candidate SNVs were ranked in descending order of the sorting score. In order to estimate the statistical significance of the sorting scores, we randomly drew 2,000 SNVs in the 500 kb neighborhood of the discovery SNP and calculated their sorting scores. Because the majority of these randomly selected SNVs are not causal variants, the distribution of their sorting scores can be used for estimating the null distribution of S. Therefore, the p value of the sorting score corresponding to the ith candidate SNV is calculated as the fraction of randomly selected SNVs with a sorting score equal to or greater than Si. We considered the candidate SNVs with p values ≤ 0.05 as the candidate causal variants.
Preferential LD Application on the 1000 Genomes Project Data
We obtained the phased genotypes from the 1000 Genomes Project35 phase I integrated variant release (March 2012 release) (Web Resources) and extracted SNVs from 379 individuals of European ancestry. SNVs with MAFs equal to zero were removed, and the coordinates of the remaining SNVs were converted from hg19 to hg18 with liftOver (Web Resources). At the end, we obtained a total of 15,845,467 autosomal SNVs. We applied the preferential LD approach to this data set by using the same parameters as indicated above.
Association Test of Candidate SNVs with HCV Treatment Response
We merged the SNV genotypes from whole-genome sequencing, whole-exome sequencing, and chip genotyping for all 479 samples (missing rate per SNV ≤ 0.1, and HWE p value > 1 × 10−6). MACH29,30 was then used for phasing the 2 Mb region centered on rs12979860 in these samples and for imputing the SNVs from the phased haplotypes to the GWAS cohort (missing rate per SNP ≤ 0.05, MAF ≥ 1%, and HWE p value > 1 × 10−6). Default MACH parameters were used, but the number of rounds was set to 50. At the step of estimating model parameters during imputation, 400 individuals randomly selected from the GWAS cohort were used. We also performed phasing and imputation by using SNVs from the whole-genome sequencing of 75 samples to impute SNVs only available through whole-genome sequencing. The association test between the imputed candidate SNVs and HCV treatment response in the GWAS cohort was performed with PLINK31 on the basis of the dosage data.
Results
In order to have the largest sample size possible with our data, we integrated the genotypes from 75 whole-genome-sequenced individuals with the genotypes from 404 whole-exome-sequenced individuals. Because most of the SNPs analyzed in GWASs are missing in exome-sequenced samples, we genotyped these samples by using Illumina high-density arrays and imputed SNPs from HapMap II and HapMap III haplotypes. We obtained a total of 13,418,055 autosomal SNVs after integrating genotypes from all of these sources (Figure 1, see Material and Methods). For SNVs found in both whole-genome and whole-exome samples, their estimated MAF can be as low as 0.10% and we have more than 80% power to detect alleles with a population frequency ≥ 0.17%. For SNVs only observed in the whole-genome samples, the lowest estimated MAF is 0.67% and we have greater than 80% power to detect alleles with a frequency ≥ 1.07% (Figure S1).
Figure 1.
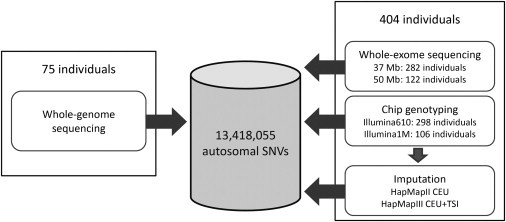
Flow Chart of Integrating SNV Genotypes from 479 Samples
The Preferential LD Approach
Our approach to identifying candidate causal variants consists of four major steps (Figure 2, see Material and Methods). The input to this approach includes a SNP reported to be significantly associated with the trait of interest (discovery SNP) in a GWAS and the genotyping platform(s) used in the GWAS, as well as the reference panel used for imputation if the GWAS is a meta-analysis. First, we identified SNVs that were in a 1 Mb interval centered on the discovery SNP, had not been evaluated in the GWAS of interest, were rarer than the discovery SNP, and were not more common than 15% (given that variants more common than this frequency range probably do not have important functions36). Second, we identified the candidate SNVs that were preferentially tagged by the discovery SNP by calculating the PLD statistic. This statistic estimates the percentage of GWAS SNPs that can tag the candidate SNV better than or as well as the discovery SNP. Third, we performed permutation tests and kept the candidate SNVs whose LD with the discovery SNP was not due to chance. Finally, we prioritized the candidate SNVs that were preferentially tagged by the discovery SNP and functionally important on the basis of a sorting score that incorporates both the preferential LD statistic and evolutionary conservation. Candidate SNVs with statistically significant sorting scores were considered to be the candidates for causal variants driving the association between the discovery SNP and the phenotype of interest.
Figure 2.
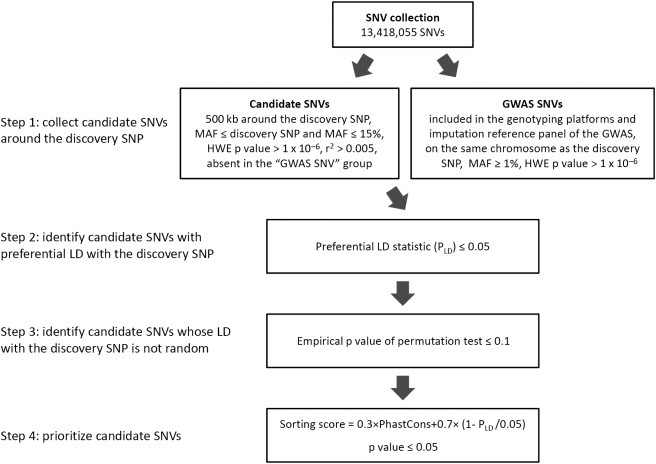
Flow Chart of the Preferential LD Approach
Proof of Concept: The Preferential LD Approach Identifies Known Causal Variants
In order to evaluate the effectiveness of our approach, we applied it to the GWAS of five different human traits for which the causal variants have been reported and are present in our samples. It is important to appreciate that in all but one case, the known causal variants not only are identified as candidates out of all identified SNVs in the relevant intervals but also receive very high priority scores—they always rank among the top ten SNVs. For the only exception where the causal variant is not conserved according to the PhastCons score, the particular variant is still among the top 25 candidates.
Crohn Disease
Crohn disease is one of the few cases for which the causal variants for GWAS signals are known; these include two nonsynonymous variants (rs2066844 and rs2066845) and one frameshift variant (rs2066847) caused by a cytosine insertion in NOD2.37–41 The GWAS carried out by the Wellcome Trust Case Control Consortium (WTCCC) detected a common SNP (rs17221417) that is significantly associated with Crohn disease at the NOD2 locus.42 However, the effect size of this discovery SNP is smaller than the effect size of any one of the three causal variants, and the genetic risk explained by this discovery SNP is significantly lower than the genetic risk explained by the three causal variants.4 We applied our preferential LD approach to search for candidate causal variants in the 1 Mb interval centered on rs17221417, which was identified by the WTCCC GWAS with the Affymetrix GeneChip Human Mapping 500K Array Set for genotyping. Because our variant collection only contained SNVs, we were not able to evaluate the causal insertion variant rs2066847. In step 1, we collected a total of 1,658 candidate SNVs in the neighborhood (500 kb in each direction) of rs17221417. Among them, 152 were found to be in preferential LD with rs17221417 in step 2. After filtering out SNVs in nonsignificant LD with rs17221417, we obtained 141 candidate SNVs in step 3. Fifty-six of these SNVs have significant sorting scores, which mean that no more than 5% of 2,000 randomly selected SNVs in the same 1 Mb neighborhood have equal or better scores. The two known causal variants, rs2066844 and rs2066845, were ranked as the top two candidates by our sorting score in the final step (Table 2). Therefore, starting from the discovery SNP (rs17221417) identified by the WTCCC GWAS, we successfully identified two causal variants of Crohn disease by using this preferential LD approach. When we applied our approach to a different discovery SNP (rs2076756) in the same locus, which was reported to be significantly associated (p value < 5 × 10−8) with Crohn disease by another GWAS,43 we also identified the two known causal variants as the top two candidates (Table 2).
Table 2.
Performance of the Preferential LD Approach on GWASs with Known Causal Variants
| Phenotype | Discovery SNP | Number of GWAS SNVs |
Number of Candidate SNVs |
Ranking of the Causal Variants in Step 4 |
Statistics of Causal Variants |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Step 1 | Step 2 | Step 3 | Step 4 | D′ | r2 | PLD | PhastCons | Sorting Score | Sorting Score p Value | ||||
| Crohn disease | rs17221417 | 12,863 | 1,658 | 152 | 141 | 56 | 1 (rs2066844) | 1 | 0.118 | 1.09 × 10−3 | 0.942 | 0.967 | 5.00 × 10−4 |
| 2 (rs2066845) | 1 | 0.055 | 2.10 × 10−3 | 0.971 | 0.962 | 1.00 × 10−3 | |||||||
| Crohn disease | rs2076756 | 8,950 | 1,523 | 161 | 133 | 54 | 1 (rs2066845) | 1 | 0.065 | 7.82 × 10−4 | 0.971 | 0.980 | 5.00 × 10−4 |
| 2 (rs2066844) | 1 | 0.14 | 3.35 × 10−4 | 0.942 | 0.978 | 1.00 × 10−3 | |||||||
| Ribavirin-induced hemolytic anemia | rs6051702 | 13,334 | 1,289 | 228 | 203 | 53 | 1 (rs1127354) | 0.593 | 0.128 | 6.75 × 10−4 | 0.998 | 0.990 | 0 |
| 2 (rs7270101) | 0.812 | 0.4 | 6.00 × 10−4 | 0.957 | 0.979 | 0 | |||||||
| Therapeutic warfarin dose | rs4917639 | 16,506 | 1,966 | 786 | 718 | 154 | 2 (rs1799853) | 1 | 0.581 | 3.03 × 10−4 | 0.937 | 0.977 | 5.00 × 10−4 |
| Bladder cancer | rs11892031 | 42,467 | 516 | 415 | 217 | 131 | 23 (rs17863783) | 1 | 0.163 | 2.35 × 10−5 | 0.2 | 0.760 | 4.50 × 10−3 |
| Hearing loss | rs870729 | 18,789 | 1,477 | 254 | 213 | 162 | 5 (rs80338945) | 1 | 0.011 | 1.39 × 10−2 | 0.949 | 0.789 | 1.50 × 10−3 |
| Hearing loss | rs7329467 | 18,789 | 999 | 354 | 93 | 82 | 6 (rs35887622) | 0.819 | 0.079 | 5.32 × 10−5 | 0.363 | 0.808 | 5.00 × 10−4 |
The following abbreviations are used: GWAS, genome-wide association study; SNV, single-nucleotide variant; and PLD, preferential linkage disequilibrium.
Ribavirin-Induced Anemia
Previously, we performed a GWAS on ribavirin-induced hemolytic anemia in individuals treated for chronic HCV infection by using the Illumina Human610-Quad BeadChip, and we identified rs6051702 to be strongly associated with treatment-induced reduction in hemoglobin levels.5 The causal variants are two nonsynonymous ITPA variants (rs1127354 and rs7270101) that cause the accumulation of inosine triphosphate, which is used in place of guanosine triphosphate during adenosine triphosphate (ATP) biosynthesis, and that therefore protect individuals from erythrocyte ATP reduction and anemia during treatment.44 By using our preferential LD approach to follow the GWAS discovery SNP, we identified a final set of 53 candidate SNVs from a total of 1,289 SNVs around the associated locus; the two causal variants, rs1127354 and rs7270101, were ranked first and second, respectively (Table 2).
Therapeutic Warfarin Dose
In a GWAS of therapeutic warfarin dose with the Illumina HumanCNV370 BeadChip, rs4917639 is observed as the strongest phenotype-associated SNP at the CYP2C9 (MIM 601130) locus.9 This association is found to be driven by two rarer nonsynonymous variants (rs1799853 and rs1057910) that impair warfarin metabolism.9,10,26 We applied the preferential LD approach to this case. Starting from 1,966 SNVs in the 1 Mb interval centered on rs4917639, we identified 154 candidates in the end, and the known causal variant, rs1799853, was the second best candidate (Table 2). The other causal variant, rs1057910, was not considered by the preferential LD approach because it is present in the HumanCNV370 chip and was tested in the original GWAS.
Bladder Cancer
Recently, Tang et al. found a rare synonymous UGT1A6 (MIM 606431) variant (rs17863783) that completely explains the association between bladder cancer and the GWAS-discovered SNP (rs11892031).11 The rare variant, rs17863783, was suggested to change UGT1A6 mRNA expression and affect the removal of carcinogens from the bladder.11 We used the preferential LD approach to follow the discovery SNP, rs11892031, identified by the bladder cancer GWAS, which used the SNPs found in both the Illumina Human1M-Duo and Human610-Quad BeadChips.45 In the end, 131 out of 516 SNVs found around the discovery SNP were identified as candidates. The causal variant, rs17863783, was ranked as the 23rd best candidate as a result of the low PhastCons score at the corresponding genomic position.
Hearing Loss
Hearing loss is a complex human disease that is largely caused by rare mutations in the GJB2 (MIM 121011)-GJB6 (MIM 604418) locus.46,47 For example, the most common causal variant in GJB2 (rs80338939; c.35delG [p.Gly12Valfs]) occurs with an allele frequency of only 1.25% in European Americans.48 In a recent hearing-loss GWAS with the Illumina HumanHap550 BeadChip, two independent common SNPs (rs870729 and rs7329467) in the GJB2-GJB6 locus were found to be significantly associated with the phenotype.24 The discovery SNP rs870729 has been found to be in high LD (D′ ≥ 0.95) with a number of known causal GJB2 variants, including c.35delG (p.Gly12Valfs), c.167delT (p.Leu56Argfs), c.109G>A (p.Val37Ile), c.269T>C (p.Leu90Pro), and c.101T>C (p.Met34Thr).4 Because c.35delG (p.Gly12Valfs) and c.167delT (p.Leu56Argfs) are deletion variants and c.109G>A (p.Val37Ile) was not observed in the 479 samples we used, they were not included in our analysis. Interestingly, when we used the preferential LD approach to follow rs870729, known causal variant c.269T>C (p.Leu90Pro) (rs80338945)49–51 was ranked as fifth out of the final 162 candidate SNVs. On the other hand, when we followed the other discovery SNP rs7329467, another causal variant, c.101T>C (p.Met34Thr) (rs35887622),51–54 was found to be the sixth best candidate among a final set of 82. Therefore, our approach has identified the hearing-loss causal variants that contribute to the reported genome-wide associations. Our results also suggest that in addition to the most significant phenotype-associated SNP, the other independent phenotype-associated SNPs are also worth investigating for the identification of additional causal variants.
We also applied the preferential LD approach to the SNV genotypes in 379 European-ancestry samples from the 1000 Genomes Project and found that the results are highly consistent for five of the eight known causal variants (Table S4). The causal variant (rs80338945) of hearing loss was absent from the 1000 Genomes Project data, whereas the causal variants of bladder cancer and hearing loss, rs17863783 and rs35887622, were ranked as 40 and 60, respectively, as opposed to 23 and 6, respectively, with our own samples. We speculated that the discrepancy might be due to the lower sample size of the 1000 Genomes Project data. The causal variant (rs80338945) not available from the 1000 Genomes Project data is the most rare (MAF = 0.2% on the basis of our samples) among all eight known causal variants, whereas the other two causal variants that showed decreased performance with the 1000 Genomes Project data are the second and third most rare (MAF ≤ 2%).
The Contribution of LD Measures
To compare the performance of different weighting schemes in identifying causal variants, we evaluated rankings of known causal variants on the basis of the magnitude of two different LD measures (D′ and r2) and compared these with PLD (Table 3). We found that D′ was the worst measure in terms of prioritizing the causal variants. PLD overall performs better than r2, especially when the causal variant is rare (e.g., MAF < 3%). For the three most rare causal variants evaluated here (rs80338945, rs35887622, and rs17863783), their rankings improved from 1,145, 55, and 102 to 150, 12, and 20, respectively, when we used PLD instead of r2. Presumably, the key reason for the superiority of PLD is that it not only identifies SNVs whose LD is the highest with the discovery variant compared to other variants interrogated in the GWAS but also excludes from consideration SNVs that are in nonspecific LD with many GWAS variants. The combination of the conservation score with the preferential LD statistic further boosted the known causal variants to the very top of the candidate list in most cases, which highlights the importance of incorporating conservation score into the discovery of causal variants.55
Table 3.
The Ranking of the Causal Variants via Different Statistics
| Phenotype | Discovery SNP | Number of Candidate SNVsa | Causal SNV | MAF |
Rank of the Causal SNVb |
||||
|---|---|---|---|---|---|---|---|---|---|
| D′ | r2 | PLD | PhastCons | Sorting Score | |||||
| Crohn disease | rs17221417 | 1,658 | rs2066844 | 0.048 | 1,306 | 26 | 34 | 42 | 1 |
| rs2066845 | 0.023 | 1,306 | 89 | 38 | 35 | 2 | |||
| Crohn disease | rs2076756 | 1,523 | rs2066845 | 0.023 | 1,195 | 68 | 27 | 31 | 1 |
| rs2066844 | 0.048 | 1,195 | 13 | 19 | 37 | 2 | |||
| Ribavirin-induced hemolytic anemia | rs6051702 | 1,289 | rs1127354 | 0.081 | 829 | 86 | 49 | 1 | 1 |
| rs7270101 | 0.128 | 776 | 22 | 39 | 10 | 2 | |||
| Therapeutic warfarin dose | rs4917639 | 1,966 | rs1799853 | 0.134 | 1,586 | 7 | 26 | 10 | 2 |
| Bladder cancer | rs11892031 | 516 | rs17863783 | 0.015 | 256 | 102 | 20 | 73 | 37c |
| Hearing loss | rs870729 | 1,477 | rs80338945 | 0.002 | 964 | 1145 | 150 | 7 | 5 |
| Hearing loss | rs7329467 | 999 | rs35887622 | 0.015 | 574 | 55 | 12 | 70 | 6 |
The following abbreviations are used: SNV, single-nucleotide variant; MAF, minor allele frequency; and PLD, preferential linkage disequilibrium.
Candidate SNVs are the ones obtained at step 1 of the preferential LD approach.
The rank of the causal variant is the number of candidate SNVs with an equal or better value of the corresponding statistic than the causal variant.
The rank of rs17863783 shown here is different from the rank shown on Table 2 because the 14 candidate SNVs with larger sorting scores than rs17863783 did not pass the permutation test at step 3.
Identifying New Candidate Causal Variants
We have so far applied the preferential LD approach to 33 independent discovery variants reported in the GWAS of three different human traits: HCV treatment response (MIM 609532), plasma level of coagulation factors, and Alzheimer disease (AD [MIM 104300]). The results of the candidate causal variants are tabulated and available at our website (Web Resources) for the community to investigate. We summarized the candidate causal variants of particular interest in Table 4 and describe them below.
Table 4.
Promising Candidate Causal Variants Identified by the Preferential LD Approach
| Phenotype | Discovery SNP | Number of Final Candidates | Candidate SNV | Rank | MAF | D′ | r2 | PLD | PhastCons | Sorting Score | Sorting Score p Value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HCV treatment response | rs12979860 | 73 | rs4803221 | 2 | 0.145 | 0.914 | 0.347 | 3.26 × 10−4 | 0.678 | 0.899 | 0 |
| rs12971396 | 19 | 0.144 | 0.923 | 0.379 | 3.26 × 10−4 | 0.004 | 0.697 | 1.20 × 10−2 | |||
| Plasma vWF level | rs687621 | 72 | rs34054981 | 2 | 0.082 | 0.747 | 0.028 | 3.58 × 10−3 | 0.748 | 0.874 | 1.00 × 10−3 |
| rs28503257 | 6 | 0.037 | 0.501 | 0.017 | 1.04 × 10−2 | 0.915 | 0.829 | 1.50 × 10−3 | |||
| rs28647808 | 7 | 0.085 | 0.751 | 0.029 | 3.37 × 10−3 | 0.532 | 0.812 | 2.50 × 10−3 | |||
| rs28641026 | 8 | 0.037 | 0.582 | 0.023 | 5.77 × 10−3 | 0.619 | 0.805 | 3.50 × 10−3 | |||
| Plasma vWF level | rs4981022 | 10 | rs6615 | 1 | 0.127 | 0.950 | 0.058 | 3.15 × 10−3 | 0.998 | 0.955 | 5.00 × 10−4 |
| rs117801489 | 3 | 0.015 | 0.848 | 0.024 | 1.92 × 10−3 | 0.862 | 0.932 | 1.50 × 10−3 | |||
| Plasma fibrinogen level | rs511154 | 21 | rs34901937 | 1 | 0.093 | 0.764 | 0.180 | 2.45 × 10−4 | 0.882 | 0.961 | 1.50 × 10−3 |
| Late-onset Alzheimer disease | rs11767557 | 16 | rs73154206 | 1 | 0.019 | 0.919 | 0.060 | 1.34 × 10−3 | 0.956 | 0.968 | 1.00 × 10−3 |
| rs11552742 | 2 | 0.008 | 1.000 | 0.029 | 5.14 × 10−3 | 0.962 | 0.917 | 2.00 × 10−3 |
The following abbreviations are used: SNV, single-nucleotide variant; MAF, minor allele frequency; PLD, preferential linkage disequilibrium; and vWF, von Willebrand factor.
HCV Treatment Response
Ge et al. previously performed a GWAS on individuals chronically infected by genotype 1 HCV by using the Illumina Human610-Quad BeadChips and observed a common SNP (rs12979860) near IL28B (MIM 607402) to be significantly associated with treatment response.56 In order to look for the causal variants driving this association signal, we followed rs12979860 by using our preferential LD approach and obtained a final set of 73 candidates (Table 4). We imputed their genotypes from our sequenced samples to the same European-American GWAS cohort used by Ge et al.,56 and we tested the association between the candidates and the phenotype. Although the second-best candidate, rs4803221 (MAF = 14.49%), was not well imputed in our GWAS cohort (MACH-estimated r2 = 0.43), it has been reported recently to predict treatment response better than the discovery SNP, rs1297986057 (MAF = 28.57%). Fifteen candidate SNVs can be well imputed (MACH-estimated r2 ≥ 0.8), and the candidate SNV, rs12971396, which was ranked 19th among all 73 candidates, is much rarer than rs12979860 but has a stronger association with the treatment response (MAF = 14.38%, p value = 5.79 × 10−26, and OR = 5.546 for rs12971396; MAF = 28.57%, p value = 1.07 × 10−25, and OR = 4.198 for rs12979860). Although rs12971396 is only weakly linked to rs12979860 (r2 = 0.38), it can explain a large portion of the association between rs12979860 and the treatment response (i.e., after rs12971396 was accounted for, the association with rs12979860 dropped from p = 1.07 × 10−25 to p = 1.17 × 10−7). Two parallel GWASs in Australian58 and Japanese59 populations have identified another SNP, rs8099917, at the IL28B locus to be significantly associated with the HCV treatment response. The candidate SNV, rs12971396, was found to be strongly linked to rs8099917 (r2 = 0.84) and completely explained the association between rs8099917 and treatment response (the p value increased from 4.07 × 10−25 to 0.88 after rs12971396 was accounted for in the GWAS cohort used by Ge et al.56).
Plasma Level of Coagulation Factors
Smith et al. carried out a meta-GWAS on the plasma levels of coagulation factors VII (FVII), VIII (FVIII), and von Willebrand factor (vWF).60 They observed rs687621 in ABO (MIM 110300) to be significantly associated with the vWF level. By using our preferential LD approach to follow this discovery SNP, we found a total of 72 candidate SNVs, of which four coding SNVs in ADAMTS13 (MIM 604134) fell within the top 10; these included two nonsynonymous SNVs (rs28503257 and rs28647808) and two synonymous SNVs (rs34054981 and rs28641026) (Table 4). The two nonsynonymous SNVs were predicted to be probably damaging by PolyPhen2.61,62 ADAMTS13 encodes a vWF-cleaving protease, which cleaves vWF at the peptide bond between Tyr842 and Met843. Such ADAMTS13-dependent proteolysis decreases the size, as well as the prothrombotic activity, of the vWF multimer. Deficiency of ADAMTS13 leaves the uncleaved and highly thrombotic vWF to accumulate in the plasma and results in thromboembolism.63 These candidate coding causal variants in ADAMTS13 might affect the efficiency of the enzyme activity and might therefore cause changes in the plasma vWF level. A common intronic SNP (rs4981022) in STAB2 (MIM 608561) was also found to significantly associate with the plasma vWF level by Smith et al. Application of the preferential LD approach to this discovery SNP led to the identification of ten candidate causal SNVs. The best candidate, rs6615, is a synonymous SNV in HSP90B1 (MIM 191175) (also called GP96 or GRP94), which encodes a chaperone in the endoplasmic reticulum (ER). Protein interaction between HSP90B1 and vWF has been documented before.64,65 HSP90B1 is also known to chaperone GPIX in the ER and is required for its proper folding and the subsequent assembly of the GPIb-IX-IV complex, which is the receptor of vWF and is essential for vWF-mediated platelet activation.66 rs6615 might directly modulate vWF levels by affecting its chaperone function on vWF, or it might indirectly alter vWF levels by affecting the assembly of the vWF receptor. The third candidate, rs117801489, is a nonsynonymous SNV in GLT8D2, and it is predicted to be probably damaging to GLT8D2 by PolyPhen2. GLT8D2 encodes a glycosyltransferase belonging to glycosyltransferase family 8. Two members (GXYLT1 and GXYLT2) of the same family can transfer xylose to O-linked glucose,67 and the O-linked glycosylation of vWF was reported to associate with plasma vWF levels.68 It is possible that vWF is a target of GLT8D2 glycosyltransferase and that rs117801489 affects the glycosylation of vWF by GLT8D2 and therefore alters the plasma vWF level.
In addition, we applied our approach to the discovery SNP identified by a meta-GWAS on plasma fibrinogen levels (rs511154),69 which is a noncoding SNP 18 kb upstream of PCCB (MIM 232050), and 21 candidate causal variants were identified. A nonsynonymous SNV (rs34901937) in PPP2R3A (MIM 604944) was ranked as the best candidate (Table 4). PPP2R3A encodes one of the regulatory subunits of the protein phosphatase 2 (PP2), which is known to regulate the platelet integrin αIIbβ3 adhesion to fibrinogen.70 Although this nonsynonymous SNV was predicted to be benign by PolyPhen2, it would still be interesting to explore whether this SNV can disrupt the regulatory PP2 subunit encoded by PPP2R3A and can therefore affect the binding of fibrinogen with integrin αIIbβ3 and change the plasma fibrinogen level.
Alzheimer Disease
A recent GWAS of late-onset AD was carried out by the Alzheimer Disease Genetics Consortium and led to the identification of four new susceptibility loci, including EPHA1 (MIM 179610).71 We followed the most significant SNP (rs11767557), which is about 3 kb upstream of EPHA1, and identified two rare SNVs in ZYX (MIM 602002) as the best two candidates (Table 4). These two candidate causal variants include one nonsynonymous SNV (rs73154206) predicted to be probably damaging by PolyPhen2 and one synonymous SNV (rs11552742). ZYX was found to be deacetylated by SIRT1,72 which can protect neurons from neurodegeneration in mouse models of AD.73 It is possible that the identified candidate causal variants affect the onset of AD through the antiapoptotic pathway involving both ZYX and SIRT1.
Discussion
We describe here a computational method, the preferential LD approach, designed to identify candidate causal variants responsible for the association between phenotypes and GWAS-discovered SNPs. Our approach seeks to make effective the combined use of the existing GWAS signals and the rapidly accumulating database of sequenced genomes. One unique advantage of this approach is its ability to identify causal variants that are rare in the population and that are only in weak LD with the discovery SNP as measured by r2. Using this approach, we were able to rediscover known causal variants for Crohn disease, HCV-treatment-induced hemolytic anemia, therapeutic warfarin dose, bladder cancer, and hearing loss. These include both cases in which the causal variant is a single rarer variant and in which multiple rarer variants contribute to the GWAS signal. Because of our constraints on the MAF of the candidate SNVs, we might miss the causal variants that are very common but are in strong LD with the GWAS discovery SNPs. Because this scenario is amenable to more “straightforward” fine-mapping efforts and the simple r2 can work quite well in prioritizing the common causal variant (Table S5), we did not attempt to address it here. Instead, we focused on the scenarios in which the causal variants are rarer because methods for detecting the causal variants in these scenarios are limited. We found that the preferential LD approach was able to identify common causal variants strongly linked with the GWAS-discovered SNP when the MAF requirements of candidate SNVs were relaxed (Table S5). Users can change the parameters of the preferential LD approach according to their specific cases.
The preferential LD approach utilized genotypes from a general population of European ancestry and can be easily applied to existing GWASs involving this population. We showed that this approach has led to the identification of a number of promising causal variant candidates, upon which testable hypotheses could be formed.
The current implementation of the preferential LD approach might fail to identify the causal variants if they are not SNVs, if the variants are too rare to be represented in our discovery samples, or if the LD structure in our samples is significantly different from the LD structure in the original GWAS cohort. The current genotype information used by the preferential LD approach includes only SNVs, and we plan to include the genotypes of insertions and deletions (indels) in the future to be able to identify indel causal variants. Our current collection of genotypes comes from 75 whole-genome-sequenced individuals and 404 whole-exome-sequenced individuals, plus additional chip genotyping and in silico imputation. As next-generation sequencing becomes routine, more and more deep-sequenced genomes will become available. We expect that increasing the sample size of the population used by the preferential LD approach will boost its power to identify rarer causal variants. For example, using 800 whole-genome samples will give us about 80% power to detect variants with MAF ≥ 0.1% in the general population (Figure S1). Currently, it is a common practice to utilize a general population for the calculation of LD between variants (e.g., the International HapMap Project and the 1000 Genomes Project).74 Therefore, we expect that the LD measures for most variants will remain similar between the general population that we used and the respective GWAS cohorts of European ancestry.
As the number of sequenced samples grows, the preferential LD approach will be able to provide a comprehensive listing of candidate causal variants for all GWAS discovery variants and represent a general tool for leveraging GWAS signals in the interpretation of human sequence data.
Acknowledgments
We thank A. Need, E. Cirulli, K. Pelak, E. Ruzzo, and P. Gaffney for comments on the manuscript. We acknowledge A. Holden, E. Behr, C. Depondt, S. Sisodiya, G. Cavalleri, N. Delanty, D. Daskalakis, D. Levy, A. Need, J. Silver, M. Silver, K. Welsh-Bohmer, C. Hulette, J. Burke, R. Ottman, R. Radtke, A. Husain, M. Mikati, W. Gallentine, S. Sinha, D. Attix, J. McEvoy, E. Cirulli, N. Walley, K. Linney, R. Brown, S. Berkovic, I. Scheffer, B. Grinton, S. Palmer, W. Lowe, and the MURDOCK Study Community Registry and Biorepository for contributing samples. Funding for this research was provided in part by the Center for HIV/AIDS Vaccine Immunology grant UO1AIO67854 from the National Institute of Allergy and Infectious Diseases, American Recovery and Reinvestment Act grant 1RC2NS070342-01, the Joseph and Kathleen Bryan Alzheimer’s Disease Research Center grant P30 AG028377 from the National Institute of Aging, National Institute of Mental Health grant RC2MH089915, and National Institute of Neurological Disorders and Stroke grant RC2NS070344. We are grateful to the individuals in the IDEAL (Individualized Dosing Efficacy vs. Flat Dosing to Assess Optimal Pegylated Interferon Therapy) trial, as well as the principal investigators, study coordinators, and nurses involved. D.B. Goldstein receives research support from UCB and Gilead. He serves as a consultant or as a member of the scientific advisory board for Hoffmann-La Roche, Biogen Idec, and Knome. D.B. Goldstein, K.V. Shianna, D. Ge, and T.J. Urban own intellectual property for their work in hepatitis C (patent numbers PCT/US2010/0555570 and 12/785,060).
Contributor Information
Qianqian Zhu, Email: qianqian.zhu@roswellpark.org.
David B. Goldstein, Email: d.goldstein@duke.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project phase I integrated variant release (March 2012 release), ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/release/20110521/
Lift Genome Annotations, http://genome.ucsc.edu/cgi-bin/hgLiftOver
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Preferential LD Approach, http://www.duke.edu/∼qz18/preferentialLD.html
UCSC Genome Browser, http://www.genome.ucsc.edu
References
- 1.Hindorff, L.A., MacArthur, J., Wise, A., Junkins, H.A., Hall, P.N., Klemm, A.K., and Manolio, T.A. A Catalog of Published Genome-Wide Association Studies. National Human Genome Research Institute, www.genome.gov/gwastudies.
- 2.Manolio T.A., Collins F.S., Cox N.J., Goldstein D.B., Hindorff L.A., Hunter D.J., McCarthy M.I., Ramos E.M., Cardon L.R., Chakravarti A. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee J.C., Parkes M. Genome-wide association studies and Crohn’s disease. Brief Funct Genomics. 2011;10:71–76. doi: 10.1093/bfgp/elr009. [DOI] [PubMed] [Google Scholar]
- 4.Wang K., Dickson S.P., Stolle C.A., Krantz I.D., Goldstein D.B., Hakonarson H. Interpretation of association signals and identification of causal variants from genome-wide association studies. Am. J. Hum. Genet. 2010;86:730–742. doi: 10.1016/j.ajhg.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fellay J., Thompson A.J., Ge D., Gumbs C.E., Urban T.J., Shianna K.V., Little L.D., Qiu P., Bertelsen A.H., Watson M. ITPA gene variants protect against anaemia in patients treated for chronic hepatitis C. Nature. 2010;464:405–408. doi: 10.1038/nature08825. [DOI] [PubMed] [Google Scholar]
- 6.Holm H., Gudbjartsson D.F., Sulem P., Masson G., Helgadottir H.T., Zanon C., Magnusson O.T., Helgason A., Saemundsdottir J., Gylfason A. A rare variant in MYH6 is associated with high risk of sick sinus syndrome. Nat. Genet. 2011;43:316–320. doi: 10.1038/ng.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rafnar T., Gudbjartsson D.F., Sulem P., Jonasdottir A., Sigurdsson A., Jonasdottir A., Besenbacher S., Lundin P., Stacey S.N., Gudmundsson J. Mutations in BRIP1 confer high risk of ovarian cancer. Nat. Genet. 2011;43:1104–1107. doi: 10.1038/ng.955. [DOI] [PubMed] [Google Scholar]
- 8.Sanna S., Li B., Mulas A., Sidore C., Kang H.M., Jackson A.U., Piras M.G., Usala G., Maninchedda G., Sassu A. Fine mapping of five loci associated with low-density lipoprotein cholesterol detects variants that double the explained heritability. PLoS Genet. 2011;7:e1002198. doi: 10.1371/journal.pgen.1002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Takeuchi F., McGinnis R., Bourgeois S., Barnes C., Eriksson N., Soranzo N., Whittaker P., Ranganath V., Kumanduri V., McLaren W. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 2009;5:e1000433. doi: 10.1371/journal.pgen.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wadelius M., Chen L.Y., Eriksson N., Bumpstead S., Ghori J., Wadelius C., Bentley D., McGinnis R., Deloukas P. Association of warfarin dose with genes involved in its action and metabolism. Hum. Genet. 2007;121:23–34. doi: 10.1007/s00439-006-0260-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang W., Fu Y.-P., Figueroa J.D., Malats N., Garcia-Closas M., Chatterjee N., Kogevinas M., Baris D., Thun M., Hall J.L. Mapping of the UGT1A locus identifies an uncommon coding variant that affects mRNA expression and protects from bladder cancer. Hum. Mol. Genet. 2012;21:1918–1930. doi: 10.1093/hmg/ddr619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donnelly, P. (2009). An overview of the relationship between genetic risk and functional variation: What the WTCCC has taught us. Proceedings of the 59th Annual Meeting of The American Society of Human Genetics, October 22, 2009, Honolulu, Hawaii.
- 13.Bouatia-Naji N., Bonnefond A., Cavalcanti-Proença C., Sparsø T., Holmkvist J., Marchand M., Delplanque J., Lobbens S., Rocheleau G., Durand E. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat. Genet. 2009;41:89–94. doi: 10.1038/ng.277. [DOI] [PubMed] [Google Scholar]
- 14.Frayling T.M., Timpson N.J., Weedon M.N., Zeggini E., Freathy R.M., Lindgren C.M., Perry J.R.B., Elliott K.S., Lango H., Rayner N.W. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316:889–894. doi: 10.1126/science.1141634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Todd J.A., Walker N.M., Cooper J.D., Smyth D.J., Downes K., Plagnol V., Bailey R., Nejentsev S., Field S.F., Payne F., Genetics of Type 1 Diabetes in Finland. Wellcome Trust Case Control Consortium Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat. Genet. 2007;39:857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pillai S.G., Ge D., Zhu G., Kong X., Shianna K.V., Need A.C., Feng S., Hersh C.P., Bakke P., Gulsvik A., ICGN Investigators A genome-wide association study in chronic obstructive pulmonary disease (COPD): Identification of two major susceptibility loci. PLoS Genet. 2009;5:e1000421. doi: 10.1371/journal.pgen.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Musunuru K., Strong A., Frank-Kamenetsky M., Lee N.E., Ahfeldt T., Sachs K.V., Li X., Li H., Kuperwasser N., Ruda V.M. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010;466:714–719. doi: 10.1038/nature09266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yeager M., Xiao N., Hayes R.B., Bouffard P., Desany B., Burdett L., Orr N., Matthews C., Qi L., Crenshaw A. Comprehensive resequence analysis of a 136 kb region of human chromosome 8q24 associated with prostate and colon cancers. Hum. Genet. 2008;124:161–170. doi: 10.1007/s00439-008-0535-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rivas M.A., Beaudoin M., Gardet A., Stevens C., Sharma Y., Zhang C.K., Boucher G., Ripke S., Ellinghaus D., Burtt N., National Institute of Diabetes and Digestive Kidney Diseases Inflammatory Bowel Disease Genetics Consortium (NIDDK IBDGC) United Kingdom Inflammatory Bowel Disease Genetics Consortium. International Inflammatory Bowel Disease Genetics Consortium Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 2011;43:1066–1073. doi: 10.1038/ng.952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Momozawa Y., Mni M., Nakamura K., Coppieters W., Almer S., Amininejad L., Cleynen I., Colombel J.-F., de Rijk P., Dewit O. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat. Genet. 2011;43:43–47. doi: 10.1038/ng.733. [DOI] [PubMed] [Google Scholar]
- 21.Nejentsev S., Walker N., Riches D., Egholm M., Todd J.A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–389. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sulem P., Gudbjartsson D.F., Walters G.B., Helgadottir H.T., Helgason A., Gudjonsson S.A., Zanon C., Besenbacher S., Bjornsdottir G., Magnusson O.T. Identification of low-frequency variants associated with gout and serum uric acid levels. Nat. Genet. 2011;43:1127–1130. doi: 10.1038/ng.972. [DOI] [PubMed] [Google Scholar]
- 23.Stacey S.N., Sulem P., Jonasdottir A., Masson G., Gudmundsson J., Gudbjartsson D.F., Magnusson O.T., Gudjonsson S.A., Sigurgeirsson B., Thorisdottir K., Swedish Low-risk Colorectal Cancer Study Group A germline variant in the TP53 polyadenylation signal confers cancer susceptibility. Nat. Genet. 2011;43:1098–1103. doi: 10.1038/ng.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dickson S.P., Wang K., Krantz I., Hakonarson H., Goldstein D.B. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8:e1000294. doi: 10.1371/journal.pbio.1000294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldstein D.B. The importance of synthetic associations will only be resolved empirically. PLoS Biol. 2011;9:e1001008. doi: 10.1371/journal.pbio.1001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takeuchi F., Kobayashi S., Ogihara T., Fujioka A., Kato N. Detection of common single nucleotide polymorphisms synthesizing quantitative trait association of rarer causal variants. Genome Res. 2011;21:1122–1130. doi: 10.1101/gr.115832.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y., Willer C.J., Ding J., Scheet P., Abecasis G.R. MaCH: Using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 2010;34:816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y., Willer C., Sanna S., Abecasis G. Genotype imputation. Annu. Rev. Genomics Hum. Genet. 2009;10:387–406. doi: 10.1146/annurev.genom.9.081307.164242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A.R., Bender D., Maller J., Sklar P., de Bakker P.I.W., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barrett J.C., Fry B., Maller J., Daly M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 33.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–121. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu Q., Ge D., Maia J.M., Zhu M., Petrovski S., Dickson S.P., Heinzen E.L., Shianna K.V., Goldstein D.B. A genome-wide comparison of the functional properties of rare and common genetic variants in humans. Am. J. Hum. Genet. 2011;88:458–468. doi: 10.1016/j.ajhg.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ogura Y., Bonen D.K., Inohara N., Nicolae D.L., Chen F.F., Ramos R., Britton H., Moran T., Karaliuskas R., Duerr R.H. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411:603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 38.Hugot J.P., Chamaillard M., Zouali H., Lesage S., Cézard J.P., Belaiche J., Almer S., Tysk C., O’Morain C.A., Gassull M. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi K.S., Chamaillard M., Ogura Y., Henegariu O., Inohara N., Nuñez G., Flavell R.A. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 40.Maeda S., Hsu L.-C., Liu H., Bankston L.A., Iimura M., Kagnoff M.F., Eckmann L., Karin M. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1β processing. Science. 2005;307:734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 41.Economou M., Trikalinos T.A., Loizou K.T., Tsianos E.V., Ioannidis J.P.A. Differential effects of NOD2 variants on Crohn’s disease risk and phenotype in diverse populations: a metaanalysis. Am. J. Gastroenterol. 2004;99:2393–2404. doi: 10.1111/j.1572-0241.2004.40304.x. [DOI] [PubMed] [Google Scholar]
- 42.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rioux J.D., Xavier R.J., Taylor K.D., Silverberg M.S., Goyette P., Huett A., Green T., Kuballa P., Barmada M.M., Datta L.W. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007;39:596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hitomi Y., Cirulli E.T., Fellay J., McHutchison J.G., Thompson A.J., Gumbs C.E., Shianna K.V., Urban T.J., Goldstein D.B. Inosine triphosphate protects against ribavirin-induced adenosine triphosphate loss by adenylosuccinate synthase function. Gastroenterology. 2011;140:1314–1321. doi: 10.1053/j.gastro.2010.12.038. [DOI] [PubMed] [Google Scholar]
- 45.Rothman N., Garcia-Closas M., Chatterjee N., Malats N., Wu X., Figueroa J.D., Real F.X., Van Den Berg D., Matullo G., Baris D. A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci. Nat. Genet. 2010;42:978–984. doi: 10.1038/ng.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kenneson A., Van Naarden Braun K., Boyle C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: A HuGE review. Genet. Med. 2002;4:258–274. doi: 10.1097/00125817-200207000-00004. [DOI] [PubMed] [Google Scholar]
- 47.Tekin M., Arnos K.S., Pandya A. Advances in hereditary deafness. Lancet. 2001;358:1082–1090. doi: 10.1016/S0140-6736(01)06186-4. [DOI] [PubMed] [Google Scholar]
- 48.Green G.E., Scott D.A., McDonald J.M., Woodworth G.G., Sheffield V.C., Smith R.J.H. Carrier rates in the midwestern United States for GJB2 mutations causing inherited deafness. JAMA. 1999;281:2211–2216. doi: 10.1001/jama.281.23.2211. [DOI] [PubMed] [Google Scholar]
- 49.Cryns K., Orzan E., Murgia A., Huygen P.L.M., Moreno F., del Castillo I., Chamberlin G.P., Azaiez H., Prasad S., Cucci R.A. A genotype-phenotype correlation for GJB2 (connexin 26) deafness. J. Med. Genet. 2004;41:147–154. doi: 10.1136/jmg.2003.013896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Löffler J., Nekahm D., Hirst-Stadlmann A., Günther B., Menzel H.-J., Utermann G., Janecke A.R. Sensorineural hearing loss and the incidence of Cx26 mutations in Austria. Eur. J. Hum. Genet. 2001;9:226–230. doi: 10.1038/sj.ejhg.5200607. [DOI] [PubMed] [Google Scholar]
- 51.Snoeckx R.L., Huygen P.L.M., Feldmann D., Marlin S., Denoyelle F., Waligora J., Mueller-Malesinska M., Pollak A., Ploski R., Murgia A. GJB2 mutations and degree of hearing loss: A multicenter study. Am. J. Hum. Genet. 2005;77:945–957. doi: 10.1086/497996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pollak A., Skórka A., Mueller-Malesińska M., Kostrzewa G., Kisiel B., Waligóra J., Krajewski P., Ołdak M., Korniszewski L., Skarzyński H., Ploski R. M34T and V37I mutations in GJB2 associated hearing impairment: Evidence for pathogenicity and reduced penetrance. Am. J. Med. Genet. A. 2007;143A:2534–2543. doi: 10.1002/ajmg.a.31982. [DOI] [PubMed] [Google Scholar]
- 53.Putcha G.V., Bejjani B.A., Bleoo S., Booker J.K., Carey J.C., Carson N., Das S., Dempsey M.A., Gastier-Foster J.M., Greinwald J.H.J., Jr. A multicenter study of the frequency and distribution of GJB2 and GJB6 mutations in a large North American cohort. Genet. Med. 2007;9:413–426. doi: 10.1097GIM.0b013e3180a03276. [DOI] [PubMed] [Google Scholar]
- 54.Houseman M.J., Ellis L.A., Pagnamenta A., Di W.-L., Rickard S., Osborn A.H., Dahl H.-H.M., Taylor G.R., Bitner-Glindzicz M., Reardon W. Genetic analysis of the connexin-26 M34T variant: Identification of genotype M34T/M34T segregating with mild-moderate non-syndromic sensorineural hearing loss. J. Med. Genet. 2001;38:20–25. doi: 10.1136/jmg.38.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cooper G.M., Goode D.L., Ng S.B., Sidow A., Bamshad M.J., Shendure J., Nickerson D.A. Single-nucleotide evolutionary constraint scores highlight disease-causing mutations. Nat. Methods. 2010;7:250–251. doi: 10.1038/nmeth0410-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ge D., Fellay J., Thompson A.J., Simon J.S., Shianna K.V., Urban T.J., Heinzen E.L., Qiu P., Bertelsen A.H., Muir A.J. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 57.Smith K.R., Suppiah V., O’Connor K., Berg T., Weltman M., Abate M.L., Spengler U., Bassendine M., Matthews G., Irving W.L., the International Hepatitis C Genetics Consortium (IHCGC) Identification of improved IL28B SNPs and haplotypes for prediction of drug response in treatment of hepatitis C using massively parallel sequencing in a cross-sectional European cohort. Genome Med. 2011;3:57. doi: 10.1186/gm273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Suppiah V., Moldovan M., Ahlenstiel G., Berg T., Weltman M., Abate M.L., Bassendine M., Spengler U., Dore G.J., Powell E. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat. Genet. 2009;41:1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- 59.Tanaka Y., Nishida N., Sugiyama M., Kurosaki M., Matsuura K., Sakamoto N., Nakagawa M., Korenaga M., Hino K., Hige S. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat. Genet. 2009;41:1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 60.Smith N.L., Chen M.-H., Dehghan A., Strachan D.P., Basu S., Soranzo N., Hayward C., Rudan I., Sabater-Lleal M., Bis J.C., Wellcome Trust Case Control Consortium Novel associations of multiple genetic loci with plasma levels of factor VII, factor VIII, and von Willebrand factor: The CHARGE (Cohorts for Heart and Aging Research in Genome Epidemiology) Consortium. Circulation. 2010;121:1382–1392. doi: 10.1161/CIRCULATIONAHA.109.869156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liu X., Jian X., Boerwinkle E. dbNSFP: A lightweight database of human nonsynonymous SNPs and their functional predictions. Hum. Mutat. 2011;32:894–899. doi: 10.1002/humu.21517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dong J.F. Cleavage of ultra-large von Willebrand factor by ADAMTS-13 under flow conditions. J. Thromb. Haemost. 2005;3:1710–1716. doi: 10.1111/j.1538-7836.2005.01360.x. [DOI] [PubMed] [Google Scholar]
- 64.Allen S., Abuzenadah A.M., Hinks J., Blagg J.L., Gursel T., Ingerslev J., Goodeve A.C., Peake I.R., Daly M.E. A novel von Willebrand disease-causing mutation (Arg273Trp) in the von Willebrand factor propeptide that results in defective multimerization and secretion. Blood. 2000;96:560–568. [PubMed] [Google Scholar]
- 65.Dorner A.J., Wasley L.C., Kaufman R.J. Protein dissociation from GRP78 and secretion are blocked by depletion of cellular ATP levels. Proc. Natl. Acad. Sci. USA. 1990;87:7429–7432. doi: 10.1073/pnas.87.19.7429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Staron M., Wu S., Hong F., Stojanovic A., Du X., Bona R., Liu B., Li Z. Heat-shock protein gp96/grp94 is an essential chaperone for the platelet glycoprotein Ib-IX-V complex. Blood. 2011;117:7136–7144. doi: 10.1182/blood-2011-01-330464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sethi M.K., Buettner F.F.R., Krylov V.B., Takeuchi H., Nifantiev N.E., Haltiwanger R.S., Gerardy-Schahn R., Bakker H. Identification of glycosyltransferase 8 family members as xylosyltransferases acting on O-glucosylated notch epidermal growth factor repeats. J. Biol. Chem. 2010;285:1582–1586. doi: 10.1074/jbc.C109.065409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Schooten C.J.M., Denis C.V., Lisman T., Eikenboom J.C.J., Leebeek F.W., Goudemand J., Fressinaud E., van den Berg H.M., de Groot P.G., Lenting P.J. Variations in glycosylation of von Willebrand factor with O-linked sialylated T antigen are associated with its plasma levels. Blood. 2007;109:2430–2437. doi: 10.1182/blood-2006-06-032706. [DOI] [PubMed] [Google Scholar]
- 69.Dehghan A., Yang Q., Peters A., Basu S., Bis J.C., Rudnicka A.R., Kavousi M., Chen M.H., Baumert J., Lowe G.D. Association of novel genetic loci with circulating fibrinogen levels: A genome-wide association study in 6 population-based cohorts. Circ Cardiovasc Genet. 2009;2:125–133. doi: 10.1161/CIRCGENETICS.108.825224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gushiken F.C., Patel V., Liu Y., Pradhan S., Bergeron A.L., Peng Y., Vijayan K.V. Protein phosphatase 2A negatively regulates integrin α(IIb)β(3) signaling. J. Biol. Chem. 2008;283:12862–12869. doi: 10.1074/jbc.M708804200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Naj A.C., Jun G., Beecham G.W., Wang L.-S., Vardarajan B.N., Buros J., Gallins P.J., Buxbaum J.D., Jarvik G.P., Crane P.K. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011;43:436–441. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fujita Y., Yamaguchi A., Hata K., Endo M., Yamaguchi N., Yamashita T. Zyxin is a novel interacting partner for SIRT1. BMC Cell Biol. 2009;10:6. doi: 10.1186/1471-2121-10-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kim D., Nguyen M.D., Dobbin M.M., Fischer A., Sananbenesi F., Rodgers J.T., Delalle I., Baur J.A., Sui G., Armour S.M. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007;26:3169–3179. doi: 10.1038/sj.emboj.7601758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Johnson A.D., Handsaker R.E., Pulit S.L., Nizzari M.M., O’Donnell C.J., de Bakker P.I.W. SNAP: A web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24:2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.