

Abstract
The “vanishing bone” syndromes represent a group of rare skeletal disorders characterized by osteolysis and joint destruction, which can mimic severe rheumatoid arthritis. Winchester syndrome was one of the first recognized autosomal-recessive, multicentric forms of the disorder. It was originally described nearly 50 years ago in two sisters with a severe crippling osteolysis. Using cultured fibroblasts from the proband, we have now identified homozygous mutations in membrane type-1 metalloproteinase (MT1-MMP or MMP14). We demonstrate that the resulting hydrophobic-region signal-peptide substitution (p.Thr17Arg) decreases MT1-MMP membrane localization with consequent impairment of pro-MMP2 activation, and we propose a structure-based mechanism for this effect.
Main Text
The inherited osteolyses, or “vanishing bone” syndromes, represent a group of genetically heterogeneous syndromes that primarily affect the bones and joints and that are clinically distinguished on the basis of anatomical distribution, severity, associated syndromic features, and inheritance patterns. Given their rarity and clinical overlap, exact diagnosis can be difficult. Identifying the genes and pathways that underlie these inherited syndromes has the potential to provide not only a defining molecular diagnostic for each but also insight into the pathogenic mechanisms that result in these disorders and the more common, sporadic forms of osteoporosis, osteolysis, and arthritis.
The first of these Mendelian disorders to be genetically defined was multicentric osteolysis, nodulosis, and arthropathy (MONA [MIM 259600]), which results from protein-inactivating mutations in MMP2.1 The then “counterintuitive” finding that loss of a gelatinase and/or collagenase would result in bone loss and joint destruction provided the first genetic evidence linking extracellular-matrix proteolysis with human growth and skeletal development.2 These findings also highlighted what appeared to be species differences between loss of MMP function in humans and mice. Specifically, Mmp2-null mice had been reported as being overtly phenotypically normal,3 whereas knockout of the metalloproteinase MT1-MMP, which functions in part as a pericellular activator of MMP-2, resulted in mice that had generalized skeletal defects more severe than those associated with MONA and that suffered from wasting and decreased survival.4 Since then, a MONA-like progressive osteolysis and arthritis phenotype has been demonstrated in Mmp2-knockout mice.5 On the other hand, no human disease has been linked to the loss of MT1-MMP function.
Winchester syndrome is a presumed autosomal-recessive disorder first described in two sisters who had severe skeletal and joint deformities and whose unaffected, healthy parents were first cousins.6 The children suffered from progressive bilateral and symmetric osteolysis of the carpals and tarsals, interphalangeal joint erosions mimicking rheumatoid arthritis, generalized osteoporosis, and eventual loss of function of the larger joints, including the shoulder, elbow, hip, and knee joints (Figure 1A). It was also noted that the siblings had gum hypertrophy, corneal opacities, and electrocardiographical findings suggestive of myocardial damage.6 MONA and Winchester syndromes have been grouped together as allelic disorders on the basis of their phenotypic overlap and the discovery of MMP2 mutations in two recently diagnosed Winchester families (reviewed in Mosig et al.8). Despite this presumed shared genetic etiology, the primary and associated features of the two syndromes nonetheless seemed distinct enough to us (on the basis of their original descriptions) to warrant a genetic analysis of the Winchester syndrome proband first described in 1969.
Figure 1.
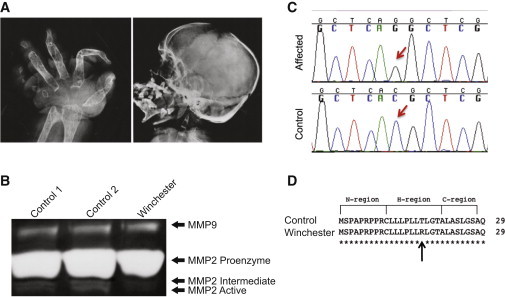
Identification of an MT1-MMP Mutation in Winchester Syndrome
(A) Radiographs of the hand and skull of the affected proband, aged 18 years. The hand radiograph demonstrates complete loss of all carpal bones, destruction of metacarpals and phalanges, and severe ankylosis of the remaining bones. The skull radiograph demonstrates the generalized nature of the osteolytic loss of bone and that the anterior fontanelle remains patent.
(B) Gelatin zymogram of conditioned medium from primary fibroblasts. Lane 3 shows less MMP2 activation in conditioned medium from Winchester cells than do lanes 1 and 2, which are medium from control cells (AG060062 and GM17071A, respectively).
(C) Chromatograms showing C50G transversion in Winchester DNA, but not in control DNA.
(D) Missense mutation results in a T>R replacement in the predicted hydrophobic region of the signal peptide.7
We therefore obtained primary cultured fibroblasts that had been established from one of the two Winchester probands (GM02295, Coriell Institute). We began our molecular characterization by analyzing first the biochemical activity and then the gene sequence of MMP2. Conditioned media from these fibroblasts had abundant levels of MMP-2 proenzyme but reduced MMP-2 activity (Figure 1B). This is in marked contrast to MONA-derived cell lines, which do not show zymographic activity of either the proenzyme or active forms of MMP-2.1 Furthermore, when we isolated genomic DNA from this patient-derived cell line, we were unable to identify mutations in either the exons or intron-exon boundaries of MMP2.
Although we could not rule out the possibility of either undetected promoter or intronic mutations, we hypothesized that loss of an upstream MMP-2 protein activator, such as MT1-MMP, could also result in decreased MMP-2 activity without affecting MMP2. Our interest in this enzyme as a potential disease candidate was based not only on an appreciation of the skeletal phenotype of the Mt1-mmp-null mouse4 but also on the fact that tissues derived from the Mt1-mmp-null mouse have an impaired ability to activate MMP-2.7 However, it should be noted that MT1-MMP is not the sole membrane-type MMP responsible for MMP-2 activation, and therefore, some level of MMP-2 activation might exist even in the absence of MT1-MMP activity. We therefore sequenced MT1-MMP (RefSeq accession number NM_004995.2) in these fibroblasts. Sequence analysis revealed a homozygous missense mutation, a C>G transversion (n.284C>G) in codon 17 of exon 1, that predicted replacement of a threonine by an arginine (p.Thr17Arg) (Figure 1C). The mutation was not present in the db132 or 1000 Genomes databases or in an additional 100 Puerto Rican control chromosomes that we screened (data not shown).
Intriguingly, the mutation occurs in the hydrophobic domain of MT1-MMP’s signal peptide (Figure 1D). As a first step in exploring the possible pathogenicity of this mutation, we used the program SignalP 4.09 to compare the wild-type (WT) and altered signal-peptide sequences. The in silico analysis suggested that the mutation might have a functional consequence because the cleavage-site score of the altered protein was lower (8.0) than that of the WT sequence (9.2).
Wishing to explore this further, we then analyzed the altered MT1-MMP in the patient-derived fibroblasts. Immunoblot of whole-cell lysates revealed that Winchester fibroblasts had dramatically lower levels of proenzyme and active forms of MT1-MMP than did control primary fibroblasts (Figure 2A). There were no concomitant decreases in MT1-MMP mRNA expression levels in Winchester cells compared to controls 1 and 2 (relative values of 1.00, 0.45, and 0.79, respectively) (Figure S1, available online). Because MT1-MMP functionality requires membrane localization and given that the signal peptide sequence is necessary for defining a protein’s correct subcellular localization and function, we next examined altered protein levels at the membrane surface. We used two complementary techniques: membrane fractionation and surface biotinylation. Unlike control cells and despite the protein’s presence in the cytoplasmic fraction, Winchester fibroblasts had almost no detectable levels of active MT1-MMP either in the membrane fraction (Figure 2B) or on their surface (Figure 2C).
Figure 2.
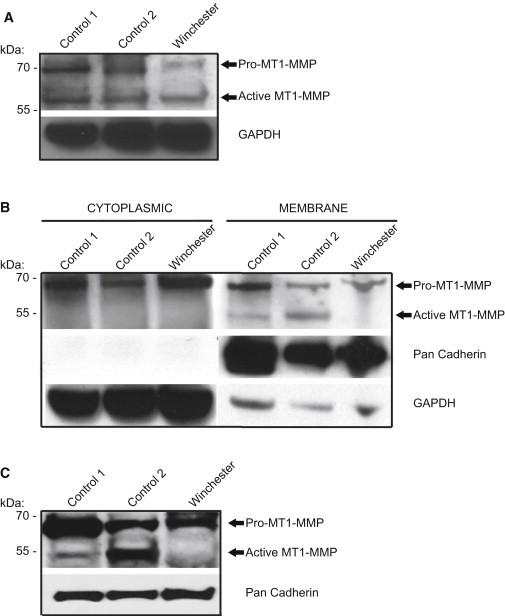
Winchester Mutation Decreases Active Form of MT1-MMP
(A) Immunoblot analysis of whole-cell lysates (30 μg) showed a ∼78% reduction in proenzyme and a ∼42% reduction in active enzyme in Winchester-patient-derived primary fibroblasts (relative proenzyme value = 1; relative active value = 1) compared to control primary fibroblasts AG060062 (proenzyme value = 5.3; active value = 2) and GM17071A (proenzyme value = 3.8; active value = 1.4).
(B) Immunoblot of cellular fractionation of primary cells (30 μg) shows no detectable active form of MT1-MMP in the membrane fraction of Winchester cells. Densitometric analysis of proenzyme between Winchester and control cells (AG060062 and GM17071A) shows comparable levels in the cytoplasmic fraction (the ratio of Winchester cells to AG060062 is 1.0 to 1.1, and the ratio of Winchester cells to GM17071A is 1.0 to 0.89). However, there is an increase in MT1-MMP proenzyme in the membrane fraction (the ratio of Winchester cells to AG060062 is 1.0 to 2.0, and the ratio of Winchester cells to GM17071A is 1.0 to 1.1.
(C) Immunblot of cell-surface-isolated MT1-MMP. Biotin labeling and isolation of cell-surface proteins show that Winchester cells lack the active form of MT1-MMP and have lower levels of the proenzyme than do control cells (the ratio of Winchester cells to AG060062 is 1.0 to 2.3, and the ratio of Winchester cells to GM17071A is 1.0 to 1.4).
Because improperly transported membrane or secreted proteins would be expected to be targeted for rapid degradation, we then investigated the stability of altered MT1-MMP. Transient transfections with expression constructs containing either WT or mutant MT1-MMP in COS-7 cells, which do not express endogenous MT1-MMP, resulted in a >100-fold increase in MT1-MMP mRNA expression levels (Figure S2A). As noted with the endogenous altered MT1-MMP in patient-derived fibroblasts (Figure 2A), the proenzyme and active forms of altered MT1-MMP were both considerably lower than WT levels in transfected cells (Figure S2B). In addition, cells expressing altered MT1-MMP had noticeably lower amounts of active MMP-2 in their conditioned media (Figure S3A). We then treated the transfected cells with the protein-synthesis inhibitor cycloheximide. Compared with the WT protein, the altered protein was quickly degraded (Figure 3A). Blocking proteasomal degradation with MG-132 increased the amount of altered proenzyme, but the active form of altered MT1-MMP remained undetectable (Figure 3B). MG-132 had no effect on WT or mutant MT1-MMP mRNA expression levels (Figure S3B). All together, the experiments on the altered protein demonstrated the deleterious effect of the mutation on subcellular localization, protein stability, and MMP-2 activation.
Figure 3.
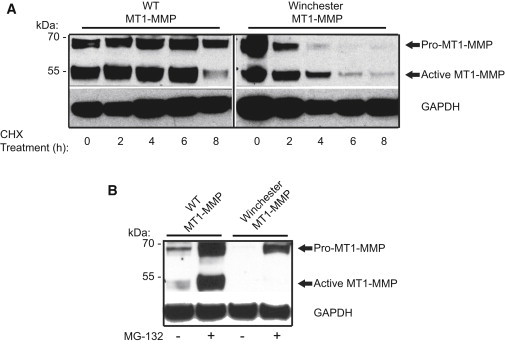
Winchester Mutation Reduces Protein Stability
Transient transfections in COS-7 cells were performed with expression constructs for WT MT1-MMP and Winchester altered MT1-MMP.
(A) Increased protein turnover in Winchester MT1-MMP-transfected cells (40 μg) incubated with 50 μg/ml cycloheximide (CHX).
(B) MG-132 treatment increases levels of the proenzyme form of MT1-MMP, but not of the active form in Winchester MT1-MMP-transfected cells (30 μg).
Mutations in the signal-peptide region have been previously associated with a number of human diseases (reviewed in Jarjanazi et al.10). One difficulty in assessing pathogenicity on the sole basis of sequence analysis is that no consensus sequence has been defined for the signal peptide. They are highly variable in both their length and amino acid sequence composition. Nonetheless, all sequences contain a tripartite domain structure that has a conservative motif ensemble: a positively charged amino N terminus, a middle hydrophobic H region containing 8–20 hydrophobic residues, and a polar carboxyl C terminus. The signal recognition particle (SRP) recognizes this nascent sequence as translated proteins emerge from the ribosome and shuttles these proteins to the endoplasmic reticulum. We hypothesized that the Winchester mutation could affect recognition and binding of the MT1-MMP signal peptide to the SRP complex. Recently, a high-resolution crystallographic structure of the SRP54 protein component of the SRP interacting with a signal peptide (Protein Data Bank ID 3KL4, Research Collaboratory in Structural Bioinformatics) was solved, allowing for the high-resolution visualization of the molecular recognition sites between the protein and peptide.11 We thereby sought to ascribe a structural correlate to the aberrant function caused by the Winchester mutation. Residues in the H region of the signal peptide—the same region in which we identified the Winchester mutation—reside in the hydrophobic groove of the SRP.11 We aligned the motifs first and then the sequence. In the crystal structure near the interface beyond the hydrophobic region, there is a threonine residue (position 459) that we mutated to arginine in silico with energy minimization and subsequently modeled with the calculated rotamer. The crystal structure of the bound peptide demonstrates a “ridges and grooves” packing between the helices (Figure 4). Replacing the polar, uncharged threonine with the large, charged arginine residue at this position appears to disrupt packing of the H domain of signal sequence to the M alpha helical domain of the protein. Therefore, we hypothesize that the ability of mutated MT1-MMP signal peptide to interact with the SRP is destabilized and therefore results in inadequate transport and, ultimately, loss of MT1-MMP’s necessary targeting to the plasma membrane.
Figure 4.
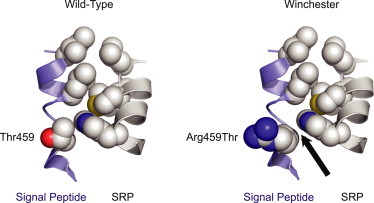
Winchester Mutation Interferes with MT1-MMP Interaction with the SRP
Molecular representation of the altered p.Thr17Arg Winchester superimposed on the Thr479 residue of the signal peptide on the basis of the X-ray crystallographic structure (Protein Data Bank ID 3KL4; Janda et al.11) for a signal peptide (blue ribbon backbone with space-filling residues) in a “ridge and groove” packing between helices with the SRP (gray ribbon backbone). On the left is the closely packed Thr459 wild-type. On the right is the p.Arg459 alteration disrupting the packing (arrow) of the signal sequence to the α-helical M4 domain protein. The figure was rendered in PyMol (v.1.5, Schrodinger, NY, NY).
We now report that an inactivating homoallelic mutation of MT1-MMP in humans results in Winchester syndrome, a rare osteolysis and arthritis disorder whose skeletal phenotype, although not as severe, parallels those previously described for the Mt1-mmp-null mouse.4 The low level of active MT1-MMP present at the surface of patient-derived fibroblasts provides evidence of at least partial activity of the enzyme and might explain the differences in skeletal findings and survival between affected humans and engineered Mt1-mmp-null mice. Extrapolating from the mouse model, we would therefore suggest that the diagnosis of Winchester syndrome be considered part of the differential diagnosis for children with osteolysis and arthritis and early lethality. Similarly, and on the basis of the original shared finding in the Winchester probands6 and its clinical relevance, all individuals suspected to be affected by Winchester syndrome and/or MONA should undergo a complete cardiac evaluation.
Previously, and on the basis of an incorrect clinical diagnosis of Winchester syndrome, Winchester syndrome and MONA were presumed to be allelic disorders arising from mutations in the same gene (reviewed in Mosig et al.8). Our studies now demonstrate that mutations in MMP2 and MT1-MMP result in related but distinct “vanishing bone” syndromes. The molecular mechanisms underlying these phenotypes and associations (if any) between substrates or pathways will need to be established. For example, although it is known that mouse fibroblasts lacking MT1-MMP are defective in their ability to degrade type I collagen,4 this will need to be experimentally established for human cells. All together, these studies highlight the difficulty in clinically distinguishing between two genetically independent MMP-based disorders. Ultimately, these findings extend the range of known MMPs involved in osteolytic and arthritic disease and should provide the basis for an increased understanding of the in vivo function of MT1-MMP in both health and disease.
Acknowledgments
The authors thank Patricia Winchester for her thoughtful insights and the radiographs used in this manuscript.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ImageJ quantification software, http://rsbweb.nih.gov/ij/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Protein Data Bank, http://www.rcsb.org/pdb/home/home.do
SignalP 4.0, http://www.cbs.dtu.dk/services/SignalP/
References
- 1.Martignetti J.A., Aqeel A.A., Sewairi W.A., Boumah C.E., Kambouris M., Mayouf S.A., Sheth K.V., Eid W.A., Dowling O., Harris J. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat. Genet. 2001;28:261–265. doi: 10.1038/90100. [DOI] [PubMed] [Google Scholar]
- 2.Vu T.H. Don’t mess with the matrix. Nat. Genet. 2001;28:202–203. doi: 10.1038/90023. [DOI] [PubMed] [Google Scholar]
- 3.Itoh T., Ikeda T., Gomi H., Nakao S., Suzuki T., Itohara S. Unaltered secretion of beta-amyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. J. Biol. Chem. 1997;272:22389–22392. doi: 10.1074/jbc.272.36.22389. [DOI] [PubMed] [Google Scholar]
- 4.Holmbeck K., Bianco P., Caterina J., Yamada S., Kromer M., Kuznetsov S.A., Mankani M., Robey P.G., Poole A.R., Pidoux I. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99:81–92. doi: 10.1016/s0092-8674(00)80064-1. [DOI] [PubMed] [Google Scholar]
- 5.Mosig R.A., Dowling O., DiFeo A., Ramirez M.C., Parker I.C., Abe E., Diouri J., Aqeel A.A., Wylie J.D., Oblander S.A. Loss of MMP-2 disrupts skeletal and craniofacial development and results in decreased bone mineralization, joint erosion and defects in osteoblast and osteoclast growth. Hum. Mol. Genet. 2007;16:1113–1123. doi: 10.1093/hmg/ddm060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winchester P., Grossman H., Lim W.N., Danes B.S. A new acid mucopolysaccharidosis with skeletal deformities simulating rheumatoid arthritis. Am. J. Roentgenol. Radium Ther. Nucl. Med. 1969;106:121–128. doi: 10.2214/ajr.106.1.121. [DOI] [PubMed] [Google Scholar]
- 7.Zhou Z., Apte S.S., Soininen R., Cao R., Baaklini G.Y., Rauser R.W., Wang J., Cao Y., Tryggvason K. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc. Natl. Acad. Sci. USA. 2000;97:4052–4057. doi: 10.1073/pnas.060037197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mosig R.A., Dowling O., Martignetti J.A. Human MMP-2 Deficiency: The Multicentric Osteolysis with Nodulosis and Arthritis (MONA) and Winchester Syndromes. In: Epstein C.J., Erickson R.P., Wynshaw-Boris A., editors. Inborn Errors of Development: The Molecular Basis of Clinical Disorders of Morphogenesis. 2nd edition. Oxford University Press; New York: 2008. pp. 1453–1461. [Google Scholar]
- 9.Petersen T.N., Brunak S., von Heijne G., Nielsen H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 10.Jarjanazi H., Savas S., Pabalan N., Dennis J.W., Ozcelik H. Biological implications of SNPs in signal peptide domains of human proteins. Proteins. 2008;70:394–403. doi: 10.1002/prot.21548. [DOI] [PubMed] [Google Scholar]
- 11.Janda C.Y., Li J., Oubridge C., Hernández H., Robinson C.V., Nagai K. Recognition of a signal peptide by the signal recognition particle. Nature. 2010;465:507–510. doi: 10.1038/nature08870. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.