

Abstract
Polymicrogyria is a malformation of the developing cerebral cortex caused by abnormal organization and characterized by many small gyri and fusion of the outer molecular layer. We have identified autosomal-recessive mutations in RTTN, encoding Rotatin, in individuals with bilateral diffuse polymicrogyria from two separate families. Rotatin determines early embryonic axial rotation, as well as anteroposterior and dorsoventral patterning in the mouse. Human Rotatin has recently been identified as a centrosome-associated protein. The Drosophila melanogaster homolog of Rotatin, Ana3, is needed for structural integrity of centrioles and basal bodies and maintenance of sensory neurons. We show that Rotatin colocalizes with the basal bodies at the primary cilium. Cultured fibroblasts from affected individuals have structural abnormalities of the cilia and exhibit downregulation of BMP4, WNT5A, and WNT2B, which are key regulators of cortical patterning and are expressed at the cortical hem, the cortex-organizing center that gives rise to Cajal-Retzius (CR) neurons. Interestingly, we have shown that in mouse embryos, Rotatin colocalizes with CR neurons at the subpial marginal zone. Knockdown experiments in human fibroblasts and neural stem cells confirm a role for RTTN in cilia structure and function. RTTN mutations therefore link aberrant ciliary function to abnormal development and organization of the cortex in human individuals.
Main Text
Malformations of the developing brain cortex are a heterogeneous group of disorders and a major cause of intellectual disability, epilepsy, and neurological symptoms, often requiring life-long support and treatment.1–3 Polymicrogyria (PMG) is in general appearance and distribution a heterogeneous disorder, considered to be the result of postmigratory abnormal cortical organization. PMG is characterized in brain imaging by many irregular small gyri and in microscopic examination by fusion of the molecular layer over multiple small gyri, which gives a festooned appearance to the cortical surface, without abnormal neuronal migration.4 The causes are also very heterogeneous, including nongenetic causes such as prenatal insufficient vascular supply (e.g., twin-twin transfusion syndrome), intoxication (e.g., fetal alcohol spectrum disorder), and viral infections (e.g., cytomegalovirus) (for review, see Leventer et al.).1
More than 100 syndromes are annotated that include PMG (London Medical Database [LMD], see Web Resources). Conversely, mutations in only a few genes, such as TUBB2B (MIM 612850), TUBA8 (MIM 605742), TUBB3 (MIM 602661), and WDR62 (MIM 613583), have been associated with isolated nonsyndromic PMG, accounting for only a minority of affected individuals.2,3 Originally, a recognizable distribution was linked to mutations in specific genes; however, subsequent observations had shown a heterogeneity of distribution and even type of cortical malformation, thus making the choice of a targeted diagnostic test more difficult.2,3 For example, mutations in GPR56 (MIM 604110) were originally described in individuals with an MRI diagnosis of PMG, but later studies of brain pathology classified the cortical abnormalities as cobblestone malformations.3 Autosomal-recessive isolated nonsyndromic PMG is extremely rare and only sporadically reported.3
Here, we report homozygous recessive mutations in RTTN (MIM 610436) in individuals from two unrelated consanguineous families presenting with bilateral PMG diagnosed via MRI of the cerebral cortex and without other major malformations.
Written informed consent for the study was obtained from the families according to the Erasmus University Medical Center (Erasmus MC) institutional review board requirement. Family 1 originates from Turkey and includes three individuals. The clinical presentation is summarized in Table 1. MRI of the brains of two individuals revealed a diffuse asymmetric PMG extending from the frontal to the temporal, parietal, and occipital areas (Figures 1A–1G). In view of the multiple consanguineous loops, we assumed that the cause would be a homozygous autosomal-recessive mutation.
Table 1.
Clinical Presentation of Individuals with RTTN Mutations
| Clinical Findings | Family 1, Individual 1 | Family 1, Individual 2 | Family 1, Individual 3 | Family 2, Individual 1 |
|---|---|---|---|---|
| Sex | male | male | female | male |
| Birth weight (gestational week) | −2 SD (38 weeks) | NK | NK | 0 SD (40 weeks) |
| Age at observation | 12 years | 14 years | 18 years | 16 years |
| Height (age) | −2 SD (12 years) | −1 SD (24 years) | NK | −2.5 SD (16 years) |
| Head circumference (age) | −2.5 SD (12 years) | −2 SD (24 years) | NK | 0 SD (16 years) |
| Cognitive development | severe ID | moderate ID | moderate ID | severe ID |
| Behavior and communication skills | temper tantrums | friendly | friendly | friendly |
| Speech | few words, dysarthria | few words, dysarthria | NK | no speech, drooling |
| Seizures | yes | yes | yes | yes |
| Findings at neurological exam | pyramidal signs, unassisted deambulation | NK | NK | spastic tetraparesis, no deambulation |
| EEG | intermittent fast activity in left centro-parietal and temporal areas; slow waves in bilateral frontal areas | NK | NK | clusters of peak waves in left fronto-central and temporal areas |
| Brain MRI (age) | + (1 year) | + (14 years) | – (permission denied) | + (7 years) |
| Polymicrogyria, distribution and cerebral findings | asymmetric R > L irregular gyral pattern that involves the posterior frontal-perisylvian and parietal regions, reduced-volume white matter beneath cortical malformation, normal third and mildly enlarged lateral ventricles, thin corpus callosum | extensive asymmetric R > L irregular gyral pattern that involves posterior frontal-perisylvian and parietal regions and possibly the occipital lobes, most severe in posterior frontal-perisylvian regions; mildly enlarged lateral ventricles; mildly short corpus callosum | – | irregular gyral pattern with microgyri in perisylvian, parietal, and superior occipital regions, most severe in parietal regions; posterior to posterior sulcus region; reduced-volume white matter in parietal and occipital regions with dysplastic infolding only; normal to mildly thick corpus callosum |
| Cerebellum | normal | mild cerebellar atrophy with reduced volume and mildly enlarged fourth ventricle | – | normal |
| Abdominal and renal ultrasound (age) | normal, situs solitus, small kidney volume (12 years) | NK | NK | normal, situs solitus (16 years) |
| Heart examination | normal (12 years) | NK | NK | normal (16 years) |
| Ophthalmological examination | normal | NK | NK | normal |
| BAEP | normal | NK | NK | normal |
| CPK | normal | NK | NK | NK |
The following abbreviations are used: NK, not known; ID, intellectual disability; EEG, electroencephalogram; R, right: L, left; BAEP, brainstem auditory evoked potentials; CPK, creatine phosphokinase.
Figure 1.
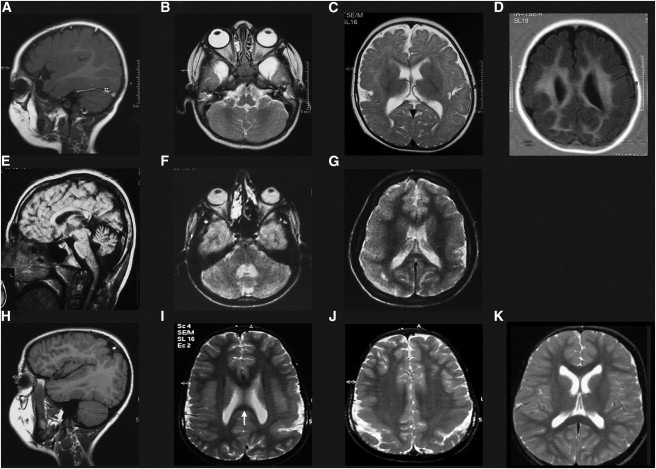
Brain Imaging of Individuals with PMG from Two Consanguineous Families
MRI of individuals 1 (A–D, at age 1 year) from family 1 shows a diffuse and asymmetric abnormality of the cortex with the thickened and irregular surface typical of PMG, most pronounced in the perisylvian (A and C), parietal (A, C, and D), and occipital (C and D) areas and moderate dilatation of lateral ventricles (C and D). Bilateral temporal arachnoidal cysts and normal cerebellum are visible (B). The MRI of individual 2 from family 1 (E–G, at age 14 years) shows small cerebellar vermis (E) and hemispheres (F) and diffuse bilateral, slightly asymmetric PMG (G). The MRI of the individual from family 2 (H–J, at age 7 years) shows a bilaterally polymicrogyric cortex in the temporal areas around the sylvian fissure (H), in the parietal (J) and occipital (I) areas, with reduced occipital white matter and thin splenium of corpus callosum (I, arrow). Age-matched control of 13 years (K).
We used Affymetrix Human GeneChip SNP 6.0 arrays to perform autozygosity mapping on the basis of a whole-genome search from the three affected members of the family.5 This technique has been used with great success for identification of regions containing the gene of interest in autosomal-recessive disorders. By this method, areas larger than 2 Mb containing high-density homozygous SNPs can confidently be considered to be identical by descent and can be explored for recessive mutations. The Affymetrix Genotyping Console (GTC4.1.1) was used for genotyping with the Birdseed v.2 algorithm and copy number and region of homozygosity analysis with the Regional GC Correction algorithm as well as a reference model file containing 12 in-house reference samples.
In the three affected individuals, we found two shared homozygous regions larger than 2 Mb on chromosomes 14q24.3-31.1, containing 25 genes, and 18q22, containing 5 genes (Figures S1 and S2 available online). We sequenced all the candidate protein-coding genes in both regions (Tables S1 and S2). We found only one homozygous nonsynonymous change in the three affected individuals in a highly conserved residue of exon 22 of RTTN, missense change c.2796A>T, which predicts an amino acid substitution, p.Leu932Phe (RefSeq accession number NM_173630.3) (Figure 2A). The change was analyzed in additional family members; it cosegregated with the phenotype in the pedigree (Figure 2A) and was heterozygous in the parents. In our cohort including 70 isolated cases of PMG,6 we identified a second consanguineous family from Cape Verde (family 2) with one affected individual showing a >2 Mb homozygous area on chromosome 18q22, overlapping the RTTN locus (Table 1). His brain MRI showed a bilateral PMG that was more pronounced in the temporal, parietal, and occipital areas (Figures 1H–1J). This proband (Figure 2B, subject 4) had a homozygous c.80G>A change in exon 2 of RTTN, causing a p.Cys27Tyr amino acid substitution (Figure 2C), whereas both parents were heterozygous for the change. The extremely low allele frequencies and the algorithm predictions point to the RTTN mutation as the cause of the disease (Figure 2).
Figure 2.
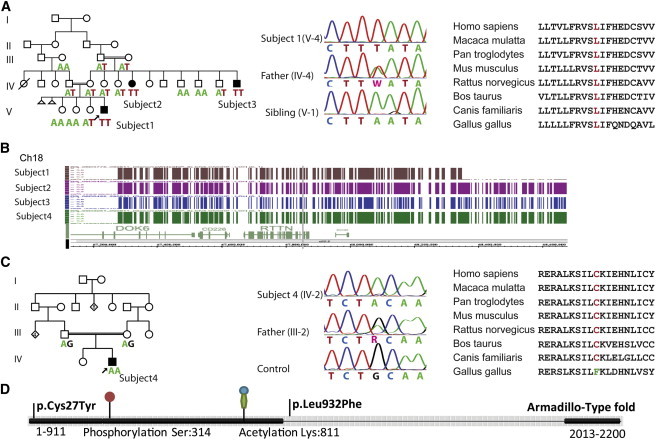
Identification of the RTTN Mutations in Families with PMG
(A) Left, pedigree of family 1. Affected subjects have black symbols, and below, the RTTN genotype is indicated, which illustrates cosegregation of the c.2796A>T change with the phenotype. Middle, electropherogram showing the homozygous mutation (subject V-4), the heterozygous father (IV-4), and the normal sequence (healthy sibling V-1). PCR products were purified with ExoSAP-IT (USB), and direct sequencing of both strands was performed with BigDye Terminator chemistry v.3.1 on an ABI PRISM 3130xl Genetic Analyzer (Applied Biosystems). Sequences were aligned and compared with consensus sequences obtained from the human genome databases (SeqScape v.2.5 software, Applied Biosystems). For annotation of DNA and protein changes, the mutation nomenclature guidelines from the Human Genome Variation Society were followed. Right, conservation of leucine 932 among species.
(B) Visualization with the Genotyping Console browser of Affymetrix SNP 6.0 Array data, showing the overlapping areas of homozygosity among individuals from family 1 (upper three) and family 2 (lowest lane).
(C) Left, pedigree of family 2. The affected subject has a black symbol, and below, the RTTN genotype at position c.80 is indicated. Middle, electropherogram showing the homozygous mutation (subject IV-2), the heterozygous father (III-2), and the normal sequence (control). Right, conservation of cysteine 27 among species.
(D) Schematic representation of Rotatin, including the position of the amino acid change, Armadillo-like domains, and posttranslational modification sites.
The c.2796A>T change was not present in 100 ethnically matched, healthy Turkish individuals and 165 individuals of European descent. The c.80G>A change was absent in 98 Cape Verdean ethnically matched individuals and 166 healthy individuals of European descent. Both the c.2796A>T and the c.80G>A changes were not annotated as polymorphic in dbSNP130, nor were they present in 679 control individuals of the 1000 Genomes database and the Exome Variant Sever (University of Washington), indicating an extremely low allele frequency in the healthy population. Algorithms PolyPhen-2, SNAP, and Mutation Taster predicted the changes as probably being deleterious (see Web Resources).
RTTN encodes Rotatin (UniProt Knowledgebase [UniProtKB], see Web Resources), a large (2,226 amino acid) protein of unknown function containing two Armadillo-like domains, highly conserved among species (Figure 2D).7,8 Armadillo domains are repeat-rich areas that mediate protein-protein interaction, as seen in proteins transducing signals of cell adhesion molecules to the cytoskeleton and wingless-type MMTV integration site family members (WNTs). The p.Cys27Tyr substitution is located in the first Armadillo domain; the p.Leu932Phe substitution is in a region of unknown function.
Rttn knockout mice, missing the murine ortholog of RTTN, show embryonic lethality, with deficient axial rotation, notochord degeneration, abnormal differentiation of the neural tube, loss of the left-right specification of the heart, and severe hydrocephalus.7 These phenotypic abnormalities are often also encountered in disorders affecting both primary and motile cilia.9–11 The no turning Rttn spontaneous mouse mutant has anteroposterior and dorsoventral patterning defects identical to Kif3a and Kif3b mutants, which lack the kinesin-II motor subunits of ciliary intraflagellar transport.8,12,13 Kif3a knockout embryos show rotational defects, lack nodal cilia, and show aberrant sonic hedgehog signaling. Cilia are complex microtubular organelles derived from centrioles, important for their role in embryonic development, signal transduction, cell-cell interaction, and human disorders, which are collectively called ciliopathies.9–11,14–17 The Drosophila melanogaster homolog of Rotatin, Ana3, is necessary for the structural integrity of centrioles and basal bodies and is essential for maintenance of sensory neurons.18 Rotatin has recently been localized to the mitotic centrosomes in HeLa and KE37 cells.18,19 We raised polyclonal antibodies against human Rotatin (rabbit anti-Rotatin, Santa Cruz Biotechnology, Sc-85129) and performed immunofluorescent staining of cilia and basal bodies in human cultured skin fibroblasts (Figure 3A).21,22 Rotatin localized at the ciliary basal bodies in control fibroblasts (Figure 3B).
Figure 3.
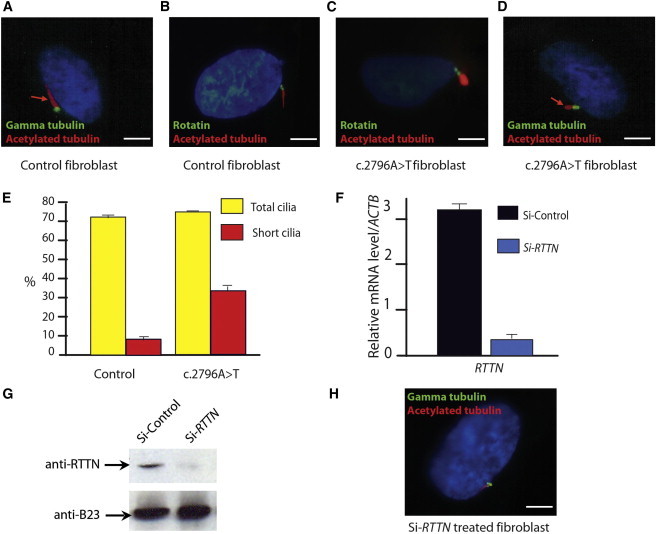
Immunofluorescent Staining of Rotatin and Cilia in Growth-Arrested Control Fibroblast, Affected-Individual Fibroblasts, and siRNA-Treated Controls
A total of 1 × 105 cells were seeded on glass coverslips in six-well cell culture plates and cultured in Dulbecco’s modified Eagle’s medium with 10% fetal calf serum (FCS) for 24 hr and for an additional 24 hr with 0.5% FCS (serum starvation), in order to arrest cell growth and allow ciliogenesis. Cells were fixed in cytoskelfix-20 (Cytoskeleton) for 5–10 min at −20°C. Afterward, cells were treated for 10 min at room temperature in blocking buffer (0.05 M Tris; 0.9 M NaCl; 0.25% gelatine; 0.5% Triton X-100; pH 7.4), then were incubated with primary antibodies for 2 hr (mouse monoclonal anti- acetylated tubulin [Sigma-Aldrich T7451;1:8,000], rabbit anti-gamma-tubulin [Sigma-Aldrich T7451;1:1,000], and rabbit anti-Rotatin [Santa Cruz Biotechnology, Sc-85129;1:200]). Cells were washed and incubated with Hoechst 33342 (Invitrogen;1:2,000), conjugated donkey anti-mouse-cy3 (Jackson ImmunoResearch; 1:200) and conjugated donkey anti-rabbit-cy2 (Jackson ImmunoResearch;1:200) for 1 hr. Fluorescence was visualized on an AXip-Axioplan2 imaging microscope (Zeiss) with a COOLSNAP-Pro camera (Zeiss). A total of 200 cells per well were examined and scored. During siRNA treatment, the cells were incubated for a longer time after trypsinization, which accounts for the higher percentage of control cells with normal cilia.
(A) Control fibroblasts show a normal cilium with basal bodies (green) and axoneme (red, arrow).
(B) Control fibroblasts stained with Rotatin antibodies (green) show localization of the protein to the basal bodies of the cilia; axoneme in red.
(C) Fibroblasts from one individual with the c.2796A>T change (subject 1 in the pedigree) show normal Rotatin localization but abnormally short and bulbous axoneme.
(D) Cell from the same individual shows normal basal bodies and abnormally short axoneme.
(E) Graphical representation of cilia abnormalities in c.2796A>T cells. The total percentage of ciliated cells is comparable with the controls (n = 10), but the percentage of structural abnormalities (in red) is significantly higher in c.2796A>T cells (35%) compared to controls (10%) (Student’s t test; p < 0.01). All the experiments were blinded and performed in parallel cultures of affected individual and controls; length and shape of the axoneme was visually scored.
(F and G) Control cells treated with RTTN-specific siRNA show downregulation of mRNA relative to housekeeping gene ACTB20 (F) and decreased Rotatin levels on western blot (G). Predesigned siRNA pools targeting transcripts of the human RTTN (L-031139-01) (Si-RTTN) and a nontarget control siRNA pool (D-001810-10-20, Dharmacon) (Si-Control) were used for knocking down RTTN in two separate control fibroblast cell lines and hNSCs (see Figure S4). siRNA (15 nM) was delivered into fibroblasts and hNSCs (3 × 105 cells) with either the siLentFect Lipid Reagent (BioRad) or nucleofectin reagent (Amaxa Nucleofector Kit V). Cells were harvested after 96 hr, seeded, and transfected again. Protein levels were examined via western blot analysis 72 hr after a second transfection. Fifty μg of cell extracts was prepared in RIPA buffer (10 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Nonidet P-40, 1% NaDOC, and 0.1% SDS) and was resolved with SDS-PAGE. Primary antibodies used for blotting were Rotatin (Santa Cruz Biotechnology; Sc-85129) and Nucleophosmin (B23) (Abcam; ab37659) as a control. Blots were developed with the enhanced chemiluminescence detection kit (Pierce). The following abbreviations are used: anti-RTTN, human Rotatin antibodies; and anti-B23, Nucleophosmin antibodies.
(H) Control cell after downregulation of RTTN expression by siRNA, showing an abnormal number of basal bodies and a short axoneme. Scale bars represent 5 μm; error bars represent SEM.
Given Rotatin localization, we investigated whether the RTTN mutations cause ciliary defects in the affected individual cells. Rotatin was detected at basal bodies in fibroblasts from an individual with c.2796A>T, suggesting that the mutation does not affect synthesis and localization of the protein (Figure 3C). However, we observed a higher percentage of structural cilia abnormalities, including short cilia, in c.2796A>T cells compared to control cell lines (n = 10). Up to 40% of the short cilia had a bulbous tip (Figures 3D and 3E and Figures S3C and S3D). Cells from the individual with c.80G>A did not show significant structural or numeric cilia anomalies (Figures S3E and S3F). To confirm the findings, we treated control fibroblast cell lines with RTTN-specific small interfering RNAs (siRNAs), which caused downregulation of RTTN mRNA and a decreased protein level (Figures 3F and 3G). In 8% of RTTN-siRNA-treated fibroblasts, we observed abnormally short cilia and multiple basal bodies, compared to 1% of the same mock-transfected control cell line (Figure 3H), indicating that Rotatin is involved in the maintenance of a normal ciliary structure.
To understand whether the Rotatin amino acid substitutions disrupt cilia-mediated signal transduction that flows through the axoneme, we compared the expression pattern of genes in c.2796A>T fibroblasts versus controls. We tested a quantitative RT-PCR (qRT-PCR)-based array containing 86 genes, whose functions are broadly associated with the sonic hedgehog signaling pathway, one of the main signaling pathways regulated at the cilium. A number of significantly deregulated genes were identified and were individually tested again by qRT-PCR for confirmation (see Tables S3, S4, and S5; Figures S4A, S4B, S4D, and S5). Among the genes with more than 2-fold deregulation, besides SHH (MIM 600725) and hedgehog-related HHIP (MIM 606178) and HHAT (MIM 605743), were WNT5A (MIM 164975), WNT2B (MIM 601968), and BMP4 (MIM 112262). Upregulation of WNT7B (MIM 601967) was also originally observed on the arrays but could not be confirmed by qRT-PCR. WNT2B is an effector of the canonical WNT/beta catenin signaling pathway, and WNT5A is a key regulator of the noncanonical WNT signaling-planar cell polarity. BMP4 is a major bone morphogenetic protein involved in patterning of mammalian telencephalon. All these genes are involved in brain development or neuronal migration in mammals and are known to be regulated at the cilium.14–17,23–31 qRT-PCR performed on c.80G>A fibroblasts confirmed downregulation of the same genes (Figures S4A, S4B, and S4D). These results indicate that even if the cilia of the c.80G>A individual appear microscopically normal, they share with the c.2796A>T cells a functional ciliary defect. Downregulation of BMP4 and WNT2B, but not WNT5A, was also observed in control fibroblasts treated with RTTN-specific siRNAs (Figures S4A, S4B, and S4D). Although we cannot explain the lack of WNT5A downregulation, the transient and partial effect of RTTN siRNA may not be sufficient to achieve a detectable downregulation of WNT5A in fibroblasts. WNT5A is a key regulator of the planar cell polarity, through polarized distribution of adhesion receptors in the cell.32 Planar cell polarity is essential for coordinating the cellular localization of the nodal cilia and for determining embryonic polarity.33 Bmp4 also plays an important role in embryonic polarity and determination of left-right asymmetry in chicken, zebrafish, and mouse embryo by controlling asymmetric expression of Nodal in the lateral plate mesoderm.26,28–30 Rttn knockout mice lose the asymmetric expression of Nodal and show abnormal heart looping and hydrocephalus, as seen in ciliopathies.7,11,12 However, the literature does not provide a link between RTTN and neural development. To gain insight in the physiological role of RTTN in human neural cells, we investigated in vitro gene expression in cultured embryonic stem cell-derived human pluripotent neural stem cells (hNSCs).34 Knockdown of RTTN expression by siRNAs in hNSCs leads to a reduction of BMP4 mRNA (Figures S4C and S4D). These results suggest that RTTN is involved in regulation of BMP signaling in neural cells.
In the developing mammalian telencephalon, WNTs and BMPs, in particular BMP4 and WNT2B, are primarily expressed at the cortical hem and are implicated in patterning of the cerebral cortex.30,31,35,36 The human cortical hem is an organizing center of the dorsomedial telencephalon that regulates choroid plexus and hippocampus development and broadly directs cerebral cortical development by giving rise to Cajal-Retzius (CR) neurons. Mutations in genes expressed by CR neurons have already been associated with human cortical malformations that are due to abnormal transmantle migration.3,37,38 Mutations in genes important in intraflagellar transport of the cilia, such as Ift88, are associated with abnormal WNT signaling and cause disorganization of the dorsal telencephalon, disappearance of the cortical hem, and cobblestone appearance of the cerebral cortex, as demonstrated by the cbs/cbs mouse mutants, which lack 80% of the normal Ift88, polaris.39 Interestingly, whereas Ift88 knockout mice are not able to build up structurally normal cilia, cbs/cbs mutants have structurally normal cilia.39
Because of the apparent role of RTTN in regulation of WNT and BMP expression in vitro, we investigated whether Rotatin is present in the same brain areas in the developing telencephalon as the CR neurons, e.g., the marginal zone, which might imply an influence on the cortical organization. We stained embryonic mouse brain at E14.5 and E16.5, ages at which the cortical hem produces migrating CR neurons, using the CR marker calretinin- and Rotatin-specific antibodies. We found that Rotatin localizes in the upper cortical layers and particularly in the marginal zone of the developing mouse cortex, similar to calretinin (Figures S6 and S7). Considering that PMG, as observed in individuals with RTTN mutation, is a postmigratory cortical organization defect, the localization of Rotatin to the marginal zone supports its involvement in the pathogenesis of the cortical malformation.
However, the mechanism by which RTTN mutation causes PMG is not yet clear. If RTTN mutation leads to abnormal cortex development through disruption of the CR neurons, one of the possible mechanisms could be disturbance of the developing leptomeninges. CR neuronal development is under the influence of leptomeninges,40,41 and BMPs are known to control the development of the roof plate.42
In conclusion, we provide evidence that autosomal-recessive missense mutations in RTTN are associated with PMG, which probably represents a “hypomorphic” phenotype, raising the question of whether RTTN null mutations might be lethal.
Rotatin is a basal body protein that localizes at the upper cortical layers of the developing telencephalon.
We hypothesize that Rotatin regulates the organization of the cerebral cortex through regulation of BMP and WNT expression and therefore provides a link between organization of the human cortex and ciliary function, hereby elucidating one of the many possible mechanisms leading to polymicrogyria, which can sometimes be the only phenotypic manifestation of a ciliopathy.43–45
Acknowledgments
We thank Lutgarde Govaerts, Department of Clinical Genetics, Erasmus MC, Rotterdam, for referring affected individuals; Robert Vries, Hubrecht Institute, Utrecht, for calretinin antibody; Rob Willemsen, Department of Clinical Genetics, Erasmus MC, Rotterdam, for support with mouse embryo sections and for critically reading the manuscript; Peter van der Spek and Andreas Kremer, Department of Bioinformatics, Erasmus MC, Rotterdam, for support with bioinformatics; and Tom de Vries Lentsch for graphical work.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/
Mutation Taster, http://www.mutationtaster.org/
National Center for Biotechnology Information (NCBI) dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/
NCBI Reference Sequences (RefSeq), http://www.ncbi.nlm.nih.gov/RefSeq/
National Heart, Lung, and Blood Institute Exome Sequencing Project, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://omim.org
PolyPhen-2.0, http://genetics.bwh.harvard.edu/pph2/
RT2 Profiler PCR Array Data Analysis, http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php
UniProtKB/Swiss-Prot Rotatin reference, http://www.uniprot.org/uniprot/Q86VV8
References
- 1.Leventer R.J., Jansen A., Pilz D.T., Stoodley N., Marini C., Dubeau F., Malone J., Mitchell L.A., Mandelstam S., Scheffer I.E. Clinical and imaging heterogeneity of polymicrogyria: a study of 328 patients. Brain. 2010;133:1415–1427. doi: 10.1093/brain/awq078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tischfield M.A., Cederquist G.Y., Gupta M.L., Jr., Engle E.C. Phenotypic spectrum of the tubulin-related disorders and functional implications of disease-causing mutations. Curr. Opin. Genet. Dev. 2011;21:286–294. doi: 10.1016/j.gde.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barkovich A.J., Guerrini R., Kuzniecky R.I., Jackson G.D., Dobyns W.B. A developmental and genetic classification for malformations of cortical development: update 2012. Brain. 2012;135:1348–1369. doi: 10.1093/brain/aws019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Judkins A.R., Martinez D., Ferreira P., Dobyns W.B., Golden J.A. Polymicrogyria includes fusion of the molecular layer and decreased neuronal populations but normal cortical laminar organization. J. Neuropathol. Exp. Neurol. 2011;70:438–443. doi: 10.1097/NEN.0b013e31821ccf1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alkuraya F.S. Autozygome decoded. Genet. Med. 2010;12:765–771. doi: 10.1097/GIM.0b013e3181fbfcc4. [DOI] [PubMed] [Google Scholar]
- 6.de Wit M.C., Lequin M.H., de Coo I.F., Brusse E., Halley D.J., van de Graaf R., Schot R., Verheijen F.W., Mancini G.M. Cortical brain malformations: effect of clinical, neuroradiological, and modern genetic classification. Arch. Neurol. 2008;65:358–366. doi: 10.1001/archneur.65.3.358. [DOI] [PubMed] [Google Scholar]
- 7.Faisst A.M., Alvarez-Bolado G., Treichel D., Gruss P. Rotatin is a novel gene required for axial rotation and left-right specification in mouse embryos. Mech. Dev. 2002;113:15–28. doi: 10.1016/s0925-4773(02)00003-5. [DOI] [PubMed] [Google Scholar]
- 8.Chatterjee B., Richards K., Bucan M., Lo C. Nt mutation causing laterality defects associated with deletion of rotatin. Mamm. Genome. 2007;18:310–315. doi: 10.1007/s00335-007-9023-7. [DOI] [PubMed] [Google Scholar]
- 9.Goetz S.C., Anderson K.V. The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Badano J.L., Mitsuma N., Beales P.L., Katsanis N. The ciliopathies: an emerging class of human genetic disorders. Annu. Rev. Genomics Hum. Genet. 2006;7:125–148. doi: 10.1146/annurev.genom.7.080505.115610. [DOI] [PubMed] [Google Scholar]
- 11.Gerdes J.M., Davis E.E., Katsanis N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 2009;137:32–45. doi: 10.1016/j.cell.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Melloy P.G., Ewart J.L., Cohen M.F., Desmond M.E., Kuehn M.R., Lo C.W. No turning, a mouse mutation causing left-right and axial patterning defects. Dev. Biol. 1998;193:77–89. doi: 10.1006/dbio.1997.8787. [DOI] [PubMed] [Google Scholar]
- 13.Marszalek J.R., Ruiz-Lozano P., Roberts E., Chien K.R., Goldstein L.S. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc. Natl. Acad. Sci. USA. 1999;96:5043–5048. doi: 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bettencourt-Dias M., Hildebrandt F., Pellman D., Woods G., Godinho S.A. Centrosomes and cilia in human disease. Trends Genet. 2011;27:307–315. doi: 10.1016/j.tig.2011.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Komatsu Y., Kaartinen V., Mishina Y. Cell cycle arrest in node cells governs ciliogenesis at the node to break left-right symmetry. Development. 2011;138:3915–3920. doi: 10.1242/dev.068833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lancaster M.A., Gopal D.J., Kim J., Saleem S.N., Silhavy J.L., Louie C.M., Thacker B.E., Williams Y., Zaki M.S., Gleeson J.G. Defective Wnt-dependent cerebellar midline fusion in a mouse model of Joubert syndrome. Nat. Med. 2011;17:726–731. doi: 10.1038/nm.2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wallingford J.B., Mitchell B. Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev. 2011;25:201–213. doi: 10.1101/gad.2008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stevens N.R., Dobbelaere J., Wainman A., Gergely F., Raff J.W. Ana3 is a conserved protein required for the structural integrity of centrioles and basal bodies. J. Cell Biol. 2009;187:355–363. doi: 10.1083/jcb.200905031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jakobsen L., Vanselow K., Skogs M., Toyoda Y., Lundberg E., Poser I., Falkenby L.G., Bennetzen M., Westendorf J., Nigg E.A. Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J. 2011;30:1520–1535. doi: 10.1038/emboj.2011.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 21.Tallila J., Jakkula E., Peltonen L., Salonen R., Kestilä M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am. J. Hum. Genet. 2008;82:1361–1367. doi: 10.1016/j.ajhg.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wheatley D.N. Primary cilia in normal and pathological tissues. Pathobiology. 1995;63:222–238. doi: 10.1159/000163955. [DOI] [PubMed] [Google Scholar]
- 23.Vivancos V., Chen P., Spassky N., Qian D., Dabdoub A., Kelley M., Studer M., Guthrie S. Wnt activity guides facial branchiomotor neuron migration, and involves the PCP pathway and JNK and ROCK kinases. Neural Dev. 2009;4:7. doi: 10.1186/1749-8104-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fischer E., Pontoglio M. Planar cell polarity and cilia. Semin. Cell Dev. Biol. 2009;20:998–1005. doi: 10.1016/j.semcdb.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 25.Fliegauf M., Benzing T., Omran H. When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 26.Fujiwara T., Dehart D.B., Sulik K.K., Hogan B.L. Distinct requirements for extra-embryonic and embryonic bone morphogenetic protein 4 in the formation of the node and primitive streak and coordination of left-right asymmetry in the mouse. Development. 2002;129:4685–4696. doi: 10.1242/dev.129.20.4685. [DOI] [PubMed] [Google Scholar]
- 27.Ross A.J., May-Simera H., Eichers E.R., Kai M., Hill J., Jagger D.J., Leitch C.C., Chapple J.P., Munro P.M., Fisher S. Disruption of Bardet-Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet. 2005;37:1135–1140. doi: 10.1038/ng1644. [DOI] [PubMed] [Google Scholar]
- 28.Chocron S., Verhoeven M.C., Rentzsch F., Hammerschmidt M., Bakkers J. Zebrafish Bmp4 regulates left-right asymmetry at two distinct developmental time points. Dev. Biol. 2007;305:577–588. doi: 10.1016/j.ydbio.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 29.Piedra M.E., Ros M.A. BMP signaling positively regulates Nodal expression during left right specification in the chick embryo. Development. 2002;129:3431–3440. doi: 10.1242/dev.129.14.3431. [DOI] [PubMed] [Google Scholar]
- 30.Shimogori T., Banuchi V., Ng H.Y., Strauss J.B., Grove E.A. Embryonic signaling centers expressing BMP, WNT and FGF proteins interact to pattern the cerebral cortex. Development. 2004;131:5639–5647. doi: 10.1242/dev.01428. [DOI] [PubMed] [Google Scholar]
- 31.Subramanian L., Tole S. Mechanisms underlying the specification, positional regulation, and function of the cortical hem. Cereb. Cortex. 2009;19(Suppl 1):i90–i95. doi: 10.1093/cercor/bhp031. [DOI] [PubMed] [Google Scholar]
- 32.Witze E.S., Litman E.S., Argast G.M., Moon R.T., Ahn N.G. Wnt5a control of cell polarity and directional movement by polarized redistribution of adhesion receptors. Science. 2008;320:365–369. doi: 10.1126/science.1151250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Antic D., Stubbs J.L., Suyama K., Kintner C., Scott M.P., Axelrod J.D. Planar cell polarity enables posterior localization of nodal cilia and left-right axis determination during mouse and Xenopus embryogenesis. PLoS ONE. 2010;5:e8999. doi: 10.1371/journal.pone.0008999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conti L., Pollard S.M., Gorba T., Reitano E., Toselli M., Biella G., Sun Y., Sanzone S., Ying Q.L., Cattaneo E., Smith A. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol. 2005;3:e283. doi: 10.1371/journal.pbio.0030283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grove E.A., Tole S., Limon J., Yip L., Ragsdale C.W. The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development. 1998;125:2315–2325. doi: 10.1242/dev.125.12.2315. [DOI] [PubMed] [Google Scholar]
- 36.Abu-Khalil A., Fu L., Grove E.A., Zecevic N., Geschwind D.H. Wnt genes define distinct boundaries in the developing human brain: implications for human forebrain patterning. J. Comp. Neurol. 2004;474:276–288. doi: 10.1002/cne.20112. [DOI] [PubMed] [Google Scholar]
- 37.Goffinet A.M. Events governing organization of postmigratory neurons: studies on brain development in normal and reeler mice. Brain Res. 1984;319:261–296. doi: 10.1016/0165-0173(84)90013-4. [DOI] [PubMed] [Google Scholar]
- 38.D’Arcangelo G., Miao G.G., Chen S.C., Soares H.D., Morgan J.I., Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 39.Willaredt M.A., Hasenpusch-Theil K., Gardner H.A., Kitanovic I., Hirschfeld-Warneken V.C., Gojak C.P., Gorgas K., Bradford C.L., Spatz J., Wölfl S. A crucial role for primary cilia in cortical morphogenesis. J. Neurosci. 2008;28:12887–12900. doi: 10.1523/JNEUROSCI.2084-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borrell V., Marín O. Meninges control tangential migration of hem-derived Cajal-Retzius cells via CXCL12/CXCR4 signaling. Nat. Neurosci. 2006;9:1284–1293. doi: 10.1038/nn1764. [DOI] [PubMed] [Google Scholar]
- 41.Siegenthaler J.A., Pleasure S.J. We have got you ‘covered’: how the meninges control brain development. Curr. Opin. Genet. Dev. 2011;21:249–255. doi: 10.1016/j.gde.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng X., Hsu C.M., Currle D.S., Hu J.S., Barkovich A.J., Monuki E.S. Central roles of the roof plate in telencephalic development and holoprosencephaly. J. Neurosci. 2006;26:7640–7649. doi: 10.1523/JNEUROSCI.0714-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Field M., Scheffer I.E., Gill D., Wilson M., Christie L., Shaw M., Gardner A., Glubb G., Hobson L., Corbett M. Expanding the molecular basis and phenotypic spectrum of X-linked Joubert syndrome associated with OFD1 mutations. Eur. J. Hum. Genet. 2012;20:806–809. doi: 10.1038/ejhg.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sattar S., Gleeson J.G. The ciliopathies in neuronal development: a clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disorders. Dev. Med. Child Neurol. 2011;53:793–798. doi: 10.1111/j.1469-8749.2011.04021.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hildebrandt F., Benzing T., Katsanis N. Ciliopathies. N. Engl. J. Med. 2011;364:1533–1543. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.