

Abstract
Meningiomas are the most common primary tumors of the CNS and account for up to 30% of all CNS tumors. An increased risk of meningiomas has been associated with certain tumor-susceptibility syndromes, especially neurofibromatosis type II, but no gene defects predisposing to isolated familial meningiomas have thus far been identified. Here, we report on a family of five meningioma-affected siblings, four of whom have multiple tumors. No NF2 mutations were identified in the germline or tumors. We combined genome-wide linkage analysis and exome sequencing, and we identified in suppressor of fused homolog (Drosophila), SUFU, a c.367C>T (p.Arg123Cys) mutation segregating with the meningiomas in the family. The variation was not present in healthy controls, and all seven meningiomas analyzed displayed loss of the wild-type allele according to the classic two-hit model for tumor-suppressor genes. In silico modeling predicted the variant to affect the tertiary structure of the protein, and functional analyses showed that the activity of the altered SUFU was significantly reduced and therefore led to dysregulated hedgehog (Hh) signaling. SUFU is a known tumor-suppressor gene previously associated with childhood medulloblastoma predisposition. Our genetic and functional analyses indicate that germline mutations in SUFU also predispose to meningiomas, particularly to multiple meningiomas. It is possible that other genic mutations resulting in aberrant activation of the Hh pathway might underlie meningioma predisposition in families with an unknown etiology.
Main Text
Meningiomas are the most common primary tumors of the CNS and account for more than one-third of all CNS tumors.1,2 Meningiomas originate from the arachnoidal cells of the leptomeninges, and slowly growing benign tumors comprise the great majority (95%) of them. Depending on the location, meningiomas can cause significant neurological deficits, but they can also be asymptomatic. Imaging and autopsy studies have shown that subclinical meningiomas occur in up to 3% of the population.3,4 Although meningiomas are most commonly observed as solitary sporadic tumors, 1%–5% are familial and fewer than 10% of individuals have multiple lesions.5 The environmental risk factors for meningiomas, except for ionizing radiation, are unclear.1 An increased risk of meningiomas has been associated with tumor-susceptibility syndromes such as neurofibromatosis type II (NF2 [MIM 101000]), Cowden syndrome (CS [MIM 601728]), and Werner syndrome (WRN [MIM 277700]), which are caused by mutations in NF2 (MIM 607379), PTEN (MIM 601728), and RECQL2 (MIM 604611), respectively. Also, a germline SMARCB1 (MIM 601607) mutation, causing predisposition to schwannomatosis and rhabdoid tumors, was recently identified in a family affected by multiple cranial and spinal meningiomas and schwannomas.6 Gene defects predisposing merely to meningiomas have not been previously reported.
Here, we report on a family of five meningioma-affected siblings, four of whom were diagnosed with multiple tumors. We utilized genome-wide linkage analysis and exome sequencing to identify the genetic predisposition in this family. The index case (III-4, Figure 1) was referred to genetic counseling after being operated on for meningiomas at different sites at the ages of 44, 48, and 59 years. Multiple meningiomas had also been removed from his three sisters: III-3 at the ages of 43 (single) and 54 (multiple) years, III-2 at the ages of 58 (single) and 64 (multiple) years, and III-1 at the age of 72 years (multiple). His fourth sister (III-5) had been operated on for a single meningioma when she was 60 years old. In addition to meningiomas, a benign myoma had been removed from III-3 at the age of 42, and III-5 was diagnosed with immature teratoma in both ovaries when she was 16 years old. Other siblings in the family had been diagnosed with lymphoma, colon adenocarcinoma, squamous cell carcinoma of the facial skin (III-9), and facial basalioma (III-11). Their cousin’s child (IV-1) had died of medulloblastoma at the age of 5 years. The father and mother of the siblings (II-1 and II-2) had died of chronic lymphocytic leukemia at the age of 58 years and of coronary thrombosis at the age of 57 years, respectively. They had not been examined for meningiomas. The index case (III-4) was screened at the clinic for NF2 mutations by direct sequencing and multiplex ligation-dependent probe amplification, but no mutations were detected. We also confirmed with array comparative genomic hybridization (CGH [Human Genome CGH Microarray 105A platform, Agilent, Santa Clara, CA]) and transcriptome analysis (GeneChip Human Exon 1.0 ST Array, Affymetrix, Santa Clara, CA) that the affected family members did not carry any larger chromosomal alterations at the NF2 locus and that their NF2 expression was not altered (fold change = 0.99). The seven available tumor samples from the affected individuals were screened by direct sequencing for somatic NF2 mutations, but none were identified.
Figure 1.
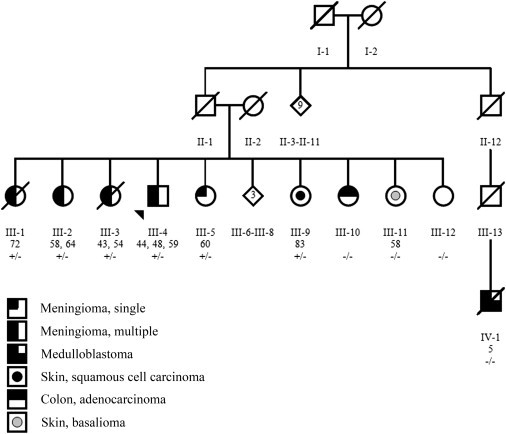
Pedigree of Family 1 with Five Siblings with Intracranial Meningiomas
The ages at meningioma operations and the c.367C>T (p.Arg123Cys) SUFU mutation status of the individuals are shown. A plus sign denotes a mutation, and a minus sign denotes a wild-type allele. The pedigree has been slightly modified for confidentiality.
Samples were obtained from four affected (III-1, III-2, III-4, and III-5) and four other (III-10, a child of III-1, a child of III-3, and a child of III-5) family members from Family 1 (Figure 1) after written informed consent was acquired. Genomic DNAs were extracted from blood and were genotyped with Illumina’s Human610-Quad DNA analysis BeadChips (Illumina, San Diego, CA). The hybridization and scanning were performed according to the manufacturer’s protocol at the Finnish Institute for Molecular Medicine (FIMM) Genome and Technology Centre. Genotype calling and quality analyses were done in BeadStudio (Illumina), and genotypes with a GenCall score less than 0.15 were excluded as unreliable.
Nonparametric linkage analysis was performed with Merlin7 (v.1.1.2) and for X chromosome with MINX. Unlikely genotypes calculated with pedwipe in MERLIN were removed before the analyses. Allele frequencies were obtained from genotypes from members of family 1 and 265 healthy Finnish controls provided by the Nordic Center of Excellence in Disease Genetics consortium. In the analysis, the family members with multiple meningiomas (III-1, III-2, and III-4) were marked as affected, the sibling with a single meningioma (III-5) and the three children (children of III-1, III-3, and III-5) were marked as unknown, and the sibling with no meningioma was marked as unaffected (Figure 1). In total, 174 chromosomal regions (1.3 Gb all together) with positive LOD scores were identified (Table S1, available online).
To study the protein-coding variants residing in the linked regions, we used the SureSelect Human All Exon Kit (v.1) (Agilent) to capture the germline exomes from III-1, III-2, and III-4. Paired-end short read sequences covering ∼38 Mb of the human genome were sequenced with Illumina Genome Analyzer II at FIMM Genome Technology Centre. NextGENe (v.2.1) software (Softgenetics, State College, PA) was used for removing bad-quality reads, for mapping the reads to human reference genome version NCBI37/Hg19, and for calling the variants. All synonymous variants, known polymorphisms obtained from dbSNP (v.132), variants present in 78 control exomes available from other projects in the laboratory, and variants residing outside the linked regions were excluded from further analyses.
After the exome data were combined with the linkage data, only 20 candidate variants remained (Table S2). These were further directly sequenced in all affected individuals in family 1, and seven were shown to segregate with the meningiomas in the family (Table S2). All seven variants were then sequenced in a set of 188 Finnish blood donor samples obtained through the Finnish Blood Transfusion Service. Three of the variants were found in the controls with a low frequency, and two were present in the screened unaffected siblings, from whom we had available DNA (Table S3).
The two remaining variants were heterozygous missense mutations c.1990A>G (p.Ser664Pro) (rs202247760) in aminoadipate-semialdehyde synthase (AASS [MIM 605113; RefSeq accession number NM_005763.3]) and c.367C>T (p.Arg123Cys) (rs202247756) in suppressor of fused homolog (Drosophila), SUFU (MIM 607035; RefSeq NM_016169.3). In silico analyses with PolyPhen28 and SIFT9 predicted only the SUFU mutation to be damaging. Both variants were sequenced in the seven available formalin-fixed paraffin-embedded (FFPE) meningiomas (one from III-1 and III-5 each, two from III-3, and three from III-4) from the members of family 1. Loss of heterozygosity (LOH) was detected at the site of the SUFU mutation (Figure 2A) in all the tumors, whereas the AASS variant site remained heterozygous in all of the seven tumors. Because SUFU mutations are found in a fraction of familial medulloblastomas, we screened the c.367C>T mutation in a distant relative (IV-1, Figure 1) who had died of medulloblastoma at 5 years of age. We utilized DNA extracted from two different FFPE samples and performed multiple sequencing reactions, but no mutations were detected. SUFU mutations have also been observed in a subset of skin tumors, and we acquired the FFPE blocks of the facial-skin lesions from the two siblings (III-9 and III-11). The sister with basalioma (III-11) did not harbor the mutation, whereas the other sister with facial-skin squamous cell carcinoma (III-9) did, but no LOH was observed in the lesion.
Figure 2.
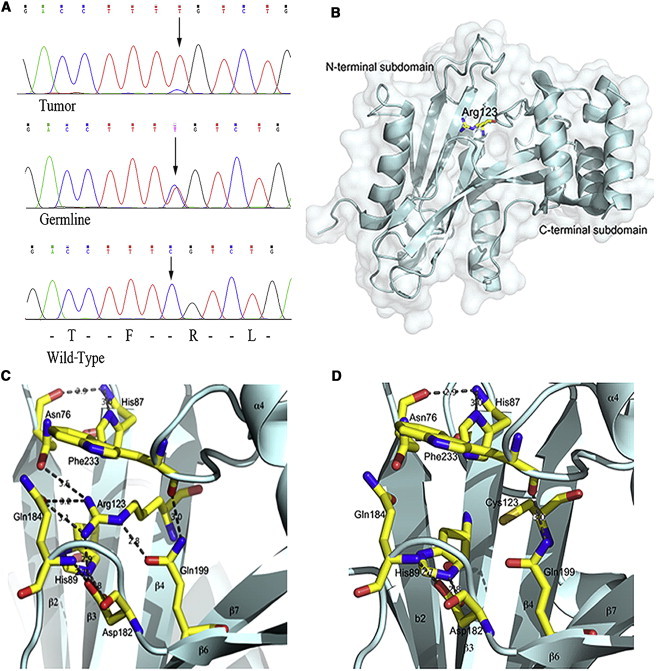
Loss of Heterozygosity and In Silico Modeling of the SUFU Mutation
(A) Heterozygous germline SUFU mutation c.367C>T (p.Arg123Cys) and loss of heterozygosity in a meningioma tumor tissue. The site of the mutation is indicated by an arrow.
(B–D) Surface representation of the N-terminal part of SUFU modeled in COOT.12
(B) Cartoon of the secondary structure. The N- and C-terminal subdomains are marked, and Arg123 is represented by a ball-and-stick model.
(C) Ribbon representation of the wild-type N-terminal part of SUFU. Secondary structure elements are labeled in accordance with Merchant et al.11 Arg123 and residues involved in interactions are represented by ball-and-stick models. Dashed lines represent hydrogen bonds and ionic contacts.
(D) The same orientation for the p.Arg123Cys altered SUFU. All contacts formed by Arg123 disappear, and loops are connected rather weakly.
To elucidate the properties of the p.Arg123Cys altered SUFU, we modeled it with the program COOT12 (Figure 2B–2D). Arg123 resides inside the core of the N-terminal subdomain of SUFU (Protein Data Bank entry 1M1L)13 (Figure 2B), and it forms a very intensive network of hydrogen bonds and ionic interactions with His89, Asp182, Gln184, and Gln199 (Figure 2C). These residues are central in the loop structures of the N-terminal subdomain of SUFU. An alteration in Arg123 might increase the flexibility of the loops and affect the formation of proper tertiary structure of the protein (Figure 2D).
SUFU is the major negative regulator of the Hh-signaling pathway in humans.10,11,14 The pathway is essential in embryogenesis, and in adult tissues, it is involved in the control of stem cell proliferation.15 Its activation has also been linked to the development of medulloblastomas, glioblastomas, and some basal cell carcinomas.15 The effect of SUFU on Hh signaling is mainly mediated by its binding to GLI1, GLI2, and GLI3 transcription factors. SUFU has also been shown to prevent the nuclear transportation of GLIs,16,17 as well as to promote their processing.
To study the effect of the observed SUFU variant on GLI function in vitro, we generated the c.367C>T mutation in a human SUFU cDNA construct cloned in a pCMV5_Myc backbone vector (Clonetech, Mountain View, CA) by utilizing the QuikChange Site-Directed mutagenesis kit (Agilent). We directly sequenced the mutated clones to ensure that only the correct mutation was created. The primers used in the site-directed mutagenesis are listed in Table S3. FuGENE HD (Roche, Basel, Switzerland) was used for transfecting human rhabdomyosarcoma cells (CRL-2061) with Myc-tagged wild-type or mutant SUFU constructs, an Hh-pathway-specific reporter plasmid “SASAKI COOPER,”18 and a control reporter Renilla luciferase phRL-TK19 (Promega, Fitchburg, WI). CRL-2061 cells are known to have constitutively activated Hh signaling as a result of a 30-fold amplification of GLI1.20 Cells were incubated for 2–3 days before the luciferase activity was measured with the Dual-Reporter Luciferase system (Promega). The luminescence was detected by the Digene DCR-1 luminometer (Digene Diagnostics/QIAGEN, Hilden, Germany). The assays were conducted with and without ShhN-conditioned medium in duplicate and were repeated at least three times. We calculated the relative luciferase activity by dividing the firefly luciferase counts by the Renilla luciferase counts. The same experiments were also conducted in Sufu-deficient (Sufu−/−) mouse embryonic fibroblasts (MEFs), and the results from both cell lines indicated that compared with the wild-type transfected cells, the altered SUFU had significantly reduced ability to suppress GLI1 activity (Figure 3A and 3B).
Figure 3.
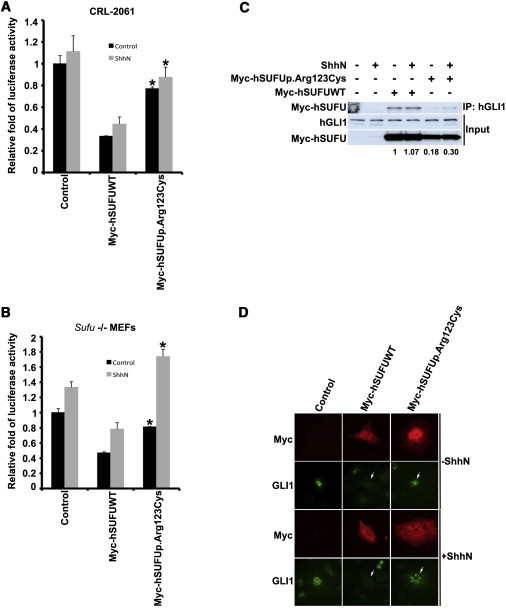
The Effect of the p.Arg123Cys Altered SUFU on Hedgehog Signaling Activity
(A and B) The p.Arg123Cys altered SUFU has decreased activity. Human rhabdomyosarcoma cells (CRL-2061) and Sufu-deficient (Sufu−/−) MEFs were transfected with Myc-tagged wild-type (Myc-SUFUWT) or altered (Myc-SUFUp.Arg123Cys) SUFU constructs, an Hh-pathway-specific reporter, and a control reporter. The cells were incubated with and without ShhN-conditioned medium, and luciferase activity was measured. Note that Myc-SUFU suppresses Hh-pathway activity in both human and mouse cell lines. ∗p < 0.05 compared with Myc-hSUFUWT (statistical analysis was done with a t test). Error bars indicate one standard deviation (duplicate samples).
(C) The p.Arg123Cys altered SUFU binds less GLI1 than does the wild-type SUFU. CRL-2061 cell lysates used in luciferase reporter assay were subjected to pull-down immunoblot analysis (IP: GLI1; WB: Myc and GLI1). In the top panel, Myc-SUFU is pulled down with antibodies against GLI1. Input of GLI1 and Myc-tagged SUFU constructs is presented in the middle and bottom panels, respectively. Quantification of GLI1 binding relative to the Myc-SUFU protein level is shown below the panel (quantitative protein analysis was performed with the Single Dimensional Electrophoretic Gel Analysis program from the ImageJ software package v.1.46g).
(D) Subcellular localization of p.Arg123Cys altered SUFU in Sufu−/− MEFs. Note that the Gli1 nuclear localization was not affected by the human SUFU mutant (Myc-SUFUp.Arg123Cys) compared with the wild-type SUFU (Myc-SUFUWT), as indicated by arrows.
To study whether this was due to reduced binding of altered SUFU to GLI, we studied the interaction of SUFU and endogenous GLI1 by pull-down immunoblot analysis in CRL-2061 cells. The cells were lysed and clarified by centrifugation, and the supernatant was removed and incubated with anti-GLI1 (H-300) agarose (Santa Cruz, Santa Cruz, CA). The agarose beads were washed three times with the lysis buffer without protease inhibitors before the SDS sample buffer was added. The samples were resolved on SDS-PAGE gels, transferred to polyvinylidene-fluoride membranes, and incubated with the following antibodies: goat anti-Myc HRP (ab1261, Abcam, Cambridge, UK) and rabbit anti-Gli1 (ab92611, Abcam). Goat anti-rabbit HRP was used as a secondary antibody. The protein level of the p.Arg123Cys altered SUFU was clearly reduced, and the altered protein bound less GLI1 than did the wild-type SUFU (Figure 3C).
To assess the effect of the p.Arg123Cys altered SUFU on the subcellular localization of GLI1, we seeded Sufu−/− MEFs on human-fibronectin-coated glass coverslips (BD, Franklin Lakes, NJ) and transfected them with the Myc-tagged wild-type and c.367C>T mutant SUFU expression plasmids. Cells were incubated for 2 days in low-serum medium with and without ShhN-conditioned medium (Figure S1) and were stained as described previously.10 Specific anti-Myc, anti-Gli1 (ab49314, Abcam), Alexa-594-conjugated anti-mouse, and Alexa-488-conjugated anti-rabbit (Molecular Probes/Life Technologies, Carlsbad, CA) antibodies were used for staining. This showed that the altered SUFU was unable to relocalize GLI1 into the cytoplasm (Figure 3D). Taken together, the functional experiments indicate that the c.367C>T (p.Arg123Cys) mutation in SUFU causes a partial loss of its repressor activity and leads to aberrant activation of the Hh-signaling pathway.
After identification of the SUFU c.367C>T (p.Arg123Cys) mutation in family 1, the coding exons and exon-intron boundaries of SUFU were sequenced in the DNA samples from a total of 162 meningioma-affected individuals (four Finnish individuals who had multiple meningiomas and from whom NF2 mutations had been screened but not identified, 77 Finnish simplex cases who had solitary meningiomas and who were enrolled in the Interphone case-control study,21 40 UK individuals with multiple meningiomas,22 and 41 FFPE meningiomas collected through the Finnish Cancer Registry [FCR] [Table 1]). Informed consent was obtained from the Interphone study subjects and from the four Finnish individuals with multiple meningiomas. The use of 41 FCR-retrieved FFPE samples was approved by the National Supervisory Authority for Welfare and Health. The study was approved by the Ministry of Social Affairs and Health in Finland, and the usage of the samples was approved by the local ethics review committees in Finland and the UK.
Table 1.
Meningioma Cases and Healthy Controls Used in the Study
| Description of the Cases and Controls | Number of Cases | Family History of Meningiomas | Lymphocyte DNA | FFPE Tumor-Tissue DNA |
|---|---|---|---|---|
| Family 1, meningioma cases | 5 | yes | 4 | 7 |
| Family 1, other family members | 11 | yes | 11 | 2 |
| Finnish meningioma cases recruited from the Interphone studya | 77 | not determined | 77 | 0 |
| Finnish multiple-meningioma casesb | 4 | no | 4 | 0 |
| UK multiple-meningioma casesb | 35 | no | 35 | 0 |
| Familialc UK meningioma cases | 5 | yes | 5 | 0 |
| Healthy blood donors | 181 | not determined | 181 | 0 |
| Meningioma Cases Identified from the Finnish Cancer Registry | ||||
| Familialc Finnish meningioma cases | 6 | yes | 1 | 5 |
| Putatively familial meningioma casesd | 20 | possibly | 0 | 20 |
| Meningioma cases from the same geographical region as family 1 | 6 | not determined | 0 | 6 |
| Early-onset (≤35 years) meningioma cases | 9 | not determined | 0 | 9 |
The following abbreviation is used: FFPE, formalin-fixed paraffin-embedded.
INTERPHONE Study Group; Bethke et al.21
These cases have been screened for NF2 mutations, but no mutations were detected.
Defined as ≥ two first-degree relatives with meningioma.
Collected on the basis of the family name at birth and birth municipality through a search into the Finnish Cancer Registry.
The primers used for blood and FFPE-tissue-derived DNAs were designed with Primer3 (Table S3). Sequences were analyzed manually and with Mutation Surveyor software v.3.30 (SoftGenetics) with an NCBI reference sequence (RefSeq NM_016169.3).
In total, 162 individuals with meningioma were screened for additional SUFU mutations, but no pathogenic variants were detected (Table S4). Of note, only 11 of the cases had a confirmed family history of the disease. Furthermore, in family 1, four of the affected individuals had been diagnosed with multiple tumors, which are usually seen in fewer than 10% of the cases. The absence of additional mutation-positive individuals might indicate the very specific nature of the mutation—which provides a “just right” level of SUFU dysfunction—and the resulting small mutational target.
Mutations in SUFU predispose to childhood medulloblastomas, and somatic mutations have been observed in medulloblastomas, as well as in a small subset of basal cell skin cancers.23 A splice-site mutation has also been reported in a family affected by basal cell nevus syndrome (BCNS [MIM 109400]) without the presence of medulloblastomas.24 The germline SUFU mutations have been predominantly truncating, and the tumors have typically shown loss of the wild-type allele, indicating that SUFU is a tumor suppressor. This notion has been validated in knockout mice experiments.14,25 In contrast to the previously reported truncating mutations, the mutation identified in the meningioma-affected family is a missense change converting arginine to cysteine. Functional analyses indicate that the activity of the altered protein is significantly reduced but not completely lost. Because SUFU is known to regulate Hh signaling in a concentration-dependent manner,16 we hypothesize that the remaining activity of the altered protein might explain the phenotypic difference between aggressive childhood medulloblastomas and meningiomas. This, however, remains to be validated in further studies.
SUFU is located in chromosomal region 10q24.32. The long arm of chromosome 10 is frequently deleted in meningiomas, especially in tumors with anaplastic, atypical, and malignant morphology. This deleted region contains various known tumor-suppressor genes, among which are PTEN and SUFU. Boström et al. (1998) studied chromosome arm 10q in glioblastomas and meningiomas and found PTEN to be mutated and deleted in a subset of gliomas but not in meningiomas, suggesting that PTEN is not the major tumor-suppressor gene at 10q in meningiomas.26 Of note, Laurendeau et al. (2010) showed that 23 of 32 (72%) genes in the Hh-signaling pathway were differentially expressed in human meningiomas compared to normal tissue, suggesting an important role for Hh-pathway activation in meningiomas.27 A germline mutation in PTCH1 (MIM 601309), another key player in Hh signaling, has also been reported in an individual with BCNS with multiple basal cell carcinoma and meningioma.28
Meningiomas are common tumors, and a subset of them displays familial aggregation. In some familial cases, the genetic predisposition is caused by an inherited syndrome, and the individuals display other syndromic manifestations as well. In other familial cases, however, the predisposing factor remains unclear, particularly if additional clinical signs pointing toward a well-characterized syndrome are lacking. Here, we report on a germline SUFU c.367C>T (p.Arg123Cys) mutation segregating with the meningiomas in a family of five affected siblings. The mutation was absent from the healthy controls, and all available tumors from the family displayed clear loss of the wild-type allele. We also showed that the altered protein’s ability to suppress Hh signaling was significantly reduced. This suggests that SUFU mutations predispose to meningiomas in addition to medulloblastomas. Additional mutations were not detected in other meningioma cases studied, and this intuitively interesting finding thus awaits confirmation in an independent material. It is also possible that other genic mutations resulting in an abnormal activation of the Hh pathway might underlie the meningioma susceptibility in some families with yet unknown etiology.
Acknowledgments
This study was supported by the Academy of Finland (Center of Excellence in Cancer Genetics and grant 21290), the Cancer Society of Finland, and the Sigrid Juselius Foundation. We would like to thank all the individuals who participated in the study. We thank Riitta Herva and Hannu Tuominen for expertise in pathology and Sini Nieminen, Mairi Kuris, Iina Vuoristo, and Inga-Lill Svedberg for excellent technical assistance. We acknowledge the Finnish Institute for Molecular Medicine Genome and Technology Centre for service, the CSC–IT Center for Science for the allocation of computational resources, and the Nordic Center of Excellence in Disease Genetics for providing the Finnish control SNP data.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
National Center for Biotechnology Information (NCBI) database, http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Primer3, http://frodo.wi.mit.edu/
SIFT, http://sift.jcvi.org/
Accession Numbers
The dbSNP accession numbers for the sequences reported in this paper are available in Tables S2 and S4.
References
- 1.Wiemels J., Wrensch M., Claus E.B. Epidemiology and etiology of meningioma. J. Neurooncol. 2010;99:307–314. doi: 10.1007/s11060-010-0386-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.CBTRUS (2011). CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2004–2007. Central Brain Tumor Registry of the United States, www.cbtrus.org.
- 3.Krampla W., Newrkla S., Pfisterer W., Jungwirth S., Fischer P., Leitha T., Hruby W., Tragl K.H. Frequency and risk factors for meningioma in clinically healthy 75-year-old patients: Results of the Transdanube Ageing Study (VITA) Cancer. 2004;100:1208–1212. doi: 10.1002/cncr.20088. [DOI] [PubMed] [Google Scholar]
- 4.Vernooij M.W., Ikram M.A., Tanghe H.L., Vincent A.J., Hofman A., Krestin G.P., Niessen W.J., Breteler M.M., van der Lugt A. Incidental findings on brain MRI in the general population. N. Engl. J. Med. 2007;357:1821–1828. doi: 10.1056/NEJMoa070972. [DOI] [PubMed] [Google Scholar]
- 5.Nahser H.C., Grote W., Löhr E., Gerhard L. Multiple meningiomas. Clinical and computer tomographic observations. Neuroradiology. 1981;21:259–263. doi: 10.1007/BF02100156. [DOI] [PubMed] [Google Scholar]
- 6.Christiaans I., Kenter S.B., Brink H.C., van Os T.A., Baas F., van den Munckhof P., Kidd A.M., Hulsebos T.J. Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J. Med. Genet. 2011;48:93–97. doi: 10.1136/jmg.2010.082420. [DOI] [PubMed] [Google Scholar]
- 7.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 8.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 10.Varjosalo M., Li S.P., Taipale J. Divergence of hedgehog signal transduction mechanism between Drosophila and mammals. Dev. Cell. 2006;10:177–186. doi: 10.1016/j.devcel.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 11.Ruel L., Thérond P.P. Variations in Hedgehog signaling: Divergence and perpetuation in Sufu regulation of Gli. Genes Dev. 2009;23:1843–1848. doi: 10.1101/gad.1838109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emsley P., Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 13.Merchant M., Vajdos F.F., Ultsch M., Maun H.R., Wendt U., Cannon J., Desmarais W., Lazarus R.A., de Vos A.M., de Sauvage F.J. Suppressor of fused regulates Gli activity through a dual binding mechanism. Mol. Cell. Biol. 2004;24:8627–8641. doi: 10.1128/MCB.24.19.8627-8641.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Svärd J., Heby-Henricson K., Persson-Lek M., Rozell B., Lauth M., Bergström A., Ericson J., Toftgård R., Teglund S. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev. Cell. 2006;10:187–197. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 15.Varjosalo M., Taipale J. Hedgehog: Functions and mechanisms. Genes Dev. 2008;22:2454–2472. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 16.Stone D.M., Murone M., Luoh S., Ye W., Armanini M.P., Gurney A., Phillips H., Brush J., Goddard A., de Sauvage F.J., Rosenthal A. Characterization of the human suppressor of fused, a negative regulator of the zinc-finger transcription factor Gli. J. Cell Sci. 1999;112:4437–4448. doi: 10.1242/jcs.112.23.4437. [DOI] [PubMed] [Google Scholar]
- 17.Kogerman P., Grimm T., Kogerman L., Krause D., Undén A.B., Sandstedt B., Toftgård R., Zaphiropoulos P.G. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat. Cell Biol. 1999;1:312–319. doi: 10.1038/13031. [DOI] [PubMed] [Google Scholar]
- 18.Taipale J., Chen J.K., Cooper M.K., Wang B., Mann R.K., Milenkovic L., Scott M.P., Beachy P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- 19.Taipale J., Cooper M.K., Maiti T., Beachy P.A. Patched acts catalytically to suppress the activity of Smoothened. Nature. 2002;418:892–897. doi: 10.1038/nature00989. [DOI] [PubMed] [Google Scholar]
- 20.Roberts W.M., Douglass E.C., Peiper S.C., Houghton P.J., Look A.T. Amplification of the gli gene in childhood sarcomas. Cancer Res. 1989;49:5407–5413. [PubMed] [Google Scholar]
- 21.Bethke L., Murray A., Webb E., Schoemaker M., Muir K., McKinney P., Hepworth S., Dimitropoulou P., Lophatananon A., Feychting M. Comprehensive analysis of DNA repair gene variants and risk of meningioma. J. Natl. Cancer Inst. 2008;100:270–276. doi: 10.1093/jnci/djn004. [DOI] [PubMed] [Google Scholar]
- 22.Hadfield K.D., Smith M.J., Trump D., Newman W.G., Evans D.G. SMARCB1 mutations are not a common cause of multiple meningiomas. J. Med. Genet. 2010;47:567–568. doi: 10.1136/jmg.2009.075721. [DOI] [PubMed] [Google Scholar]
- 23.Taylor M.D., Liu L., Raffel C., Hui C.C., Mainprize T.G., Zhang X., Agatep R., Chiappa S., Gao L., Lowrance A. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002;31:306–310. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- 24.Pastorino L., Ghiorzo P., Nasti S., Battistuzzi L., Cusano R., Marzocchi C., Garrè M.L., Clementi M., Scarrà G.B. Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am. J. Med. Genet. A. 2009;149A:1539–1543. doi: 10.1002/ajmg.a.32944. [DOI] [PubMed] [Google Scholar]
- 25.Lee Y., Kawagoe R., Sasai K., Li Y., Russell H.R., Curran T., McKinnon P.J. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene. 2007;26:6442–6447. doi: 10.1038/sj.onc.1210467. [DOI] [PubMed] [Google Scholar]
- 26.Boström J., Cobbers J.M., Wolter M., Tabatabai G., Weber R.G., Lichter P., Collins V.P., Reifenberger G. Mutation of the PTEN (MMAC1) tumor suppressor gene in a subset of glioblastomas but not in meningiomas with loss of chromosome arm 10q. Cancer Res. 1998;58:29–33. [PubMed] [Google Scholar]
- 27.Laurendeau I., Ferrer M., Garrido D., D’Haene N., Ciavarelli P., Basso A., Vidaud M., Bieche I., Salmon I., Szijan I. Gene expression profiling of the hedgehog signaling pathway in human meningiomas. Mol. Med. 2010;16:262–270. doi: 10.2119/molmed.2010.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tate G., Li M., Suzuki T., Mitsuya T. A new germline mutation of the PTCH gene in a Japanese patient with nevoid basal cell carcinoma syndrome associated with meningioma. Jpn. J. Clin. Oncol. 2003;33:47–50. doi: 10.1093/jjco/hyg005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.