

Abstract
DUF1220 domains show the largest human-lineage-specific increase in copy number of any protein-coding region in the human genome and map primarily to 1q21, where deletions and reciprocal duplications have been associated with microcephaly and macrocephaly, respectively. Given these findings and the high correlation between DUF1220 copy number and brain size across primate lineages (R2 = 0.98; p = 1.8 × 10−6), DUF1220 sequences represent plausible candidates for underlying 1q21-associated brain-size pathologies. To investigate this possibility, we used specialized bioinformatics tools developed for scoring highly duplicated DUF1220 sequences to implement targeted 1q21 array comparative genomic hybridization on individuals (n = 42) with 1q21-associated microcephaly and macrocephaly. We show that of all the 1q21 genes examined (n = 53), DUF1220 copy number shows the strongest association with brain size among individuals with 1q21-associated microcephaly, particularly with respect to the three evolutionarily conserved DUF1220 clades CON1(p = 0.0079), CON2 (p = 0.0134), and CON3 (p = 0.0116). Interestingly, all 1q21 DUF1220-encoding genes belonging to the NBPF family show significant correlations with frontal-occipital-circumference Z scores in the deletion group. In a similar survey of a nondisease population, we show that DUF1220 copy number exhibits the strongest correlation with brain gray-matter volume (CON1, p = 0.0246; and CON2, p = 0.0334). Notably, only DUF1220 sequences are consistently significant in both disease and nondisease populations. Taken together, these data strongly implicate the loss of DUF1220 copy number in the etiology of 1q21-associated microcephaly and support the view that DUF1220 domains function as general effectors of evolutionary, pathological, and normal variation in brain size.
Introduction
DUF1220 protein domains are approximately 65 amino acids in length and have undergone unusually rapid and extensive copy-number expansion during recent primate evolution and most strikingly in the human lineage.1 The first DUF1220-encoding gene identified as a dramatic human-lineage-specific (HLS) increase in copy number was MGC8902 (cDNA IMAGE 843276) and was the result of a genome-wide survey of gene copy-number change among human and great-ape lineages.2 The same gene family (now termed the neuroblastoma breakpoint family [NBPF]) was independently identified when one of its members was found to be disrupted by a rearrangement in an individual with neuroblastoma (MIM 613017).3 Analysis of DUF1220 sequences indicated that the domain shows an exceptional HLS copy-number expansion that decreases generally with evolutionary distance from humans, shows signs of positive selection at the coding-sequence level, and in the brain, exhibits neuron-specific expression.1,4 Analysis of DUF1220 copy number in the genomes of 36 mammalian species confirmed that the highest copy number is found in humans (272).5 In other species, copy number ranges from a high of 125 in chimps to 99 in gorillas, 92 in orangutans, 35 in macaques, 30 in marmosets, 4 in dolphins, only 1 in mice, and none outside of mammals.
DUF1220 sequences exist within two very distinct genomic environments: as a single (ancestral) domain in PDE4DIP (myomegalin) and as multiple tandem copies in the NBPF multigene family. Only the second form has undergone an evolutionarily rapid and recent copy-number expansion. Excluding the ancestral DUF1220 domain, the remaining DUF1220-domain-encoding DNA sequences can be divided into six subgroups, or clades, designated HLS1, HLS2, HLS3, CON1, CON2, and CON3.5 The copy number of DUF1220 sequences that belong to HLS clades has increased markedly in the human lineage, whereas the copy number of DUF1220 sequences belonging to CON clades is more similar across primates and nonprimate mammals.5
Numerous copy-number variations (CNVs) in the 1q21.1-1q21.2 region, where most DUF1220 sequences map, have been implicated in an increasingly large number of recurrent human diseases (Table S1, available online). Interestingly, two independent reports have found that deletions within this region are associated with microcephaly (MIM 612474) and reciprocal duplications with macrocephaly (MIM 612475),6,7 indicating that the dosage of one or more sequences in this interval affects human brain size. We have noted that although these implicated 1q21 CNVs contain a number of non-NBPF genes, they also encompass or immediately flank DUF1220 sequences,8 which, because of their highly duplicated character, were not directly examined in these previous studies. To address this issue, we developed custom, high-resolution, targeted 1q21 oligonucleotide arrays that included highly-duplicated sequences (e.g., of DUF1220) in order to identify causal genomic sequences underlying 1q21-associated brain-size variations in disease and nondisease populations.
Material and Methods
DUF1220 Copy Number versus Brain Graphs
DUF1220 association with copy number, brain weight, and cortical neuron counts were graphed with Excel. The relationships were evaluated by ordinary least-squares (simple linear) regression with R version 2.10.1. Brain weights were taken from Falk,9 Kouprina et al.,10 Roth and Dicke,11 and Ponce de Leon,12 and neuron counts were from Roth and Dicke.11
Genomic DNA Samples
The Medical Genetics Laboratories (Cytogenetic and Microarray Laboratories) at Baylor College of Medicine provided DNA isolated from the blood of individuals affected with microcephaly or macrocephaly. The samples provided included 28 individuals with previously reported6 1q21 deletions or duplications and microcephaly or macrocephaly and were ascertained from a larger survey of more than 16,000 individuals. From the same laboratory, an additional 14 samples, which were not previously assayed on low-resolution arrays, were included in this study. Collaborating labs (of A.R., J.G., and J.R.) from the National Institute for Mental Health provided DNA samples (extracted from immortalized cell lines according to standard methods [QIAGEN, MD, USA]) from normal individuals at the extremes of high and low brain size. The presence of extreme brain size was determined on the basis of residual volumes of total brain, total gray matter, and total white matter after age and sex were accounted for in a large (n > 300) brain structural magnetic resonance image (sMRI) database. Brain sMRI scans were all T-1 weighted images with contiguous 1.5 mm axial slices and 2.0 mm coronal slices and were obtained on the same 1.5-T General Electric (Milwaukee, WI) Signa scanner with a three-dimensional spoiled gradient-recalled echo sequence. The aforementioned volumetric indices were derived for each scan with the use of a previously described,13 well-validated, and fully automated image-processing pipeline. All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national), and proper informed consent was obtained.
Design of Custom High-Density 1q21.1-1q21.2 DNA Microarrays
In order to detect CNVs in DUF1220-domain-encoding sequences in the 1q21.1-1q21.2 region of the genome among individuals, we designed high-density custom human microarrays by using Agilent Technologies 8x60K platform. The array was enriched for both unique and nonunique 60 mer probe sequences that mapped to Chr 1: 141–150 Mb. The arrays were designed prior to the release of the 2009 human genome assembly (hg19) and were therefore generated with the 2006 human genome assembly (hg18). The nonunique probes from chromosome 1, including DUF1220-domain-encoding sequences, were specific to 1q21 gene regions. On average, the probe coverage on chromosome 1 included 8–9 probes per 1 kb. A total of 43,010 probes were located on chromosome 1. The remaining 15,500 probes were randomly chosen and mapped to unique regions from the rest of the genome (hg18).
aCGH Assays
Oxford Gene Technology (OGT) performed the array comparative genomic hybridization (aCGH) as an Agilent certified service provider. OGT utilized the following protocols for preparation of the test and reference samples: Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis, Enzymatic Labeling for Blood, Cells, or Tissues (with a High Throughput option) version 6.3, October 2010 (G4410-90010); and Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis, Enzymatic Labeling for Blood, Cells, or Tissues (with a High Throughput option) version 6.2.1, February 2010 (G4410-90010).
The Agilent Genomic DNA Enzymatic Labeling Kit (Agilent p/n 5190-0449) was used for labeling the samples. Protocols were adhered to with the restriction-digest option and with the 96-well clean-up option (AutoScreen-96A Well plates GE Healthcare p/n 25-9005-98). A Little Dipper (Scigene) was used for automated slide washing under ozone-controlled conditions. Slides were acetonitrile dipped for 1 min after slide washing (wash 1 and wash 2) for the reduction of slide background. Because high-background slide quality-control (QC) metrics were observed with the chromosome 1 array design, all slides were washed twice before scanning. Slides were scanned on an Agilent C scanner under ozone-controlled conditions. Data were extracted from the TIFF images with Agilent Feature Extraction software (version 10.7.1). All sample- and slide-processing steps and QC metrics were recorded in OGT’s Laboratory Information Management System. So that all test samples could be compared to one another, the same single unaffected male DNA sample was used as a reference sample for all experiments.
Analysis of aCGH Data
The custom 1q21 microarray was composed of probes corresponding to both unique and nonunique sequences, and approximately 75% of the probes were located on chromosome 1. The nonunique probes have sequences that are found at multiple loci in the human genome, and the majority of them are located within the highly duplicated DUF1220-domain-encoding regions. The original design of the microarray probe sequences was based on the hg18 2006 assembly, which was converted to the hg19 build as follows. All 57,897 genomic probe sequences on the microarray were aligned to the hg19 2009 assembly with BLAT. If a BLAT search resulted in a match of at least 59 of 60 nucleotides of each probe sequence, that probe sequence was retained and mapped according to the 2009 assembly and was used in the aCGH analysis. This remapping of the original 57,897 probes resulted in 267,841 genomic probe loci in the data set so that the nonunique probes were remapped to all loci determined by the BLAT alignment results. The log2 ratios for the nonunique array probes were assigned to each newly mapped nonunique probe locus. In summary, a specific probe that was originally assigned to one genomic locus was remapped to multiple loci. The log2 ratios for probes that mapped to the same location were averaged.
Specific steps of the aCGH analysis are described in Figure S1A and are as follows. For each array experiment, the TIFF image of the array was imported into Agilent Feature Extraction Software version 10.7.3.1 for image analysis, which included dye-bias normalization and data extraction (Figure S1A, step 1).
Dye-Bias Normalization
The dye-bias normalization excluded all probes on chromosomes 1, X, and Y. The chromosome 1 probes were omitted for normalization methods because the majority of these probes were not unique on the basis of the array design. Additionally, probes on chromosomes X and Y were omitted for alleviating any bias created from test and reference samples that were not matched for sex.
Across-Array Normalization
So that data could be compared between experiments, an across-array normalization was carried out as follows. After the dye-normalized log10 ratio for each probe was converted to a linear ratio, all probes corresponding to a given target sequence or region of interest (e.g., DUF1220 clade, non-NBPF gene, etc.) were averaged for an average linear ratio for that gene or region (Figure S1A, step 2). Next, all single-locus probes (i.e., probes corresponding to single-copy genome sequences excluding chromosomes 1, X, and Y) were averaged (Figure S1A, step 3). An adjusted score, for which the average linear ratio for a target region (from step 2) was divided by the average linear ratio of all single-locus probes (from step 3) (Figure S1A, step 4), was then generated. Finally, the resulting normalized linear ratio was then converted into a log2 ratio, which was then used for statistical analyses. Cytosure Interpret Software was used for viewing segmentations across chromosome 1 and for generating Figure 1.
Figure 1.

CytoSure Images of Disease-Population aCGH Data on 1q21.1-1q21.2
aCGH data for the disease population subdivided into class I deletions (smaller), class II deletions (larger), and duplications. Regions highlighted in blue denote copy-number aberrations.
So that bias could be reduced from cross-hybridizing probes aligning to multiple clade types (e.g., HLS1 and CON3), in the clade analysis all nonunique probes mapping to multiple clade types (21%) were removed from the analysis. The nonunique probes that mapped to only a single clade type were retained, and the values assigned to each clade type represent an average of the signals obtained for each probe within a clade type. However, some cross-hybridization of probes to multiple genomic regions of the same clade type, such as to multiple HLS1 regions, remained.
The region from each 1q21 gene’s transcription start coordinate to each gene’s transcription stop coordinate was divided into nonoverlapping windows of 500 bases so that the array results could be viewed for each 1q21 gene (for example, Figures 2B–2D). For each sample tested, the log2 ratio values for all probes whose coordinates mapped within each window were averaged and plotted for each gene.
Figure 2.
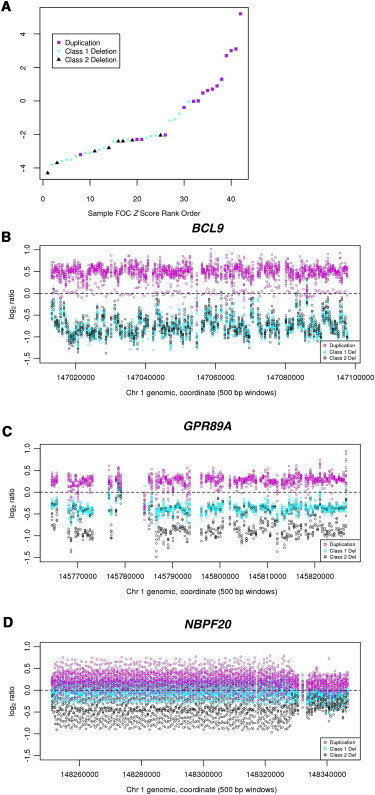
aCGH Data for Disease Samples from Three Genes in the 1q21.1-1q21.2 Region
Class I duplications are shown in magenta, class I deletions are shown in teal, and class II deletions are shown in black.
(A) Graph of rank-ordered FOC Z scores for samples within each disease class.
(B) BCL9.
(C) GPR89.
(D) NBPF20.
For viewing the DUF1220 regions on the basis of clade classification, the sequence spanning each DUF1220 repeat (clade span; Figure S1B) located in the 1q21 region (Figure 3B) was used as a separate window and the average log2 ratio was calculated on the basis of all probes that aligned within each window. Each DUF1220 window was first ordered by its clade classification and then by its genomic coordinate within each clade classification (Figure 3B).
Figure 3.
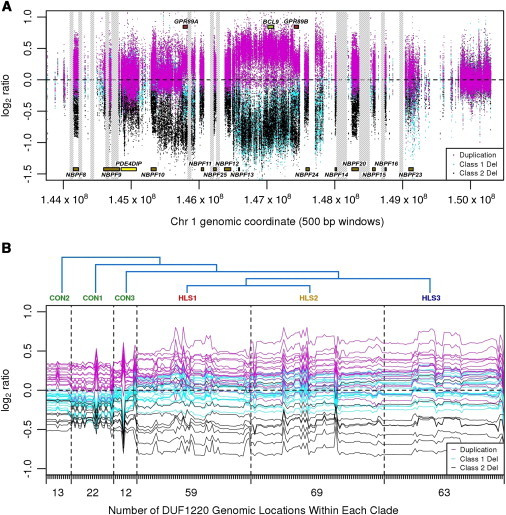
aCGH Data for Three Types of Disease Samples
Class I duplications are shown in magenta, class I deletions are shown in teal, and class II deletions are shown in black.
(A) Array data for the entire 1q21.1-1q21.2 region. Sequence gaps are displayed as vertical gray bars. Because of space limitations, only the NBPF gene family, BCL9, and GPR89 are labeled.
(B) DUF1220 clade array profile. Log2 ratios corresponding to DUF1220 signals are shown for individual samples from the three classes of the disease population. aCGH data for each individual are plotted as a continuous horizontal line so that results from individual samples can be distinguished from one another. For each sample tested, data points are first grouped according to clade and then within each clade, they are grouped in the order in which they occur across the 1q21 region. The numbers of DUF1220 domains belonging to each clade are represented as tick marks along the x axis. The number displayed below represents the number of DUF1220 domains found within each clade. The marks represent 238 clade-specific DUF1220 domains located in the 1q21 region and present on the custom array. Phylogeny of the six clades (CON1–CON3 and HLS1–HLS3) is displayed at the top.
qPCR Analysis
Quantitative real-time PCR (qPCR), with the use of Taqman master mix on an Applied Biosystems 7300 Real-Time PCR system, was carried out on genes for each individual with optimal primer and fluorogenic probe sets that are unique to the DNA sequence of the gene of interest. Optimal primers and probes were designed with PrimerExpress (Applied Biosystem software). The amplicon sequence was used as a BLAT query against the human March 2006 (hg18) assembly for verifying that the primer and designs were sound. The functionality of each primer pair was then verified with the UCSC database for in silico PCR. Primer oligos were obtained from Thermofisher/Operon (Huntsville, AL).
Relative copy-ratio estimates were generated with the standard curve method, normalized to CFTR, cystic fibrosis transmembrane conductance regulator (ATP-binding cassette subfamily C, member 7), an ATP-binding cassette that was used as a control gene thought to represent one gene per haploid human genome.14 Reactions were carried out in triplicate per plate run and replicated in at least three separate plate reaction runs. In total, DNA was assayed at least nine times per individual. Copy ratios of all assays were averaged for the final ratio measure. Additionally, qPCR-derived copy ratio of DUF1220 domains was compared to aCGH copy ratio on 26 individuals with known CNVs in the 1q21.1-1q21.2 region. The qPCR primer and probe sequences are listed in Table S2.
Statistical Analyses
To test our primary hypothesis that DUF1220 copy-number ratio is associated with head circumference, we tested for association between the copy number of each of the sequences of interest and frontal occipital circumference (FOC) Z scores via linear regression models. Because of a priori hypotheses regarding the biologic relevance of DUF1220 copy number to brain size, our primary inference was based on six tests that we performed by using the 42 samples from individuals with either microcephaly or macrocephaly. One test was conducted for each of the three conserved DUF1220 clades (CON1, CON2, and CON3), and one was conducted for each of the HLS clades (HLS1, HLS2, HLS3). In each case, the average of the log2 ratio of the individual sequences was used as the measure for a clade. Secondary analyses were stratified according to whether an individual had a deletion (n = 26) or a duplication (n = 16) in the 1q21.1-1q21.2 region. On the basis of the results of the stratified analyses, we conducted tertiary (exploratory) analyses to test for association between each of the other genes assayed by aCGH and the FOC Z scores among the deletion subgroup. The ethnic distribution of individuals with known deletions or duplications was categorized as 26 white, 12 Hispanic, and 3 African American or other. Differences of copy ratio between ethnic groups were tested with a one-way ANOVA. We assessed population stratification in the group with a known deletion by including ethnicity as a covariate in a linear regression model of FOC Z score and CON1. There was no evidence of (1) a significant difference of copy ratio between ethnic groups (p > 0.10), (2) confounding by ethnicity, or (3) an association between ethnicity and FOC Z scores. Given the lack of ethnic associations, results presented below are from unadjusted regression models.
For aCGH studies of a nondisease population, a sample of 59 unrelated non-Hispanic white individuals was selected from a cohort of greater than 300 individuals for extremes in brain size. For this selection, gray-matter residual volumes were obtained from a regression controlling for sex and age. The 59 individuals selected for analysis had gray-matter residual volumes greater than 0.5 (large group, n = 29) and less than −0.5 (small group, n = 30). We selected extremes in phenotypes to potentially increase our power to detect differences in copy ratio between groups. We selected non-Hispanic white individuals to control for potential population stratification. This population was 69.5% male and had a mean age of 10.9 years and a standard deviation (SD) of 3.0 years. We first tested for association between each of the six DUF1220 clades and having a large or small gray-matter residual volume (on the basis of the selection criteria described above) via t tests. Lastly, in an exploratory analysis, we tested for association of large or small gray-matter volume versus aCGH-predicted copy ratio of other genes in the 1q21 region. Analyses were conducted with R version 2.10.1.
Results
Microcephaly and Macrocephaly
To directly investigate the possible involvement of DUF1220 copy number in 1q21.1-1q21.2 microcephaly and macrocephaly, we designed custom 1q21 microarrays to more comprehensively cover the 4.4 Mb 1q21.1-1q21.2 interval, as well as the sequences flanking the interval. In addition, we developed new bioinformatic tools that allowed the copy number of different types of DUF1220 sequences to be independently assessed. The arrays were used for high-resolution aCGH analysis (Figure S1) of 42 individuals (Table S3) with either 1q21.1-1q21.2 deletions (class I [smaller] or class II [larger]) or duplications. As originally defined by Brunetti-Pierri et al.,6 class I deletions include a deletion in the distal 1q21.1-1q21.2 region (hg18), whereas class II deletions are larger and include the TAR syndrome (MIM 274000) region in addition to the 1q21.1-1q21.2 region. Of these individuals, 28 harbor 1q21 CNVs associated with microcephaly and macrocephaly and were identified from a previous, low-resolution genome-wide aCGH survey of >16,000 individuals.6 Also tested were an additional 14 individuals who had similar 1q21 CNVs and who were not previously described. In all cases, the same reference sample, a single unaffected male, was used, allowing all test samples to be indirectly compared to one another. For those samples (n = 28) that had been previously analyzed by lower-resolution aCGH, resulting custom 1q21 array data confirmed what had been previously reported but also allowed specific measurement of DUF1220 copy number and more detailed coverage of the 1q21 region (Figures 1 and 3A).
In order to visualize DUF1220-specific aCGH signals, we plotted data for each of the 241 human DUF1220-domain-encoding copies in the 1q21.1-1q21.2 region in six clade-specific groupings (Figure 3B). Resulting data profiles for each individual tested from the disease population show that, overall, the class II deletion group lost more DUF1220 copies than did the class I deletion group for all six DUF1220 clades, whereas the duplication group gained DUF1220 sequences. qPCR independently confirmed these trends (Figure S2 and Table S4).
aCGH-predicted copy-number values of DUF1220-domain-encoding sequences (included in NBPF genes) and non-NBPF sequences within the 1q21.1-1q21.2 region were next compared to head-circumference values (FOC Z scores) for each sample. The resulting data from the entire disease population (class I and class II deletion groups and the duplication group) indicate that the copy number of DUF1220 sequences (clades CON1–CON3 and HLS1–HLS3) shows a strong correlation with FOC Z scores (Table 1 and Figure 4). In addition, a number of other genes within the region also exhibit a significant correlation with FOC Z scores (Table S5), confirming results from the previous low-resolution aCGH analysis of these samples.6
Table 1.
Correlation between aCGH-Predicted DUF1220 Clade Copy Number and FOC Z Scores for the Entire Disease Population
| Clade | Beta | SE | R2 | p Value |
|---|---|---|---|---|
| CON1 | 7.301 | 1.151 | 0.501 | 1.56 × 10−7 |
| CON2 | 7.125 | 1.136 | 0.496 | 1.97 × 10−6 |
| CON3 | 6.826 | 1.172 | 0.459 | 8.29 × 10−7 |
| HLS1 | 4.067 | 1.021 | 0.284 | 0.0003 |
| HLS2 | 3.703 | 0.978 | 0.264 | 0.0005 |
| HLS3 | 3.599 | 0.969 | 0.256 | 0.0006 |
Significant (p ≤ 0.05) associations are shown in bold. Beta is the effect estimate of 1 unit increase in copy ratio from a linear regression model. The following abbreviation is used: SE, standard error of beta.
Figure 4.
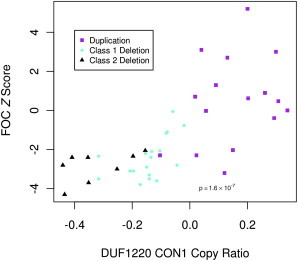
Correlation between FOC Z Scores and aCGH Copy Ratios for DUF1220 CON1 Clade Sequences
Correlation between FOC Z scores and aCGH-based copy ratio of CON1 DUF1220 sequences within the disease population. CON1 copy ratio and FOC Z scores are displayed on the x axis and y axis, respectively. Individuals with class I deletions (aqua), class II deletions (black), and duplications (purple) are plotted against their FOC Z score.
To determine which sequences were driving this association, we stratified the samples by deletion (class I and class II combined) or duplication groups in the 1q21 region. Analysis of the duplication group alone demonstrated no evidence of association between head circumference and any 1q21.1-1q21.2 sequences, including those of DUF1220 (data not shown). However, using array data from the combined class I and class II deletion groups, we found an association between head circumference and the copy number of each of the six DUF1220 clades. The strongest association was obtained with the three evolutionarily conserved DUF1220 clades (CON1, p = 0.0079; CON2, p = 0.0134; and CON3, p = 0.0116), whereas a significant, though more modest, association was found for the three HLS clades (HLS1, p = 0.0476; HLS2, p = 0.0431; and HLS3, p = 0.0444) (Table 2 and Figure 4). Interestingly, all NBPF (i.e., DUF1220-encoding) genes that map to 1q21 showed a significant association (p < 0.044) with head circumference (on the basis of FOC Z scores) in the deletion group (Table 3). Except for C1orf54, no significant association was found (p < 0.05) for any of the 40 non-DUF1220-encoding genes in the critical region or adjacent to the implicated deletion interval (Table S6). Although C1orf54 shows a correlation with head circumference in the deletion group, this gene is found outside the critical region for microcephaly and macrocephaly, as previously defined by Brunetti-Pierri, et al.,6 and shows no significant association with FOC Z scores across the full disease population (i.e., 1q21-associated microcephaly and macrocephaly) (Table S5). It also does not exhibit correlative evolutionary evidence as seen in the dramatic copy-number increase of DUF1220 sequences. For the above reasons, C1orf54 is most likely a false-positive association. Taken together, these findings implicate loss of DUF1220 copy number in 1q21-associated microcephaly. In addition, it should be noted that these results do not eliminate an increase in DUF1220 copy number as the likely cause of 1q21-associated macrocephaly in these samples, although definitive proof of its involvement will most likely require analysis of additional samples and/or finer copy-number measurements.
Table 2.
Correlation of aCGH-Predicted DUF1220 Clade Copy Number with FOC Z Scores for the Combined Class I and II Deletion Groups
| Clade | Beta | SE | R2 | p Value |
|---|---|---|---|---|
| CON1 | 4.128 | 1.425 | 0.259 | 0.0079 |
| CON2 | 3.421 | 1.282 | 0.229 | 0.0134 |
| CON3 | 3.108 | 1.137 | 0.237 | 0.0116 |
| HLS1 | 1.472 | 0.705 | 0.154 | 0.0476 |
| HLS2 | 1.430 | 0.669 | 0.160 | 0.0431 |
| HLS3 | 1.376 | 0.648 | 0.158 | 0.0444 |
Significant (p ≤ 0.05) associations are shown in bold. Beta is the effect estimate of 1 unit increase in copy ratio from a linear regression model. The following abbreviation is used: SE, standard error of beta.
Table 3.
Association between Average aCGH-Predicted Copy Ratios for NBPF Genes in the 1q21 Region and FOC Z Scores in the Deletion Group
| Gene | Beta | SE | R2 | p Value |
|---|---|---|---|---|
| NBPF8 | 1.431 | 0.673 | 0.159 | 0.044 |
| NBPF9 | 2.741 | 1.087 | 0.210 | 0.019 |
| NBPF10 | 1.615 | 0.740 | 0.165 | 0.039 |
| NBPF11 | 2.708 | 1.080 | 0.208 | 0.019 |
| NBPF12 | 2.280 | 0.955 | 0.192 | 0.025 |
| NBPF13 | 2.557 | 1.047 | 0.199 | 0.022 |
| NBPF14 | 1.662 | 0.754 | 0.168 | 0.037 |
| NBPF15 | 3.219 | 1.206 | 0.229 | 0.013 |
| NBPF16 | 3.052 | 1.154 | 0.226 | 0.014 |
| NBPF20 | 1.465 | 0.684 | 0.161 | 0.042 |
| NBPF23 | 4.621 | 1.617 | 0.254 | 0.009 |
| NBPF24 | 2.649 | 1.063 | 0.203 | 0.020 |
| NBPF25 | 1.815 | 0.810 | 0.173 | 0.035 |
Significant (p ≤ 0.05) associations are shown in bold. Beta is the effect estimate of 1 unit increase in copy ratio from a linear regression model. The following abbreviation is used: SE, standard error of beta.
The change in FOC-Z-score distribution across these disease samples reflects a gradual, rather than abrupt, profile (Figure 2A), suggesting that any gene (or domain) whose dosage underlies these changes should also show a similar distribution. Indeed, unlike single- or low-copy sequences, the high copy number of DUF1220 sequences allows them to be incrementally reduced over an extremely broad copy-number range. For example, single-copy 1q21 genes, such as BCL9 (Figure 2B), show only two discrete array profiles: one for the deletion groups and one for the duplication group. Genes, such as GPR89 (Figure 2C), with a copy in the intervals for both the class I and class II deletion groups, show only three primary levels of copy number. In contrast, DUF1220 clades, such as those from NBPF20 (Figure 2D), show a continuous copy-number profile that closely resembles the gradual distribution of FOC Z scores across these samples (Figure 2A).
Nondisease Population
To investigate whether DUF1220 copy number might contribute to population variation in a brain-size-related phenotype in a nondisease population, we implemented the same custom 1q21.1-1q21.2 aCGH analysis on 59 individuals with extremes of normal variation of brain gray-matter volume. Although this phenotype is distinct from that found in the disease population, it provides an analysis of a related brain phenotype in an independently generated, nondisease cohort. After classifying individuals into large and small gray-matter groups, we tested DUF1220 sequences for mean copy-number differences between groups by using t tests. The mean CON1 and mean CON2 copy-number values were both significantly greater in the large gray-matter group than in the small gray-matter group (p = 0.0246 and p = 0.0334, respectively) (Figure S3 and Table S7). Notably, the CON1 clade also showed the strongest association with head circumference in the deletion group of the disease population.
Subsequent analysis, which included 40 additional genes not related to NBPF genes but located in the 1q21.1-1q21.2 region, resulted in the ancestral DUF1220-containing gene, PDE4DIP, and four non-DUF1220-containing genes—SEC22B, NUDT17, SV2A, and SF3B4—showing a significant association between gray-matter volume and aCGH-predicted copy number (p < 0.048; Table S8). However, three of these genes (NUDT17, SV2A, and SF3B4) show a negative correlation with gray matter and thus exhibit an inverse relationship with brain size. Additionally, SEC22B, SV2A, and SF3BF lie outside the critical 1q21.1-1q21.2 region previously linked to microcephaly and macrocephaly, and none of the four genes show an association with brain size in the disease population. Finally, unlike the close parallel that DUF1220-domain copy number shows with evolutionary changes in brain size, none of these genes exhibit such complementary evolutionary evidence (all four genes are single copy across primate lineages; Table S9). These observations strongly suggest that these genes most likely represent false-positive correlations. Importantly, none of the other non-DUF1220-containing genes that map within the critical region show any correlation with head circumference in the group of healthy individuals and are probably not influencing human brain size under normal conditions.
The very strong association identified for CON1 (p = 1.56 × 10−7), CON2 (p = 1.97 × 10−6), and CON3 (p = 8.29 × 10-7) in the entire disease population is most likely indicative, in part, of the substantial range in FOC Z scores (>±4 SDs) observed in this group. In contrast, the sample of healthy individuals, does not exhibit such wide variation, which ranges only from <±3 SDs, even though it shows head-circumference extremes. For these reasons, the more modest, but still significant, association between gray-matter residual values and CON1 and CON2 (p = 0.0246 and p = 0.0334, respectively; Table S7) could be expected given the relatively small sample size and the comparably lower range of brain volumes in a group of healthy individuals.
Discussion
Here, we have shown that of all the 1q21 genes examined (n = 53), only DUF1220 sequences exhibit a significant direct correlation with brain-size phenotypes in both pathological and normal human populations. Although we provide data implicating the loss of DUF1220 copy number in 1q21-associated microcephaly, the data are also fully consistent with the view that increases in DUF1220 copy number underlie 1q21-associated macrocephaly.
We also note that the microcephaly and macrocephaly samples tested here were identified with lower-resolution aCGH platforms that restricted analyses to single-copy sequences and therefore did not directly examine DUF1220 copy number. As a result, the disease samples that were tested were all biased to contain only large 1q21 CNVs with imprecise breakpoint estimates. Because each DUF1220 domain repeat is on average only 1.8 kb,5 it is likely that gain and loss of DUF1220 sequences, accompanied by negligible changes in flanking single-copy sequences, frequently occur and are invariably missed with current aCGH approaches. These observations suggest that there are likely to be a substantial number of microcephaly and macrocephaly cases that are due to DUF1220 CNVs that have yet to be identified.
Although the studies described here link DUF1220 copy number to pathological and normal variation in brain size, independent evidence is also consistent with its involvement in human brain evolution. Among primate lineages, there is a high correlation between DUF1220 copy number (the highest copy number, >270, was found in Homo sapiens [human and Neanderthal]) and increased brain size (R2 = 0.98; p = 1.8 × 10−6) (Figure S4A), as well as an increased number of cortical neurons (R2 = 0.98; p = 0.0011) (Figure S4B). Unlike DUF1220-domain-encoding sequences, other genes in the 1q21 region are virtually all single to low copy among primates, and none show a strong correspondence between copy number and primate brain size (Table S9). Taken together, these observations support the view that DUF1220-domain copy number, i.e., DUF1220-domain dosage, functions as a general effector of evolutionary, pathological, and normal variation in brain size.
Although the copy number of all NBPF (i.e., DUF1220-encoding) genes showed significant association with head circumference in the complete disease sample set, as well as in the combined deletion group, we also looked for associations by using DUF1220 domains that had been separated into specific phylogenetic clades. Resulting copy-number profiles of the six DUF1220 clades indicated that the copy number of conserved DUF1220 clades CON1 and CON2 exhibited a stronger correlation with brain size in the disease and nondisease populations than did that of the HLS clades. This bias might be related to the finding that a specific clade order is maintained within each NBPF gene: the CON1 and CON2 clades are found nearest to the NBPF gene region encoding the N terminus of the predicted protein, whereas the other clade sequences are more distally located.5 Thus, loss of CON1 and CON2 sequences could be expected to frequently result in complete loss of gene function (even if downstream DUF1220 sequences were not deleted) because the promoter region and/or transcription start site are more likely to be included in a loss of sequence that contains the gene regions encoding the N terminus of the predicted protein. This might result in a more pronounced effect on phenotype compared to loss of other DUF1220 clade sequences, which lie in the middle and C-terminal regions of the predicted proteins. Alternatively the bias might be a reflection of technical issues, e.g., accurately measuring the higher copy number of the HLS clades, compared to the CON clades, is typically more difficult. Finally it is also possible that the CON and HLS clades carry out distinct functional roles and, as a result, influence different brain phenotypes.
The capacity of DUF1220 sequences to undergo frequent duplications and deletions is most likely related, at least in part, to their organization in the genome. DUF1220 sequences that lie within the 1q21 region are generally found in two distinct arrangements: (1) within the 13 predicted NBPF genes, which are primarily interspersed throughout the 1q21 region and are separated by single- or low-copy-number non-NBPF genes, and (2) tandemly ordered within individual NBPF genes. Such an organization provides a number of different options through which copy number can be easily altered. One or a combination of mechanisms could underlie these changes; such mechanisms include nonallelic homologous recombination (NAHR), DNA-replication-based mechanisms, and possibly mitotic recombination. Specifically, NAHR events could occur during meiosis and cause either duplications or deletions of a few or many DUF1220 sequences or even of entire gene regions. Alternatively, DNA-replication-based mechanisms might also be involved in changes in DUF1220 copy number. As a result of the numerous DUF1220 tandem duplications found in the human genome,5 replication slippage might occur during DNA synthesis in which either the template or a new DNA strand loops out and causes deletions or duplications, respectively; another possibility might be long-distance template-switching mechanisms.15 These processes could produce rapid localized increases in DUF1220 copy number, such as the human-specific evolutionary hyperamplifications noted by O’Bleness, et al.,5 as well as in the variations in DUF1220 copy number observed here within human populations.
Utilizing lower-resolution bacterial artificial chromosome (BAC)-based arrays, Brunetti-Pierri et al.6 postulated that HYDIN might be a plausible candidate gene important to brain-size differences seen between individuals with 1q21-related microcephaly and macrocephaly. Our data, obtained with higher-density custom 1q21 arrays, show no suggestive evidence that this is the case, either in disease or nondisease populations. Because Brunetti-Pierri et al. used conventional BAC arrays with large probes that targeted only unique sequences, small regions and highly-duplicated sequences that might be important to causing microcephaly and macrocephaly could have been missed. Also, from an evolutionary standpoint, no genes, including HYDIN, show a high correlation between copy number and primate brain size. Likewise, no genes, except those encoding DUF1220 domains, show a correlation with both pathological-associated head size and human-population variation extremes for brain gray-matter volume. Although HYDIN might be involved in hydrocephaly (MIM 610813), our data provide no support for its involvement in the brain-size differences that have occurred over the course of primate evolution.
Regarding the mechanism by which DUF1220 acts, we have previously proposed links among DUF1220 domains, microcephaly, and the centrosome.4,8 The centrosome is a key mitotic regulator that determines when cells switch from symmetric to asymmetric cell division. The timing of this switch is essential for neuronal migration during brain development and can profoundly influence neuron number and brain size. Almost all known microcephaly-associated genes encode centrosomal proteins and one, CDK5RAP2, is a homolog of PDE4DIP, which encodes a centrosomal protein that contains a copy of the ancestral form of DUF1220. In addition, the EVI5 promoter has recently been recruited to the regulatory region of several NBPF genes.16 Interestingly, EVI5 is known to encode a centrosomal protein,17 suggesting a possible means by which DUF1220 domains might be targeted to the centrosome.
Finally, the data presented here provide additional support to a previously described model that links DUF1220’s recent evolutionary copy-number burst to the large number of 1q21 CNVs that have been reported to be associated with disease (Figure 5 and Table S1). The model proposes that the adaptive value conferred by increasing DUF1220 copy number has resulted in favoring the retention of the high instability of the 1q21.1-1q21.2 region; this, in turn, has given rise to the unusually high number of recurrent disease-associated CNVs that have been reported for this region.8 There are an estimated 13 NBPF genes (encoding a total of 240 DUF1220 repeats) that map to the 1q21.1-1q21.2 region; these are often separated by several single-copy, non-DUF1220 genes, producing a highly disease-prone genome architecture (Figure S5). In such an environment, the large number of nontandem, DUF1220-repeat-rich NBPF genes in this region can be expected to produce frequent NAHR events between NBPF genes that will often carry along (i.e., duplicate or delete) the intervening single-copy genes. In this sense, although DUF1220 copy number by itself appears to be directly related to evolutionarily advantageous increases in brain size, the dosage imbalance (generated by illegitimate DUF1220-driven recombination events) of the single-copy 1q21.1-1q21.2 genes might produce the unusually diverse pathologies that have been reported for disease-associated 1q21.1-1q21.2 CNVs. In summary, although hyperamplification of DUF1220 copy number is among the most remarkable HLS changes found in the genome and has ramifications related to human brain evolution, it has come—and continues to come—at an expensive price given that it has been largely responsible for creating and maintaining one of the most unstable and disease-prone regions of the human genome.
Figure 5.
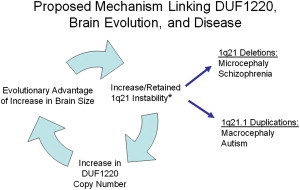
DUF1220 Model Linking 1q21.1-1q21.2 Instability, DUF1220-Domain Copy Number, Brain Evolution, and Recurrent Disease
∗Twelve genomic diseases have been linked to CNVs in the 1q21.1-1q21.1 region. They are listed in Table S1 and include autism, congenital heart disease, congenital anomaly of the kidney and urinary tract, epilepsy, intellectual disability, intermittent explosive disorder, macrocephaly, Mayer-Rokitansky-Küster-Hauser syndrome, microcephaly, neuroblastoma, schizophrenia, and thrombocytopenia-absent-radius syndrome.
Acknowledgments
We thank Pawel Stankiewicz, Martin Kennedy, John Hokanson, and Mark Johnston for helpful comments; Scott Vacha and Amir Ben-Dor for help with aCGH analyses; Andrew Fortna for programming expertise; Gunter Scherer and Elaine Spector for blood draws and DNA isolations; and Jake Saunders for graphics help. We also thank Oxford Gene Technology for high-quality aCGH services. This work was supported by National Institutes of Health (NIH) grants R01 MH081203-1 and R01 AA11853-11 to J.M.S. and a Butcher Foundation grant to J.M.S. and T.F. J.D. was supported in part by JFK Partners, University of Colorado funding from the Maternal Child Health Bureau, and Leadership Education in Neurodevelopmental Disabilities grant T73MC11044. M.O. was supported by a postdoctoral fellowship from the National Institute on Alcohol Abuse and Alcoholism/National Institute on Drug Abuse (5T32AA007464-32). C.M.D. was supported by American Recovery and Reinvestment Act grant R01 AA011853-12S1 and by an NIH Computational Bioscience Program Training Grant (5T15 LM009451-05). N.A. was supported by National Institute of Mental Health Supplementary Grant R01 MH081203-02S1; J.K. was supported in part through a Graduate Assistantship from the Coleman Institute for Cognitive Disabilities. J.M.S. is a founder and shareholder of GATC Science. J.R.L. is a consultant for Athena Diagnostics, holds stock ownership of 23andMe and Ion Torrent Systems, and is a coinventor on multiple United States and European patents for DNA diagnostics. The Baylor College of Medicine and Department of Molecular and Human Genetics derive revenue from molecular genetics testing clinical services provided by the Medical Genetics Laboratories (https://www.bcm.edu/geneticlabs).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Baylor College of Medicine Medical Genetics Laboratories, https://www.bcm.edu/geneticlabs
Human BLAT Search, http://genome.ucsc.edu/cgi-bin/hgBlat
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
R version 2.10.1, http://cran.r-project.org/
UCSC In Silico PCR, http://genome.ucsc.edu/cgi-bin/hgPcr?command=start
References
- 1.Popesco M.C., Maclaren E.J., Hopkins J., Dumas L., Cox M., Meltesen L., McGavran L., Wyckoff G.J., Sikela J.M. Human lineage-specific amplification, selection, and neuronal expression of DUF1220 domains. Science. 2006;313:1304–1307. doi: 10.1126/science.1127980. [DOI] [PubMed] [Google Scholar]
- 2.Fortna A., Kim Y., MacLaren E., Marshall K., Hahn G., Meltesen L., Brenton M., Hink R., Burgers S., Hernandez-Boussard T. Lineage-specific gene duplication and loss in human and great ape evolution. PLoS Biol. 2004;2:E207. doi: 10.1371/journal.pbio.0020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vandepoele K., Van Roy N., Staes K., Speleman F., van Roy F. A novel gene family NBPF: Intricate structure generated by gene duplications during primate evolution. Mol. Biol. Evol. 2005;22:2265–2274. doi: 10.1093/molbev/msi222. [DOI] [PubMed] [Google Scholar]
- 4.Dumas L., Kim Y.H., Karimpour-Fard A., Cox M., Hopkins J., Pollack J.R., Sikela J.M. Gene copy number variation spanning 60 million years of human and primate evolution. Genome Res. 2007;17:1266–1277. doi: 10.1101/gr.6557307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Bleness M., Dickens C.M., Dumas L., Kehrer-Sawatzki H., Wyckoff G.J., Sikela J.M. Evolutionary History and Genome Organization of DUF1220 Protein Domains. Genes, Genomes, Genetics. 2012;2 doi: 10.1534/g3.112.003061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brunetti-Pierri N., Berg J.S., Scaglia F., Belmont J., Bacino C.A., Sahoo T., Lalani S.R., Graham B., Lee B., Shinawi M. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mefford H.C., Sharp A.J., Baker C., Itsara A., Jiang Z., Buysse K., Huang S., Maloney V.K., Crolla J.A., Baralle D. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N. Engl. J. Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dumas L., Sikela J.M. DUF1220 domains, cognitive disease, and human brain evolution. Cold Spring Harb. Symp. Quant. Biol. 2009;74:375–382. doi: 10.1101/sqb.2009.74.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Falk D. Endocranial casts and their significance for primate brain evolution. In: Swindler D.R., Erwin J., editors. Comparative Primate Biology, Volume 1: Systematics, Evolution, and Anatomy. Alan R. Liss; New York: 1986. pp. 477–490. [Google Scholar]
- 10.Kouprina N., Pavlicek A., Mochida G.H., Solomon G., Gersch W., Yoon Y.H., Collura R., Ruvolo M., Barrett J.C., Woods C.G. Accelerated evolution of the ASPM gene controlling brain size begins prior to human brain expansion. PLoS Biol. 2004;2:E126. doi: 10.1371/journal.pbio.0020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roth G., Dicke U. Evolution of the brain and intelligence. Trends Cogn. Sci. 2005;9:250–257. doi: 10.1016/j.tics.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 12.Ponce de León M.S., Golovanova L., Doronichev V., Romanova G., Akazawa T., Kondo O., Ishida H., Zollikofer C.P. Neanderthal brain size at birth provides insights into the evolution of human life history. Proc. Natl. Acad. Sci. USA. 2008;105:13764–13768. doi: 10.1073/pnas.0803917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lenroot R.K., Gogtay N., Greenstein D.K., Wells E.M., Wallace G.L., Clasen L.S., Blumenthal J.D., Lerch J., Zijdenbos A.P., Evans A.C. Sexual dimorphism of brain developmental trajectories during childhood and adolescence. Neuroimage. 2007;36:1065–1073. doi: 10.1016/j.neuroimage.2007.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hallast P., Rull K., Laan M. The evolution and genomic landscape of CGB1 and CGB2 genes. Mol. Cell. Endocrinol. 2007;260–262:2–11. doi: 10.1016/j.mce.2005.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee J.A., Carvalho C.M., Lupski J.R. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell. 2007;131:1235–1247. doi: 10.1016/j.cell.2007.11.037. [DOI] [PubMed] [Google Scholar]
- 16.Vandepoele K., Andries V., van Roy F. The NBPF1 promoter has been recruited from the unrelated EVI5 gene before simian radiation. Mol. Biol. Evol. 2009;26:1321–1332. doi: 10.1093/molbev/msp047. [DOI] [PubMed] [Google Scholar]
- 17.Faitar S.L., Dabbeekeh J.T.S., Ranalli T.A., Cowell J.K. EVI5 is a novel centrosomal protein that binds to alpha- and gamma-tubulin. Genomics. 2005;86:594–605. doi: 10.1016/j.ygeno.2005.06.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.