

Abstract
Whole-exome sequencing (WES), which analyzes the coding sequence of most annotated genes in the human genome, is an ideal approach to studying fully penetrant autosomal-recessive diseases, and it has been very powerful in identifying disease-causing mutations even when enrollment of affected individuals is limited by reduced survival. In this study, we combined WES with homozygosity analysis of consanguineous pedigrees, which are informative even when a single affected individual is available, to identify genetic mutations responsible for Walker-Warburg syndrome (WWS), a genetically heterogeneous autosomal-recessive disorder that severely affects the development of the brain, eyes, and muscle. Mutations in seven genes are known to cause WWS and explain 50%–60% of cases, but multiple additional genes are expected to be mutated because unexplained cases show suggestive linkage to diverse loci. Using WES in consanguineous WWS-affected families, we found multiple deleterious mutations in GTDC2 (also known as AGO61). GTDC2’s predicted role as an uncharacterized glycosyltransferase is consistent with the function of other genes that are known to be mutated in WWS and that are involved in the glycosylation of the transmembrane receptor dystroglycan. Therefore, to explore the role of GTDC2 loss of function during development, we used morpholino-mediated knockdown of its zebrafish ortholog, gtdc2. We found that gtdc2 knockdown in zebrafish replicates all WWS features (hydrocephalus, ocular defects, and muscular dystrophy), strongly suggesting that GTDC2 mutations cause WWS.
Main Text
Walker-Warburg syndrome (WWS, also known as MDDGA1–7 [MIM 236670, 613150, 253280, 253800, 613153, 613154, and 614643]) is a severe neuromuscular disorder characterized by congenital muscular dystrophy, ocular malformations, smooth appearance of the cortical surface accompanied by neuronal migration defects (cobblestone lissencephaly), and ventricular enlargement (hydrocephalus).1,2 Mutations in seven genes (POMT1 [MIM 607423], POMT2 [MIM 607439], POMGNT1 [MIM 606822], FKTN [MIM 607440], FKRP [MIM 606596], LARGE [MIM 603590], and ISPD [MIM 614631]) are known to cause WWS2–5 but are only identified in 50%–60% of cases.4,6–8 Identification of additional genes mutated in WWS has been hindered by the small size of the available pedigrees, from which DNA is only available from one or two affected individuals. To increase our power to understand the genetic architecture of WWS and to identify mutations in additional genes, we collected a cohort of affected individuals from consanguineous marriages (n = 19; Table S1, available online). All subjects were enrolled after informed consent was obtained, and research was conducted according to protocols approved by the institutional review board at Boston Children’s Hospital. All affected individuals were diagnosed with classic WWS on the basis of the following criteria: (1) brain malformation characterized by cobblestone lissencephaly on neuroimaging, severe hydrocephalus, and cerebellar hypoplasia; (2) the presence of ocular malformations (microphthalmia and macrophthalmia, retinal dysplasia, optic nerve hypoplasia, and anterior chamber defects); and (3) a clinical diagnosis, usually based on severe hypotonia, of congenital muscular dystrophy.1,9 Serum creatine kinase and histopathological confirmation of muscular dystrophy were not available in most cases because of the clinical severity and reduced lifespan associated with this condition.
We first determined the pattern of homozygosity in the consanguineous pedigrees by hybridizing genomic DNA from the probands and all available family members on whole-genome SNP arrays—Affymetrix 250K StyI or Illumina 610-Quad—at the W.M. Keck Foundation Biotechnology Resource Laboratory at Yale University. Genotyping data were analyzed with in-house-developed software for the identification of regions of homozygosity (ROHs) throughout the genome, and genes known to be mutated in WWS were sequenced whenever they were contained in a ROH present only in the affected individual. As we previously reported,6 homozygous mutations were found in eight families (8 of 19; 42%) (Table S1). Homozygosity analysis of the remaining 11 families identified an average percentage of homozygosity of 9.3% (1.3% ± standard error of the mean), which is larger than the expected 6% for a first-cousin union10 and suggests increased background consanguinity that could not be captured by the available nuclear pedigrees (Table S1). Multiple regions were shared between at least two families, as expected by chance in this type of cohort, but the fact that no region overlapped in more than three families indicates that multiple additional genes remain to be identified.
The percentage of homozygosity in the cohort was high but was sufficient to reduce the number of candidate genes to a few hundred or thousand per sample. Therefore, we performed whole-exome sequencing (WES) analysis to study all candidate genes at once.11 Seven of the remaining probands were suitable for WES, and whole-exome DNA libraries were prepared from genomic DNA by hybridization with the Agilent SureSelect All Exon 37Mb kit and were run on an Illumina HiSeq2000 machine either at the Duke Center for Human Genome Variation in Durham, NC or at the Broad Institute of MIT and Harvard in Cambridge, MA. Sequencing reads were aligned to the Human Reference Genome (UCSC hg19) with Burrows-Wheeler Aligner software.12 Variant calling was performed with SAMtools software,12 and either SequenceVariantAnalyzer or ANNOVAR13 was used for annotating variants. Sequencing yielded an average of 49,538 variants per individual (see Table S2 for WES statistics). Variants were then filtered with custom MySQL queries for the identification of recessive mutations, which were (1) most likely pathogenic (missense or nonsense variants, frameshift indels, or splicing errors within 10 bp of the splice site), (2) novel or rare (minor allele frequency < 0.5%) in a control cohort of more than 7,000 control individuals (1,000 Genomes November 2011 release, 5,379 exomes from the National Heart, Lung, and Blood Institute [NHLBI] Grand Opportunity Exome Sequencing Project, 69 genomes from Complete Genomics, and 821 in-house Walsh lab exomes), (3) found in a ROH in the propositus, and (4) found to be inherited in an autosomal-recessive pattern in the family upon Sanger sequencing of the parents and unaffected siblings. Filtered variants from Middle Eastern individuals, who are not widely represented in the exome databases, were also genotyped in at least 384 ethnically matched individuals with mass-spectrometry-based Sequenom SNP genotyping at the Intellectual and Developmental Disabilities Research Center (IDDRC) Molecular Genetics Core at Boston Children’s Hospital for confirming that the variants were not rare population-specific polymorphisms. This approach narrowed the number of variants to an average number of five candidates per family (Table S2).
Homozygous mutations in genes known to be involved in WWS were identified in affected individuals in two families (Figure S1); these were a homozygous 1 bp insertion leading to a frameshift change (c.642dup [p.Asp215∗]) (NM_006731.2) in FKTN in family 82, a first-cousin union of Hispanic ethnicity from the United States (Table 1 and Figures S1A and S1B), and a homozygous missense change (c.1433A>G [p.His478Arg]) (NM_013382.4) in POMT2 in family 144, a second-cousin union from Sudan (Table 1 and Figures S1C and S1D). The missense change obtained the highest pathogenicity score in two independent pathogenicity prediction algorithms, Polyphen214 and SIFT15 (Polyphen2 = 1 [probably damaging]; SIFT = 0 [damaging]). Despite being present in a large ROH in the affected individuals, these genes had not been previously sequenced because in each family, a smaller homozygous region with an identical haplotype was present over the gene of interest in one of the parents and was most likely due to a common haplotype in the population. These findings illustrate the power of WES to identify disease-causing mutations in an unbiased manner because all known disease genes can be tested independently.
Table 1.
Clinical Features of Individuals with FKTN, POMT2, or GTDC2 Mutations
| Person | Age at Exam | Gene | Cobblestone Lissencephaly | Enlarged Ventricles | Cerebellar Hypoplasia | Ocular Defects | Muscular Defects |
|---|---|---|---|---|---|---|---|
| 82-1 | 2 months | FKTN | + | + | + | retinal dysplasia, glaucoma | CK = 44,566 U/la |
| 144-1 | 6 months | POMT2 | + | + | + | glaucoma | hypotonia |
| 86-1 | 23 weeks | GTDC2 | + | + | + | − | − |
| 86-2 | 20 weeks | + | + | + | N/A | N/A | |
| 90-1 | 2 months | GTDC2 | + | + | + | retinal dysplasia | hypotonia |
| 100-1 | 1 month | GTDC2 | + | + | + | microphthalmia and macrophthalmia | hypotonia |
The following abbreviations are used: CK, creatine kinase; and N/A, not available.
Normal CK range: 40–180 U/l.
We then analyzed the variants in the remaining WWS-affected families and identified two independent homozygous changes in glycosyltransferase-like domain-containing 2 (GTDC2), a single-exon gene on the short arm (3p22.1) of chromosome 3 (Figure 1). The first variant was a nonsense change (c.1333C>T [p.Arg445∗]) (NM_032806.4) in a male from a Jordanian first-cousin union (family 90, Figure 1C) and led to a possible early truncation of GTDC2 (Figure 1G); the second variant was a missense change predicted to be very deleterious and affected a conserved amino acid (c.473G>A [p.Arg158His]; Polyphen2 = 1 [probably damaging] and SIFT = 0 [damaging]) in a female from a Saudi family (family 100, Figures 1D and 1E). Each change was contained in a large ROH: 25.8 Mb and 28.1 Mb in families 90 and 100, respectively (Figure 1G). The phenotypes of the two families were undistinguishable from each other and from that of classic WWS cases—they included cobblestone lissencephaly, severe hydrocephalus, severe hypoplasia of the cerebellar vermis, and muscle hypotonia (Table 1 and Figures 1A and 1B). No muscle biopsy was performed because all affected individuals died within a few days or weeks of birth.
Figure 1.
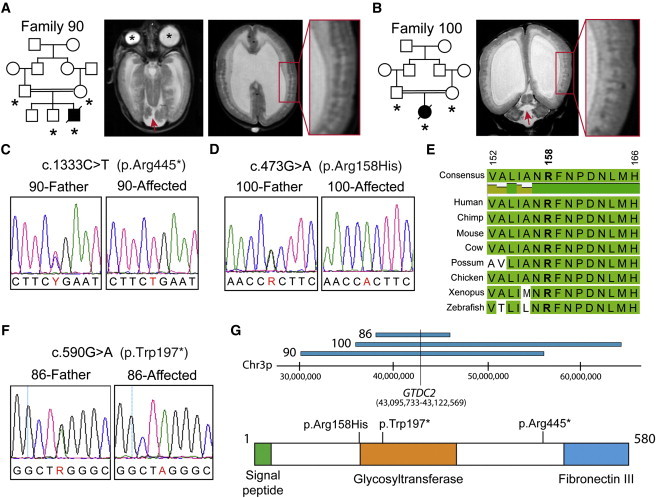
Three Independent Alleles in GTDC2 Cause WWS
(A) Pedigree structure of family 90 (DNA was available for individuals marked by asterisks) and T2-weighted axial magnetic resonance images (MRIs) of the propositus. The MRI on the left shows microphthalmia and macrophthalmia (asterisks) and cerebellar hypoplasia (red arrow), and the MRI on the right shows severe ventricular enlargement and cortical-migration defects characteristic of cobblestone lissencephaly (inset).
(B) Pedigree structure of family 100 and coronal MRIs of the propositus. These images show a degree of malformation similar to that seen in family 90.
(C–D) Sanger sequencing confirming the mutations identified by WES in families 90 and 100.
(E) Conservation of arginine 158 and the surrounding region in vertebrates.
(F) Sanger sequencing of an additional GTDC2 nonsense mutation identified in family 86.
(G) Schematics showing overlapping regions of homozygosity in the three families affected by GTDC2 mutations and the GTDC2 protein structure including predicted functional domains. Locations of the mutations are listed above the protein.
WWS is the most severe of a spectrum of dystroglycan-glycosylation disorders affecting the muscle, eyes, and brain. It has variable severity and is commonly defined as muscular-dystrophy dystroglycanopathies (MDDGB1–6 [MIM 613155, 613156, 613151, 613152, 606612, and 608840] and MDDGC1–5 and MDDGC7 [MIM 609308, 613158, 613157, 611588, 607155, and 613818]).7,9,16 We sequenced GTDC2 in a validation cohort of nonconsanguineous cases (n = 52) spanning the dystroglycanopathy phenotypic spectrum to identify additional alleles. We identified an additional nonsense allele in family 86, an Indian family in which two pregnancies were terminated as a result of a diagnosis of ventricular enlargement during gestation. Both fetuses were female and were diagnosed with WWS upon autopsy (Table 1). DNA was available from one of the two affected individuals, and sequencing revealed a homozygous GTDC2 mutation (c.590G>A) generating a stop codon at amino acid 197 (p.Trp197∗; Figure 1F). The family displayed a small total amount of homozygosity (1%)—only two regions were larger than 5 cM, and GTDC2 was contained in the largest (7.5 Mb and 5.9 cM) of these regions on chromosome 3 (Figure 1G)—suggesting that the parents might be distantly related.
GTDC2 is an ideal candidate gene for WWS because it is predicted by multiple protein-domain prediction algorithms to contain an uncharacterized glycosyltransferase domain (Figure 1G). All genes in which mutations have been identified to date are involved in glycosylation of the transmembrane protein dystroglycan, which must be glycosylated in order to interact with extracellular matrix components such as laminins, and this binding is essential for brain, eye, and muscle integrity.2,17–19 GTDC2 (which is alternatively called AGO61) is predicted by homology to belong to glycosyltransferase family 61, and, similar to LARGE (another gene involved in dystroglycanopathies), it is also predicted to encode a xylosyltransferase.20 We studied expression of GTDC2 mRNA in multiple human tissues by using quantitative PCR (qPCR) both during fetal development and in the adult. cDNA samples from postmortem fetal and adult human tissues were obtained commercially (BioChain Institute and Clontech). qPCR was performed with SYBR Green reagents (Applied Biosystems) on an ABI 7500 qPCR platform as previously described21 with primers hGTDC2_F 5′-GCGGAGTCCAGGCTTCAGGCTTTC-3′ and hGTDC2_R 5′-CCGCCGAGAGGTGCATCCTAATG-3′ and normalizing expression values to beta-actin (ACTB).
At both fetal and adult stages, GTDC2 expression was especially high in the brain, muscle, heart, and kidneys (Figure 2A), which are all variably affected in dystroglycanopathy cases.6,22 Brain expression was highest in the cortex and cerebellum (Figure 2A), and very high expression was also noted in the pancreas (Figure S2A). To analyze possible changes in cellular distribution, we analyzed expression of the murine ortholog of GTDC2, Gtdc2, by in situ hybridization (performed at the In Situ Hybridization Core Facility at the University of North Carolina School of Medicine in Chapel Hill, NC) with a probe amplified from an embryonic day (E)12.5 mouse brain cDNA library (a kind gift of Byoung-Il Bae at Boston Children’s Hospital) (primers were mGtdc2-forward 5′-TTCCCTTACGCTGTCAATCC-3′ and mGtdc2-reverse 5′-TACGTGTTCTCCCCTTGCTC-3′). We found that Gtdc2 is highly expressed during brain and eye development; it peaks in the cerebral cortex during the last week of gestation in both neuronal progenitors in the ventricular zone and migrating and differentiating neurons but declines around birth, supporting a role for this gene during neuronal development (Figure 2B), but we did not observe differential expression in neuronal progenitors or neuronal subtypes.
Figure 2.
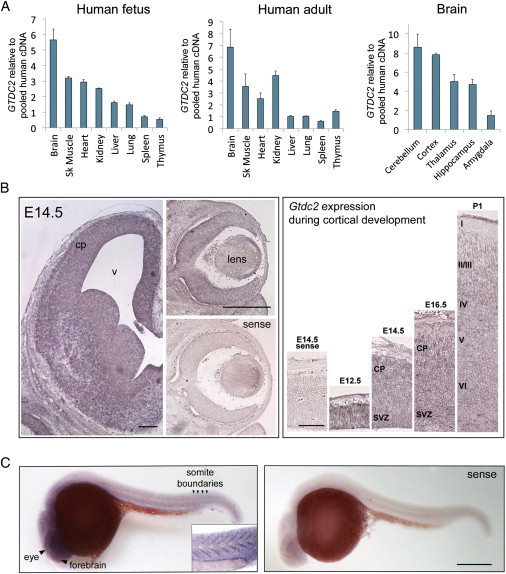
Expression of GTDC2 and Its Orthologs in Mice and Zebrafish
(A) qPCR analysis of GTDC2 mRNA expression in fetal and adult human tissues. High expression was found at both stages in the brain, muscle, kidneys, and heart. In the adult brain, the cerebellum and cortex had the highest expression. Error bars represent the standard error across three sample repeats normalized to beta-actin.
(B) Murine Gtdc2 is expressed during brain development. At E14.5 (left panels), expression is observed throughout the cortical plate (cp) and the developing brain, as well as throughout the eye, including the lens and cornea. No staining was evident in the sense control. The scale bars in the left panels represent 200 μm. Cortical expression (right panels) peaks between E14.5 and E16.5 and is reduced at birth (P0). The scale bar in the right panel represents 100 μm.
(C) In situ hybridization of zebrafish gtdc2 at 1 dpf shows expression throughout the body; the highest staining is in the brain and eye and at the boundaries between somites (arrowheads and inset). The scale bar represents 150 μm.
To test how GTDC2 loss of function would affect brain and muscle development, we used the zebrafish, which has been recently established as an animal model for dystroglycanopathies.23–26 The mRNA for the zebrafish ortholog (gtdc2) of GTDC2 was expressed in the zebrafish embryo and larva throughout development; there was especially high expression in the developing brain, eyes, and somites (Figure 2C and Figure S2B), consistent with expression of the zebrafish orthologs of other genes mutated in WWS.27,28 Zebrafish in situ hybridization was performed according to standard protocols29 with a probe amplified with a mixed 20-somite to 5 day postfertilization (dpf) zebrafish cDNA library (a kind gift of Peter Wang at Boston Children’s Hospital) (primers were zfgtdc2-forward 5′-ATGCACATTTTCCACGATGA-3′ and zfgtdc2-reverse 5′-AACGACTGCTCCTCCAAAGA-3′). To knock down gtdc2, we tested two different antisense morpholino oligonucleotides (MOs) targeted to interfere with gtdc2 translation by either blocking the start codon (gtdc2Start: 5′-CCGGCAGGTTCATCCTACACCCGAT-3′) or the splice donor site in the only intron present in this gene in the fish (gtdc2Splice: 5′-TTATCATAACCCTGTGCACCT GTGA-3′). When these MOs were injected into fertilized zebrafish oocytes, they provided similar results (gtdc2Start at 3–5 ng and gtdc2Splice at 7–10 ng). Therefore, data from only gtdc2Start will be shown. The standard control MO provided by Gene Tools was used as a control at the same concentration used for the experimental MO injections. Injected embryos/larvae and uninjected clutchmate controls were analyzed at 0, 1, 2, and 3 dpf for survival and morphology of the tail, head, and eyes. Developmental defects in gtdc2 morphants were evident from 1 dpf—one quarter of the embryos were severely affected with a very short body, thick and poorly developed tail, and no discernible eyes or head structures. Half of the morphants were mildly affected and were scored on a combination of phenotypes: thick tail, U-shaped somites, reduced mobility inside the chorion, a small head, and underdeveloped eyes, where ventral fusion of the retina was delayed (Figure 3A). We asked whether coinjection of the in-vitro-synthesized human GTDC2 mRNA with the gtdc2 MO could improve these phenotypes and found this to be the case (Figure 3B). However, almost no rescue effect was observed when we tested the mRNA mutated to include the p.Arg158His allele (Figure 3C). The Arg158 residue is highly conserved in GTDC2, and it is located at the beginning of the glycosyltransferase domain; therefore, it is possible that this change might severely impair protein function.
Figure 3.
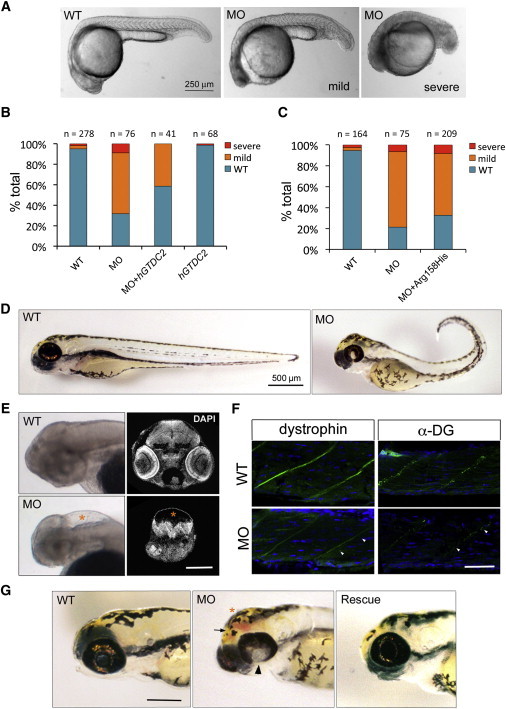
Loss of gtdc2 in the Zebrafish Embryo Recapitulates Phenotypes Observed in WWS
(A) Mild (center) and severe (right) phenotypes observed in the 1 dpf zebrafish embryo after injection of a MO designed against the start site of gtdc2.
(B) Phenotypes observed at 1 dpf can be rescued by coinjection of the gtdc2 MO and 150 pg of GTDC2 mRNA, whereas no effect is observed by injection of the mRNA alone (hGTDC2).
(C) Coinjection of 150 pg of GTDC2 mRNA containing the p.Arg158His alteration is not able to rescue the morphant phenotype. Representative experiments are shown.
(D) Surviving morphants at 3 dpf show several developmental defects: reduced length, bent tail, failure of ventral retinal fusion, and dysmorphic head shape.
(E) 3 dpf morphants also display hydrocephalus (lower left panel: whole-mount of nonpigmented larva [asterisk]) and ventral compression of the remaining brain tissue (lower right panel: DAPI staining on histological section at a location matching the wild-type [WT] section above). The scale bar represents 200 μm.
(F) Immunohistochemistry in sections of the WT and morphant muscle shows that expression of both dystrophin (green, left) and glycosylated dystroglycan (green, right) are reduced at the myosepta (marked by arrowheads). Nuclei are labeled in DAPI (blue). The scale bar represents 50 μm.
(G) Rescue with GTDC2 mRNA (on right) ameliorates the most severe head phenotypes observed in the morphant (center). These phenotypes are failure of retinal fusion (arrowhead), hydrocephalus (asterisk), and hemorrhage (arrow). The scale bar represents 200 μm.
gtdc2 knockdown severely affected survival: 43.4% of embryos died within the first 3 days after injection. However, clutchmates that were uninjected or injected with a control MO displayed survival rates of 98.4% and 89.0%, respectively (Figure S3A). Surviving morphants at 3 dpf were shorter than controls and often had a bent tail, impaired motility, smaller eyes in which the retina failed to fuse ventrally, and a domed appearance of the top of the head (Figures 3D and 3G and Figure S3B). When pigmentation was blocked at 1 dpf with the addition of 0.003% 1-phenyl 2-thiourea to the water, external analysis of the head revealed extreme ventricular enlargement (hydrocephalus), often accompanied by hemorrhaging (Figure 3G), in the most affected larvae (Figure 3E). Ventral compression and reduction in brain volume were evident upon histological analysis of coronal cryosections stained with DAPI (Figure 3E). Disorganization of the retinal epithelium was also observed in a few morphants; there was variable severity in ultrathin plastic-embedded sections stained with 1% toluidine blue and 1% sodium borate (Figure S3C) at the Conventional Electron Microscopy Facility at Harvard Medical School. Finally, muscle development was severely disrupted by a loss of both dystrophin and glycosylated dystroglycan from the myosepta, the junction of muscle fibers in which the extracellular matrix is enriched (Figure 3F). Disorganization of the muscle fibers was also observed upon histology (Figure S3D). These phenotypes (hydrocephalus and retinal and muscle defects) are the hallmarks of WWS and are consistent with the eye, muscle, and brain phenotypes observed in morphants of genes (such as POMT1, POMT2, FKTN, FKRP, and ISPD) previously found to be mutated in WWS.24–26,28 All phenotypes were ameliorated by GTDC2 mRNA injections when embryos were analyzed at 2 and 3 dpf (Figure 3G and Figure S3E).
In summary, we have combined whole-exome sequencing, homozygosity mapping, and functional analysis in zebrafish to identify and validate GTDC2 mutations as a cause of WWS. In addition, we have reduced the number of candidate variants in other families to a manageable number of five variants per family. This approach will most likely yield additional mutated genes in the future to help unravel the pathways involved in WWS and to determine the role of different glycosyltransferases in brain and muscle development.
Acknowledgments
We thank the families who enrolled in our studies and the physicians who have contributed individuals to our validation cohort. The EuroBioBank and Telethon Network Genetic Biobanks (GTB07001F) are also acknowledged for providing biological samples. We are also grateful to A.J. Barkovich for help with the review of the magnetic resonance imaging scans. M.C.M. was supported by a Junior Faculty Career Development Award from the Manton Center for Orphan Disease Research and a K99/R00 Transition to Independence award from the National Institutes of Health (NIH) (National Institute of Child Health & Human Development, K99HD067379). T.W.Y. was supported by a NIH T32 grant (T32NS007484-08), the Clinical Investigator Training Program at Harvard-MIT Health Science and Technology, and the Nancy Lurie Marks Junior Faculty MeRIT Fellowship. Research was supported by grants from the NIH (National Institute of Neurological Disorders and Stroke R01NS035129 to C.A.W. and National Institute of Arthritis and Musculoskeletal and Skin Diseases R01AR044345 to A.H.B), the Muscular Dystrophy Association to M.C.M. and A.H.B., the William Randolph Hearst Fund to M.C.M. and V.A.G., and the Manton Center for Orphan Disease Research to M.C.M., A.H.B., and C.A.W. Sequencing and Sequenom genotyping at Boston Children’s Hospital were supported by the Intellectual and Developmental Disabilities Research Centers (P30HD19655). Sequencing at the Broad Institute was supported by a grant from the NIH and the American Recovery & Reinvestment Act (National Institute of Mental Health RC2MH089952). C.A.W. is an Investigator of the Howard Hughes Medical Institute.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Carbohydrate-Active Enzymes Database, www.cazy.org
dCHIP Software, http://biosun1.harvard.edu/complab/dchip/
NHLBI Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
References
- 1.Dobyns W.B., Pagon R.A., Armstrong D., Curry C.J., Greenberg F., Grix A., Holmes L.B., Laxova R., Michels V.V., Robinow M. Diagnostic criteria for Walker-Warburg syndrome. Am. J. Med. Genet. 1989;32:195–210. doi: 10.1002/ajmg.1320320213. [DOI] [PubMed] [Google Scholar]
- 2.Godfrey C., Foley A.R., Clement E., Muntoni F. Dystroglycanopathies: Coming into focus. Curr. Opin. Genet. Dev. 2011;21:278–285. doi: 10.1016/j.gde.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 3.Barresi R., Campbell K.P. Dystroglycan: From biosynthesis to pathogenesis of human disease. J. Cell Sci. 2006;119:199–207. doi: 10.1242/jcs.02814. [DOI] [PubMed] [Google Scholar]
- 4.Roscioli T., Kamsteeg E.J., Buysse K., Maystadt I., van Reeuwijk J., van den Elzen C., van Beusekom E., Riemersma M., Pfundt R., Vissers L.E. Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan. Nat. Genet. 2012;44:581–585. doi: 10.1038/ng.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Willer T., Lee H., Lommel M., Yoshida-Moriguchi T., de Bernabe D.B., Venzke D., Cirak S., Schachter H., Vajsar J., Voit T. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat. Genet. 2012;44:575–580. doi: 10.1038/ng.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manzini M.C., Gleason D., Chang B.S., Hill R.S., Barry B.J., Partlow J.N., Poduri A., Currier S., Galvin-Parton P., Shapiro L.R. Ethnically diverse causes of Walker-Warburg syndrome (WWS): FCMD mutations are a more common cause of WWS outside of the Middle East. Hum. Mutat. 2008;29:E231–E241. doi: 10.1002/humu.20844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mercuri E., Messina S., Bruno C., Mora M., Pegoraro E., Comi G.P., D’Amico A., Aiello C., Biancheri R., Berardinelli A. Congenital muscular dystrophies with defective glycosylation of dystroglycan: A population study. Neurology. 2009;72:1802–1809. doi: 10.1212/01.wnl.0000346518.68110.60. [DOI] [PubMed] [Google Scholar]
- 8.Bouchet C., Gonzales M., Vuillaumier-Barrot S., Devisme L., Lebizec C., Alanio E., Bazin A., Bessières-Grattagliano B., Bigi N., Blanchet P. Molecular heterogeneity in fetal forms of type II lissencephaly. Hum. Mutat. 2007;28:1020–1027. doi: 10.1002/humu.20561. [DOI] [PubMed] [Google Scholar]
- 9.Cormand B., Pihko H., Bayés M., Valanne L., Santavuori P., Talim B., Gershoni-Baruch R., Ahmad A., van Bokhoven H., Brunner H.G. Clinical and genetic distinction between Walker-Warburg syndrome and muscle-eye-brain disease. Neurology. 2001;56:1059–1069. doi: 10.1212/wnl.56.8.1059. [DOI] [PubMed] [Google Scholar]
- 10.Lander E.S., Botstein D. Homozygosity mapping: A way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570. doi: 10.1126/science.2884728. [DOI] [PubMed] [Google Scholar]
- 11.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang K., Li M., Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 16.Clement E., Mercuri E., Godfrey C., Smith J., Robb S., Kinali M., Straub V., Bushby K., Manzur A., Talim B. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann. Neurol. 2008;64:573–582. doi: 10.1002/ana.21482. [DOI] [PubMed] [Google Scholar]
- 17.Michele D.E., Barresi R., Kanagawa M., Saito F., Cohn R.D., Satz J.S., Dollar J., Nishino I., Kelley R.I., Somer H. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature. 2002;418:417–422. doi: 10.1038/nature00837. [DOI] [PubMed] [Google Scholar]
- 18.Hu H., Li J., Gagen C.S., Gray N.W., Zhang Z., Qi Y., Zhang P. Conditional knockout of protein O-mannosyltransferase 2 reveals tissue-specific roles of O-mannosyl glycosylation in brain development. J. Comp. Neurol. 2011;519:1320–1337. doi: 10.1002/cne.22572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manzini M.C., Walsh C.A. What disorders of cortical development tell us about the cortex: One plus one does not always make two. Curr. Opin. Genet. Dev. 2011;21:333–339. doi: 10.1016/j.gde.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inamori K., Yoshida-Moriguchi T., Hara Y., Anderson M.E., Yu L., Campbell K.P. Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science. 2012;335:93–96. doi: 10.1126/science.1214115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maynard T.M., Meechan D.W., Dudevoir M.L., Gopalakrishna D., Peters A.Z., Heindel C.C., Sugimoto T.J., Wu Y., Lieberman J.A., Lamantia A.S. Mitochondrial localization and function of a subset of 22q11 deletion syndrome candidate genes. Mol. Cell. Neurosci. 2008;39:439–451. doi: 10.1016/j.mcn.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bello L., Melacini P., Pezzani R., D’Amico A., Piva L., Leonardi E., Torella A., Soraru G., Palmieri A., Smaniotto G. Cardiomyopathy in patients with POMT1-related congenital and limb-girdle muscular dystrophy. Eur. J. Hum. Genet. 2012 doi: 10.1038/ejhg.2012.71. Published online May 2, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta V., Kawahara G., Gundry S.R., Chen A.T., Lencer W.I., Zhou Y., Zon L.I., Kunkel L.M., Beggs A.H. The zebrafish dag1 mutant: A novel genetic model for dystroglycanopathies. Hum. Mol. Genet. 2011;20:1712–1725. doi: 10.1093/hmg/ddr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin Y.Y., White R.J., Torelli S., Cirak S., Muntoni F., Stemple D.L. Zebrafish Fukutin family proteins link the unfolded protein response with dystroglycanopathies. Hum. Mol. Genet. 2011;20:1763–1775. doi: 10.1093/hmg/ddr059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kawahara G., Guyon J.R., Nakamura Y., Kunkel L.M. Zebrafish models for human FKRP muscular dystrophies. Hum. Mol. Genet. 2010;19:623–633. doi: 10.1093/hmg/ddp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thornhill P., Bassett D., Lochmüller H., Bushby K., Straub V. Developmental defects in a zebrafish model for muscular dystrophies associated with the loss of fukutin-related protein (FKRP) Brain. 2008;131:1551–1561. doi: 10.1093/brain/awn078. [DOI] [PubMed] [Google Scholar]
- 27.Moore C.J., Goh H.T., Hewitt J.E. Genes required for functional glycosylation of dystroglycan are conserved in zebrafish. Genomics. 2008;92:159–167. doi: 10.1016/j.ygeno.2008.05.008. [DOI] [PubMed] [Google Scholar]
- 28.Avsar-Ban E., Ishikawa H., Manya H., Watanabe M., Akiyama S., Miyake H., Endo T., Tamaru Y. Protein O-mannosylation is necessary for normal embryonic development in zebrafish. Glycobiology. 2010;20:1089–1102. doi: 10.1093/glycob/cwq069. [DOI] [PubMed] [Google Scholar]
- 29.Detrich H.W. III, Westerfield M., Zon L.I., editors. Essential Zebrafish Methods: Cell and Developmental Biology. Elsevier; Oxford: 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.