

Abstract
Mice with somatotrope-specific deletion of the Janus kinase binding site in leptin receptors are GH deficient as young adults and become obese by 6 months of age. This study focused on the metabolic status of young (3–4.5 month old) preobese mutant mice. These mutants had normal body weights, lean body mass, serum leptin, glucose, and triglycerides. Mutant males and females showed significantly higher respiratory quotients (RQ) and lower energy output, resulting from a higher volume of CO2 output and lower volume of O2 consumption. Deletion mutant females were significantly less active than controls; they had higher levels of total serum ghrelin and ate more food. Mutant females also had lower serum insulin and higher glucagon. In contrast, deletion mutant males were not hyperphagic, but they were more active and spent less time sleeping. Adiponectin and resistin, both products of adipocytes, were increased in male and female mutant mice. In addition, mutant males showed an increase in circulating levels of the potent lipogenic hormone, glucose-dependent insulinotropic peptide. Taken together, these results indicate that mutant mice may become obese due to a reduction in lipid oxidation and energy expenditure. This may stem from GH deficiency. Reduced fat oxidation and enhanced insulin sensitivity (in females) are directly related to GH deficiency in mutant mice because GH has been shown by others to increase insulin sensitivity and fat oxidation and reduce carbohydrate oxidation. Gender-dependent alterations in metabolic signals may further exacerbate the future obese phenotype and affect the timing of its onset. Females show a delay in onset of obesity, perhaps because of their low serum insulin, which is lipogenic, whereas young males already have higher levels of the lipogenic hormone, glucose-dependent insulinotropic peptide. These findings signify that leptin signals to somatotropes are vital for the normal metabolic activity needed to optimize body composition.
Leptin, a product of white fat cells (1), is an important cytokine that is best known for its regulation of appetite and food intake by way of specific receptors (LEPR) in the hypothalamic neurons. Evidence, based on both in vivo and in vitro studies, has shown that leptin also regulates other cell types, including pituitary somatotropes. Animals, which cannot make leptin (2, 3), show reduced numbers of somatotropes (2–5). Humans with impaired leptin signaling, due to a mutation in LEPRb (exon 16), have impaired growth (6).
Leptin receptors are expressed in the majority of somatotropes in rats (7, 8) and sheep (9) or pigs (10), and Cai and Hyde (11) reported increased expression of LEPRb in pituitaries of mice with somatotrope hyperplasia [human GHRH transgenic mice (12)]. Our recent studies of both rats and mice agree that most somatotropes bear leptin receptors (13–15).
However, the importance of leptin to somatotrope function is still not well understood. The evidence showing that leptin deficiency reduced the numbers of somatotropes (16, 17) was questioned because the reduction could have been caused by the metabolic disease itself or the hypogonadal condition (18–20). Luque et al. (21) reported that leptin restored GH secretion and increased pituitary GHRH receptors in ob/ob mice that were infused with exogenous leptin for 7 d, which could not be attributed to changes in hypothalamic GHRH expression. Our recent in vitro studies reported that exposure to 10–100 pg/ml leptin increased the percentages of somatotropes reduced by 24 h of fasting, and 1–10 pg/ml of leptin stimulated an increase in GH mRNA in normal pituitary somatotropes (14). Collectively these observations pointed to the importance of direct interactions of leptin on somatotropes.
To better understand the impact of leptin signaling on somatotropes, we recently used a selective knockout strategy to ablate the signaling portion of leptin receptors in pituitary somatotropes (22). Mice bearing Cre-recombinase driven by the rat growth hormone promoter (rGHp-Cre) (23) were crossed with mice bearing leptin receptor gene alleles in which exon 17 was flanked by loxP (Lepr exon 17 flox/flox) (24, 25). Somatotrope-specific loss in leptin signaling reduced the population of immunolabeled somatotropes and serum GH by 60%. The GH deficiency (GHD) was detected as early as 3 months of age, but body weights did not significantly differ between mutant and control mice until 5 months in males and 6.5 months in females. dual-energy x-ray absorptiometry analysis of the older animals showed a clear increase in fat mass, with no change in lean mass. Serum leptin levels were normal, although they increased appropriately with increasing fat mass.
The delay in fat mass accumulation in the mutant mice, presents the opportunity to identify behaviors and factors that might eventually drive the changes in body composition, observed in later life. To this end, a comprehensive laboratory animal monitoring system (CLAMS) was used to assess activity level, sleep episodes, and food/water intake as well as O2 consumption and CO2 output by indirect calorimetry. These data were correlated with changes in circulating adipokines and proinflammatory cytokines known to be associated with obesity.
Materials and Methods
Production of deletion mutant mice lacking LEPR exon 17 in somatotropes
Breeding cages were set up to produce N6 and N7 generation deletion mutants and littermate controls (FVB/NJ) as previously described (22). Mice were housed five per cage and maintained at 10-h light,14-h dark cycle until they were old enough for testing (3–4.5 months of age). The animals were fed Teklad 8640 or Teklad 22/5 rodent diet (Tekland, Madison, WI), which is 22% protein, 5% fat, and 4.5% fiber. While breeding, the mice were fed Teklad Global 19% protein diet 2019, which is 19% protein, 9% fat, and 5% fiber. The Animal Care and Use Committee approved all protocols, annually (protocol 3014).
Comprehensive laboratory animal monitoring system
The indirect calorimetry, activity levels, and food intake were determined using the Oxymax CLAMS (Columbus Instruments, Columbus, OH) over a period of 72 h. This unit is in the same room that houses the breeding colonies, which facilitates acclimation. Animals were acclimated in the CLAMS unit for 48 h before the 72-h test period and received water and food (powdered Teklad 8604 diet), ad libitum. A total of four different groups (four animals per group) of male or female mutant and littermate control mice were evaluated. Within and between groups, they were age and weight matched. For example, in a given experiment, if we had a 3-month-old and a 4-month-old deletion mutant, we matched this with a 3-month-old and a 4-month-old control. For each animal, we calculated lean body mass, which was body weight (M) to the ¾ power, according to Kleiber's law (26, 27). This formula is a well-established surrogate for metabolic mass. Their weight was loaded into the CLAMS program and the conversion to lean body mass was built into the software used by the CLAMS, which normalized the volume of oxygen, carbon dioxide, and the resulting respiratory quotient (RQ) to lean body mass.
The activity monitors in CLAMS also are an indirect means of detecting sleep. A sleep epoch was defined as a 60-sec bout of no activity. The computer-generated sleep analysis recorded the number of consecutive sleep epochs and the total percent of time sleeping for each mouse. This system has been shown to give excellent agreement with other sleep detection approaches, including electromyographic/electroencephalographic (EEG) and video monitoring (28).
The output from the software analysis program (CLAX, Columbus Instruments) was analyzed using InStat3 or PRISM 5.0 software (both from GraphPad Inc., San Diego, CA). One-way ANOVA was run to detect significant differences between mutant and control mice within gender. Bonferroni and Neuman Keuls multiple comparisons tests were used as post hoc tests. The percent relative cumulative frequency approach (29) was used to determine the frequency of RQ values. The differences between mutant and control values were assessed by nonlinear regression curve fitting and Mann Whitney and D'Agostino and Pearson omnibus normality tests.
Multiplex enzyme immunoassays for cytokines and adipokines
Trunk blood samples were collected from the mice at the time the animals were killed between 0900 and 1100 h to avoid diurnal variations. The animals were 3.5–4 months of age. In addition, serum from 20–30 additional age and weight-matched mice were taken at the same time of day. These mice were littermates or from litters born during the same time period. This group provided serum from animals that had not been run in the CLAMS to determine differences that might have come from having been in the CLAMS environment. No differences were detected in their weights or in the values themselves. Therefore, the serum values are averaged with those from mice exposed to the CLAMS. All serum samples were assayed in the Luminex LX200 xPONENT 3.1 with Millipore Multiplex adiopcyte, adipokine, or metabolic disease panels designed for mouse analytes (Millipore, Bedford, MA). The analytes included leptin, ghrelin, adiponectin, resistin, glucose insulinotropic peptide (GIP), IL-6, insulin, glucagon, plasminogen activator inhibitor-1 (PAI-1), monocyte chemotactic protein-1 (MCP-1), and TNF-α. Sera were also tested for glucose with a glucometer and triglycerides with the Cayman Chemical colorimetric assay kit (Ann Arbor, MI).
Values for male or female mice were averaged and subjected to one-way ANOVA, followed by Tukey's, Neuman-Keul's, and Bonferroni's multiple comparison post hoc tests. In some cases, a Student's t test with Welch's correction was used to compare values from control and deletion mutant animals (P < 0.05 was considered significant).
Results
Young deletion mutant mice are not obese and have normal leptin levels
As shown in Fig. 1, total mean body weights, lean body mass (Fig. 1A), and circulating leptin (Fig. 1B) did not differ between 3- and 4.5-month-old mutant mice within a given sex. The average leptin levels in females were significantly lower than that in males. With respect to food intake, the system reported the cumulative amount of food eaten over the 72-h period, with the option of reporting cumulative food eaten in only the two to three light or dark periods during that time. The system also reported the amount of food eaten at each feeding bout.
Fig. 1.

A, No differences are seen between 3- and 4.5-month-old control and mutant animals in total mean body weights and average lean body mass. Lean body mass (M3/4) was calculated according to Kleiber's law (26, 27). Statistical analysis was done in Prism (GraphPad) with one-way ANOVA followed by Newman-Keuls and Bonferroni multiple comparisons tests. B, There are no changes in serum leptin in deletion mutant males or females, although leptin levels are lower in the control and deletion mutant females than they are in the males (P < 0.0001, Bonferroni). The levels for both males (n = 42 controls, n = 38 mutants) and females (n = 27 controls, n = 45 mutants) are within the normal range reported for the mouse, however. C, There was a higher food intake during all dark phases of the 72-h test period in males but no differences between the two groups (control vs. deletion mutant). D, Deletion mutant females ate significantly more food during all light and dark phases of the 72-h test period (P < 0.0001, Bonferroni). E, During the dark phases of the test period, mutant females ate more food at each feeding bout (P = 0.0023, Bonferroni). F, A significant increase in ghrelin in females is shown, which correlates with their increased food intake (P = 0.0087 Bonferroni n = 6) but no changes in serum ghrelin in deletion mutant males (n = 9, Bonferroni). *, Significantly greater than control values; °, significantly different from values in light phase or significantly different from males
Consistent with previous reports, male and female mice consumed more food during the dark phase, independent of genotype (Fig. 1, C and D). In addition, deletion mutant males showed no differences in the cumulated grams of food eaten during dark or light phases (Fig. 1F). In contrast, the cumulated amount of food eaten by the deletion mutant females was higher than that eaten by the controls during both the dark and light phases (Fig. 1D), and mutant females ate more food/feeding bout during the dark phase (Fig. 1E). Half of the females were 3–3.5 months old and the others were 4.5 months old. When the data were separated by age, the higher food consumption was significantly different in the population of older females (4.5 months, data not shown) but not in the 3-month-old group. Mutant females also showed a significant increase in circulating total ghrelin levels, which correlated with their increased food consumption (Fig. 1F).
Preobese deletion mutants have a higher respiratory quotient and lower energy expenditure
Indirect calorimetry for the four groups of males and females demonstrated that deletion mutant females (Fig. 2, A, C, and D) and males (Fig. 2, B, E, and F) had higher RQ than littermate controls. The increase in RQ in both sexes was associated with an increase in CO2 production (VCO2) (Fig. 2, G and I) and a decrease in O2 consumption (VO2) (Fig. 2, H and J) and energy expenditure (Fig. 2, and L), in both the light and dark phases. Overall, female mutant mice showed a larger shift in RQ values (Fig. 2C, RQ EC50 controls: 0.886 vs. mutants: 0.93; D'Agostino and Pearson omnibus normality test P < 0.0001; Mann Whitney test, P < 0.02), compared with male mutants (Fig. 2E, RQ EC50, controls 0.946 vs. mutants 0.967;D'Agostino and Pearson omnibus normality test P < 0.0001; Mann-Whitney test P < 0.01). Of note, the more profound differences in RQ in females occurred during the dark phase, when mice are consuming more food.
Fig. 2.

Indirect calorimetry in four sets of males and females (16 animals/set) showing changes in RQ (RQ = VCO2/VO2), VCO2, VO2, and energy expenditure. A–F, Deletion mutants have higher RQ, especially during the nighttime feeding cycle with females. C and E, Higher EC50 of individual RQ values is shown on the percentile curve after the use of the percent relative cumulative frequency approach (D'Agostino and Pearson omnibus normality test P < 0.0001; Mann-Whitney t test, P = 0.01, males, and P = 0.024, females). Statistics in the remaining figures were done with the t test with Welch's correction and the InSTAT3 software (GraphPad). D and F, There is higher RQ in both night and day cycles (P < 0.0001). G and I, The higher RQ is due to higher VCO2/lean body mass, averaged for light and dark periods (P < 0.0001). Figures 2 h and 2jH and J, The higher RQ is also associated with a lower VO2 averaged for light and dark periods (P < 0.0001). K and L, Illustration of the lower energy output averaged during light and dark phases (P < 0.0001). Closed star, Significantly greater than control values; open star, significantly different from values in light phase.
Preobese mutant females show reduced activity and males have reduced sleep
When activity was measured by infrared beam breaks, both control and mutant female mice showed enhanced activity during the dark phase (Fig 3, A–C). However, deletion mutant females showed reduced total activity, compared with female controls, which included both ambulation and grooming (total activity, Fig. 3A), walking (Fig. 3B), and rearing or jumping (Fig. 3C). In contrast, deletion mutant males showed no significant difference in number of beam counts when the total activity in the x-axis was detected (data not shown). However, there was a significant increase in ambulatory (Fig. 3D) or rearing or jumping activity (Fig. 3E). The sleep analysis, based on absence of activity, showed that deletion mutant males spent less time sleeping than littermate controls (Fig. 3F).
Fig. 3.

Activity analysis in the CLAMS monitored by infrared beam breaks. All data were analyzed with the t test with Welch's correction and the InSTAT3 software (GraphPad). A, Total activity (walking and grooming) in the deletion mutant and control females with a significant decrease in deletion mutants, during both dark and light phases (P < 0.0001). B, Ambulatory activity in the x-axis (walking) is also reduced in mutant females (P < 0.0001). C, There is a reduction in z-axis activity (rearing or jumping) in mutant females (P < 0.0001). D, Slight increases in x-axis activity (ambulation) is shown in mutant males (P < 0.0001). E, Graphs of the increased z-axis activity seen in the mutant males (P < 0.0001). F, Illustration of the sleep analysis indicating that mutant males spend a lower percent of time sleeping than controls (P < 0.001). Closed star, Significantly greater than control values; open star, significantly different from values in light phase or, in the case of panel F, significantly different from males.
Glucose regulation and triglycerides
Glucose-regulatory hormones (insulin and glucagon) and C-peptide, a proinsulin, were assayed in sera from animals collected after the CLAMS experiments as well as that from additional animals collected during this period. All animals had glucose levels that were within the normal ranges for young adult mice (females-controls 137 ± 23 mg/dl; mutants, 153.5 ± 9 mg/dl; males-controls, 165 ± 6 mg/dl; mutants, 173 ± 29 mg/dl). There was no difference between controls and mutants. Similarly, there were no differences in triglyceride levels between controls (males, 249 ± 54 mg/dl; females 296 ± 35 mg/dl) and mutants (males, 257 ± 114 mg/dl, females, 225 ± 25 mg/dl). The triglyceride values for both groups are slightly higher, but within the range reported for FVB strains at the Jackson Laboratory Mouse Phenome Database (http://jaxmice.jax.org/support/phenotyping/FVBNJdata001800.pdf).
Insulin was assayed with adipokine and metabolic disease panels. Figure 4A shows that there was no significant difference in serum insulin levels in the males. In contrast, deletion mutant females had significantly lower insulin levels when compared with the male populations. Furthermore, deletion mutant females showed a significantly lower level of insulin by Student's t test when compared with control females (P < 0.02). Levels of the proinsulin, C-peptide, followed insulin levels. C-peptide levels in females were 53% of those in the control and significantly lower than in all other populations (Fig. 4B). There were also no significant differences in glucagon levels in the two groups of males. In contrast, deletion mutant females showed 1.6-fold higher glucagon levels (Fig. 4C).
Fig. 4.

Serum levels of insulin, C-peptide, and glucagon. The statistical analysis was done by one-way ANOVA followed by Newman-Keuls and Bonferroni's multiple comparison tests. Figure 4a A, Both sets of females had lower insulin than either group of males (34 control and 36 deletion mutants) (P < 0.0001, Bonferroni). In addition, insulin levels in the 36 deletion mutant females were reduced in comparison with the 30 control females (P < 0.0001, Bonferroni). B, Only the deletion mutant females showed a significant reduction in C-peptide (P = 0.0001, Bonferroni). C, Only deletion mutant females expressed higher serum glucagon (P = 0.0028, Bonferroni). *, Significantly different from controls; °, significantly different from male groups.
Deletion mutant mice have higher levels of adipokines, whereas alteration in inflammatory cytokines are sexually dimorphic
Circulating adiponectin and resistin levels were significantly elevated in both male and female mutant mice compared with controls (Fig. 5, A and B). Sexually dimorphic changes were observed for circulating GIP and IL-6 in mutant mice. Specifically, GIP was elevated in males, whereas IL-6 was reduced in females (Fig. 5, C and D). No differences were observed between mutants and controls for circulating PAI-1, TNF-α, and MCP-1 (data not shown).
Fig. 5.
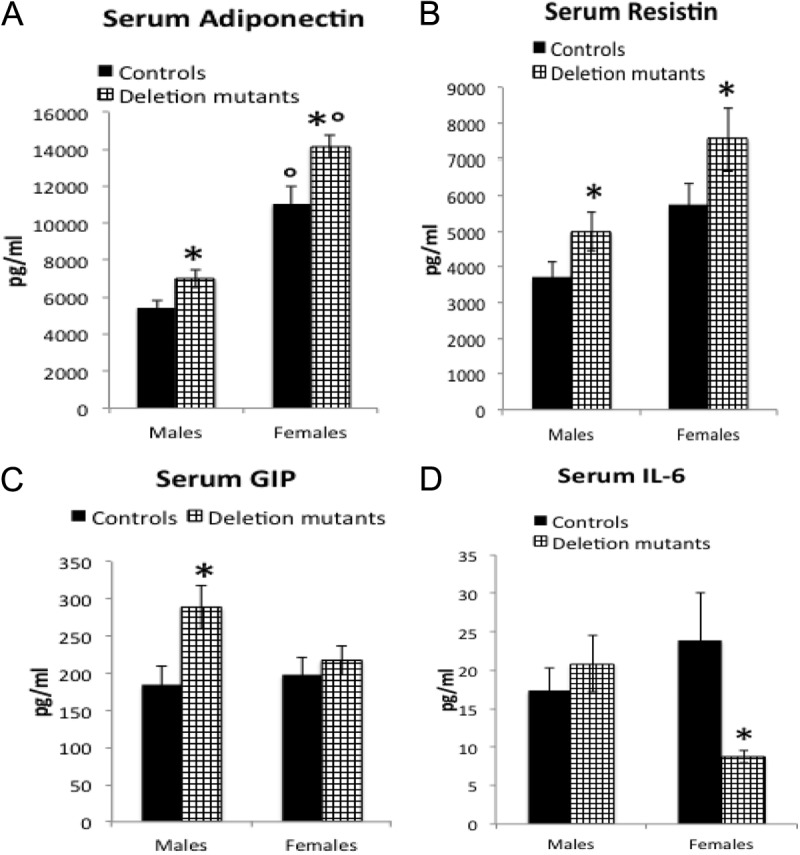
Deletion mutants showed changes in adipokines. The statistical analysis was done by one-way ANOVA followed by Newman-Keuls and Bonferroni's multiple comparison tests. A, The 22 deletion mutant females showed higher levels of adiponectin (P < 0.0001) when compared with controls (n = 18). Furthermore, both groups of females expressed higher adiponectin than males (P < 0.0001). Deletion mutant males (n = 17) showed higher levels of adiponectin than controls (n = 16, unpaired t test with Welch's correction, P = 0.0064). B, Higher levels of serum resistin in both groups of deletion mutants are shown (males, unpaired Student's t test with Welch's correction, P = 0.034, n = 40 controls, 44 mutants; females, ANOVA followed by Newman-Keuls, P = 0.0004, n = 40 controls and 45 mutants). C Serum GIP is higher only in males (Bonferroni's P = 0.0179, n = 26 control and 27 mutant males; 26 control and 32 mutant females). D, The proinflammatory IL-6 is lower in deletion mutant females (Newman-Keuls, P = 0.03) than controls, but values in deletion mutant males and controls are not different (n = 35 control males, 50 deletion mutant males, 24 control females, 34 deletion mutant females). *, Significantly different from controls; °, significantly different from male groups.
Discussion
Our laboratory has previously reported that somatotrope-specific, Cre-recombinase-mediated excision of LEPRb exon 17, which codes for the portion of the molecule in the cytoplasmic tail that contains the Janus kinase binding site (22), blocks phosphorylation of signal transducer and activator of transcription 3 in response to leptin stimulation (22). Lack of leptin-mediated Janus kinase/signal transducer and activator of transcription signaling in somatotropes resulted in mice that grow normally but are GHD as adults.
As in cases of human adult-onset GHD (30–32), the deletion mutant mice became obese later in adult life. In the current study, we examined the metabolic profile of 3- to 4.5-month-old mutant mice before they became obese as indicated by their normal body weights and leptin levels. The objective was to determine whether the young, preobese mutants would exhibit metabolic changes and behaviors that could eventually lead to obesity. These preobese mutants showed no changes in serum glucose or triglycerides. We also observed that male mutant mice were not hyperphagic, which is consistent with their normal leptin levels and our previous report showing that leptin receptors in the hypothalamus remained intact (22). In contrast, mutant female mice ate more than controls. Whereas their leptin levels were also not different from controls, they did have significantly higher ghrelin levels. The following discussion will point to differences in metabolic activity, behaviors, and hormones, which may predispose the onset of obesity in these animals, highlighting sex differences where appropriate.
Mutant males and females burn less fat and more carbohydrates
The strongest predisposition to obesity was seen in the calculation of respiratory quotients (RQ = VCO2/VO2) for these mutants. Indirect calorimetry demonstrated that deletion mutant males and females had higher RQ than the littermate controls. In relation to the amounts of CO2 produced, the oxidation of fat requires more oxygen than the oxidation of carbohydrates; therefore, a higher RQ indicates that animals are oxidizing carbohydrates rather than fat (33) This, in the context of their reduced energy expenditure, would lead to excess fat accumulation. These changes may be directly caused by their GHD (22). In previous reports, male mice with adult-onset isolated GHD had higher RQ values, particularly during the dark cycle (34), and were fatter than littermate controls. In humans, an increased RQ is both predictive of obesity (35–37) and associated with the obesity-inducing effects of ghrelin (38). The association between GH deficiency and fat vs. carbohydrate oxidation was shown in studies of normal and GHD humans. In the first study, GH treatment of normal humans and tissues resulted in a significant reduction in RQ and a 29% increase in fat oxidation by muscle mitochondria coupled with a 69% decrease in carbohydrate oxidation (39). Similarly, GH treatment of GHD subjects resulted in a significant decrease in hepatic carbohydrate oxidation (40). The higher RQ indicating preferential burning of carbohydrates is thus consistent with the GHD of the mutant animals in the present study. Future studies would be needed to evaluate the fat and carbohydrate oxidation in muscle and liver of these animals.
GHD and changes in pancreatic hormones
Whereas obesity promotes insulin resistance and dysregulation of glucose transport, GHD is often associated with increased insulin sensitivity (34). As reported in our previous studies, the preobese male and female deletion mutant mice had normal levels of serum glucose. To determine any changes in pancreatic hormones in these animals, we assayed glucagon, insulin, and the insulin prohormone C-peptide. The preobese deletion mutant males showed no changes in any of these hormones. However, all three hormones were significantly changed in deletion mutant females. Both insulin and C-peptide were significantly reduced, whereas glucagon was significantly increased. These changes may reflect increased insulin sensitivity as a result of the GH deficiency (34). In addition, their lower insulin may explain why the female deletion mutant mice have a delay in the onset of obesity compared with the male deletion mutant mice because insulin is a potent lipogenic hormone (41, 42).
GHD and reduced activity or sleep as risk factors for obesity
GHD in humans is also associated with loss of lean body mass and muscle strength, which can be restored by GH therapy (43–45). In the present model, the loss of muscle strength or mass may be the basis behind the reduced activity seen in the deletion mutant females. The reduced activity levels also correlate with the higher ghrelin seen in these females (Fig. 1C) because recent studies have shown that ghrelin is involved in reducing spontaneous physical activity (46). Another contributing factor in females may be the reduction in IL-6, which is secreted by adipose tissue, immune cells, fibroblasts, and endothelial cells (47). Recent studies by Faldt et al. (35) have reported that IL-6 deficiency was associated with a higher RQ and a reduced endurance in young, preobese animals. The investigators suggested that endogenous IL-6 is important for high oxygen consumption that allows the animal to maintain skeletal muscle work during prolonged exercise. The lower IL-6 in the deletion mutant females thus also correlates with their reduced activity and high RQ.
In contrast, preobese male deletion mutants remain active and in fact exhibit a slight increase in activity levels over control males. This is correlated with a significant reduction in the percent of time sleeping, which may be another response to GHD. Sleep and GH secretion are related as a burst of GH secretion is seen during deep non-rapid eye movement (REM) sleep in humans and other animals (48–51). However, GH itself may be more directly involved in REM sleep. Obal et al. (52–54) reported sleep alterations in the dwarf rat (dw/dw) or mouse (lit/lit), which has defective GHRH signaling. They were able to separate regulatory pathways and reported that non-REM sleep was mediated centrally via the GHRH receptor, and REM sleep was regulated via GHRH-GH pathways. There is also significant association between sleep loss and onset of obesity and type 2 diabetes (55–58). Thus, reduced sleep could be an important factor that leads to the obesity seen in the older (5–6 months old) males (22), although it is clear that the quality of sleep may need to be monitored by EEG recordings to fully evaluate its impact. It is worthwhile to note that Pack et al. (28) reported an 88–94% agreement between EEG recordings, videography, and the activity readings in the Oxymax CLAMS unit in mice. Their study showed that the mean difference per 2-h interval between inactivity-defined sleep (used in our study) and EEG/electromyogram defined sleep was only 1 min.
Adipokine secretion as risk factors in preobese mutants
Adiponectin is secreted by adipocytes and increases with adipogenesis (59). However, it is also secreted in inverse proportion to the body mass index (60–62). It has antidiabetic, antiatherogenic, and antiinflammatory properties and is thus protective against the damaging inflammatory effects of obesity (63, 64). Both male and female deletion mutant mice had higher adiponectin levels than controls (Fig. 4A). And in both sets of females, adiponectin levels were higher than those in males. This may be associated with adipogenesis, signifying that the animals are beginning to build excess fat stores. In addition, plasma adiponectin is inversely correlated with cytokines IL-6 and TNF-α (47), and deletion mutant females do show reduced IL-6 (Fig. 4D).
Resistin is a cytokine that is also expressed primarily by adipocytes and is positively correlated with fat mass. Its function is to antagonize insulin action by inhibiting glucose uptake in skeletal muscle and impairing insulin actions on hepatic glucose production (65–67). It may be an important risk factor for the development of insulin resistance and, therefore, we reasoned that animals might show higher resistin levels. In the present studies, the preobese deletion mutant males did show higher resistin levels than controls by Student's t test. Mice with developmental GHD or GH resistance as well as adult-onset isolated GHD mice have been shown to be more insulin sensitive relative to controls (34), consistent with the fact that GH, at least at high levels, reduces insulin sensitivity in humans (68–70) and mice (71). Future studies will be needed to correlate their higher resistin with changes in insulin resistance in these animals, especially after they become obese.
GIP, also known as the glucose-dependent insulinotropic peptide, is a 42-amino acid peptide synthesized by K cells in the mucosa of the duodenum and the jejunum (72, 73). GIP is clearly elevated in obese humans and animals (74). There are functional GIP receptors on adipocytes and GIP has been shown to increase fatty acid synthesis (75), stimulate lipoprotein lipase activity (76), and stimulate glucose transport into adipocytes and intestinal epithelial cells (77, 78). All of these actions may predispose an animal to obesity and GIP is thus a target for therapeutic modulation of activity via vaccination or receptor modulation (73, 74). In the present study, GIP levels were significantly higher in deletion mutant males than in controls. This may be seen as a risk factor for males and the fact that it appears elevated before they become obese may explain the earlier onset of obesity in the deletion mutant males.
Inflammatory responses are not evident in preobese mutants
Finally, because obesity is associated with low-grade inflammatory changes (79), we also tested proinflammatory cytokines to determine whether a rise in their levels might predict the eventual onset of increased fat mass. These included PAI-1, a serine protease inhibitor that is involved in angiogenesis and atherogenesis; TNF-α, a proinflammatory cytokine secreted by macrophages (80); and adipocytes (81) and MCP-1, a chemokine that recruits monocytes to sites of inflammation (79). There were no significant increases in serum levels of PAI-1, MCP-1, and TNF-α.
Mechanisms behind the GHD
Our previous study showed that the GHD was associated with a lower number of cells immunolabeled for GH (22), which pointed to the importance of leptin signals needed to maintain an optimal number of somatotropes. However, more recent studies from our laboratory have discovered that the full somatotrope population in these mutants can be detected by their expression of GH mRNA (82), suggesting that the leptin-mediated optimization functions at the level of protein translation or storage. Furthermore, we discovered that the GH stores and secretion in this population can be fully restored if one exposes them, in vitro to ghrelin and GHRH (82). These findings suggest roles for leptin in pathways that optimize GH translation or storage, and future studies will be needed to explore these roles.
Summary and conclusions
We originally hypothesized that, without leptin signals, somatotropes are unable to respond appropriately to increasing adiposity by secreting sufficient GH, which is needed to maintain optimal levels of fat stores (22) by both lipolytic activity and preferential oxidation of fat. Our more recent studies (82) suggested that this may be due to defects in pathways leading to GH protein translation or storage. Whatever the mechanism causing GHD, we postulated that, in these GHD mice, conditions that prevent adiposity are left unchecked. Indirect calorimetry was run to identify metabolic and behavioral factors in preobese animals that might eventually cause fat accumulation. The preobese animals showed increased RQ, which is a significant risk factor for obesity. In addition, there were differences in patterns of behavior and hormone levels when males and females were compared. These differences (high GIP in males and low insulin in females) may actually explain the differences in the timing of onset of obesity in this group of mutants. Future studies are needed to determine the impact of these changes on insulin resistance and glucose tolerance. It is clear from these studies that the normal signaling circuitry involving leptin from adipocytes to somatotropes is vital to help balance the metabolic activity needed to optimize body composition.
Acknowledgments
This work was supported by Grants National Institutes of Health (NIH) R03 HD059066 and NIH 1R01HD059056 (to G.V.C.) and core facilities funded by NIH Grants NCRR P20 RR020146 and NIH P30 NS047546 (at University of Arkansas for Medical Sciences), and NIH Grant P01 DK26687 (to S.C.), Department of Veterans Affairs, Office of Research and Development, Veterans Administration Merit Awards BX001114 and NIH grant RO1 DK088133 (to R.D.K.).
Disclosure Summary: The authors have no conflicts to disclose.
Footnotes
- CLAMS
- Comprehensive laboratory animal monitoring system
- EEG
- electroencephalographic
- GHD
- GH deficiency
- GIP
- glucose insulinotropic peptide
- LEPR
- leptin receptor
- MCP-1
- monocyte chemotactic protein-1
- PAI-1
- plasminogen activator inhibitor-1
- REM
- rapid eye movement
- RQ
- respiratory quotient
- VCO2
- CO2 production
- VO
- O2 consumption.
References
- 1. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. 1994. Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432 [DOI] [PubMed] [Google Scholar]
- 2. Lloyd RV, Jin L, Tsumanuma I, Vidal S, Kovacs K, Horvath E, Scheithauer BW, Couce ME, Burguera B. 2001. Leptin and leptin receptor in anterior pituitary function. Pituitary 4:33–47 [DOI] [PubMed] [Google Scholar]
- 3. Urbanski HF. 2001. Leptin and puberty. Trends Endocrinol Metab 12:428–429 [DOI] [PubMed] [Google Scholar]
- 4. Mann DR, Plant TM. 2002. Leptin and pubertal development. Semin Reprod Med 20:93–102 [DOI] [PubMed] [Google Scholar]
- 5. Yura S, Ogawa Y, Sagawa N, Masuzaki H, Itoh H, Ebihara K, Aizawa-Abe M, Fujii S, Nakao K. 2000. Accelerated puberty and late-onset hypothalamic hypogonadism in female transgenic skinny mice overexpressing leptin. J Clin Invest 105:749–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clément K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM, Basdevant A, Bougnères P, Lebouc Y, Froguel P, Guy-Grand B. 1998. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 392:398–401 [DOI] [PubMed] [Google Scholar]
- 7. Sone M, Nagata H, Takekoshi S, Osamura RY. 2001. Expression and localization of leptin receptor in the normal rat pituitary gland. Cell Tissue Res 305:351–356 [DOI] [PubMed] [Google Scholar]
- 8. Sone M, Osamura RY. 2001. Leptin and the pituitary. Pituitary 4:15–23 [DOI] [PubMed] [Google Scholar]
- 9. Iqbal J, Kurose Y, Canny B, Clarke IJ. 2006. Effects of central infusion of ghrelin on food intake and plasma levels of growth hormone, luteinizing hormone, prolactin, and cortisol secretion in sheep. Endocrinology 147:510–519 [DOI] [PubMed] [Google Scholar]
- 10. Lin J, Barb CR, Matteri RL, Kraeling RR, Chen X, Meinersmann RJ, Rampacek GB. 2000. Long form leptin receptor mRNA expression in the brain, pituitary, and other tissues in the pig. Domest Anim Endocrinol 19:53–61 [DOI] [PubMed] [Google Scholar]
- 11. Cai A, Hyde JF. 1998. Upregulation of leptin receptor gene expression in the anterior pituitary of human growth hormone-releasing hormone transgenic mice. Endocrinology 139:420–423 [DOI] [PubMed] [Google Scholar]
- 12. Cai A, Hyde JF. 1999. The human growth hormone-releasing hormone transgenic mouse as a model of modest obesity: differential changes in leptin receptor (OBR) gene expression in the anterior pituitary and hypothalamus after fasting and OBR localization in somatotrophs. Endocrinology 140:3609–3614 [DOI] [PubMed] [Google Scholar]
- 13. Akhter N, Johnson BW, Crane C, Iruthayanathan M, Zhou YH, Kudo A, Childs GV. 2007. Anterior pituitary leptin expression changes in different reproductive states: stimulation, in vitro, by gonadotropin releasing hormone (GnRH). J Histochem Cytochem 55:151–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crane C, Akhter N, Johnson BW, Iruthayanathan M, Syed F, Kudo A, Zhou YH, Childs GV. 2007. Fasting and glucose effects on pituitary leptin expression: is leptin a local signal for nutrient status? J Histochem Cytochem 55:1059–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McDuffie IA, Akhter N, Childs GV. 2004. Regulation of leptin mRNA and protein expression in pituitary somatotropes. J Histochem Cytochem 52:263–273 [DOI] [PubMed] [Google Scholar]
- 16. Isozaki O, Tsushima T, Miyakawa M, Demura H, Seki H. 1999. Interaction between leptin and growth hormone (GH)/IGF-I axis. Endocr J 46(Suppl):S17–S24 [DOI] [PubMed] [Google Scholar]
- 17. Popovic V, Damjanovic S, Dieguez C, Casanueva FF. 2001. Leptin and the pituitary. Pituitary 4:7–14 [DOI] [PubMed] [Google Scholar]
- 18. Childs GV, Iruthayanathan M, Akhter N, Johnson BW. 2006. Estrogen mediated cross talk between the ovary and pituitary somatotrope. Pre-ovulatory support for reproductive activity. Mol Cell Endocrinol 247:60–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Childs GV, Iruthayanathan M, Akhter N, Unabia G, Whitehead-Johnson B. 2005. Bipotential effects of estrogen on growth hormone synthesis and storage in vitro. Endocrinology 146:1780–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Iruthayanathan M, Zhou YH, Childs GV. 2005. Dehydroepiandrosterone restoration of growth hormone gene expression in aging female rats, in vivo and in vitro: evidence for actions via estrogen receptors. Endocrinology 146:5176–5187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luque RM, Huang ZH, Shah B, Mazzone T, Kineman RD. 2007. Effects of leptin replacement on hypothalamic-pituitary growth hormone axis function and circulating ghrelin levels in ob/ob mice. Am J Physiol Endocrinol Metab 292:E891–E899 [DOI] [PubMed] [Google Scholar]
- 22. Childs GV, Akhter N, Haney A, Syed M, Odle A, Cozart M, Brodrick Z, Gaddy D, Suva LJ, Akel N, Crane C, Benes H, Charlesworth A, Luque R, Chua S, Kineman RD. 2011. The somatotrope as a metabolic sensor: deletion of leptin receptors causes obesity. Endocrinology 152:69–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Luque RM, Amargo G, Ishii S, Lobe C, Franks R, Kiyokawa H, Kineman RD. 2007. Reporter expression, induced by a growth hormone promoter-driven Cre recombinase (rGHp-Cre) transgene, questions the developmental relationship between somatotropes and lactotropes in the adult mouse pituitary gland. Endocrinology 148:1946–1953 [DOI] [PubMed] [Google Scholar]
- 24. Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua SC, Jr, Elmquist JK, Lowell BB. 2004. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron 42:983–991 [DOI] [PubMed] [Google Scholar]
- 25. McMinn JE, Liu SM, Dragatsis I, Dietrich P, Ludwig T, Eiden S, Chua SC., Jr 2004. An allelic series for the leptin receptor gene generated by CRE and FLP recombinase. Mamm Genome 15:677–685 [DOI] [PubMed] [Google Scholar]
- 26. Kleiber M. 1932. Body size and metabolism. Hilgardia 6:315–351 [Google Scholar]
- 27. Kleiber M. 1947. Body size and metabolic rate. Physiol Rev 27:511–541 [DOI] [PubMed] [Google Scholar]
- 28. Pack AI, Galante RJ, Maislin G, Cater J, Metaxas D, Lu S, Zhang L, Von Smith R, Kay T, Lian J, Svenson K, Peters LL. 2007. Novel method for high-throughput phenotyping of sleep in mice. Physiol Genomics 28:232–238 [DOI] [PubMed] [Google Scholar]
- 29. Riachi M, Himms-Hagen J, Harper ME. 2004. Percent relative cumulative frequency analysis in indirect calorimetry: application to studies of transgenic mice. Can J Physiol Pharmacol 82:1075–1083 [DOI] [PubMed] [Google Scholar]
- 30. Abs R, Mattsson AF, Bengtsson BA, Feldt-Rasmussen U, Góth MI, Koltowska-Häggström M, Monson JP, Verhelst J, Wilton P. 2005. Isolated growth hormone (GH) deficiency in adult patients: baseline clinical characteristics and responses to GH replacement in comparison with hypopituitary patients. A sub-analysis of the KIMS database. Growth Horm IGF Res 15:349–359 [DOI] [PubMed] [Google Scholar]
- 31. Doga M, Bonadonna S, Gola M, Mazziotti G, Giustina A. 2006. Growth hormone deficiency in the adult. Pituitary 9:305–311 [DOI] [PubMed] [Google Scholar]
- 32. Randeva HS, Murray RD, Lewandowski KC, O'Callaghan CJ, Horn R, O'Hare P, Brabant G, Hillhouse EW, Shalet SM. 2002. Differential effects of GH replacement on the components of the leptin system in GH-deficient individuals. J Clin Endocrinol Metab 87:798–804 [DOI] [PubMed] [Google Scholar]
- 33. Lusk G. 1928. The elements of the science of nutrition. 4th ed Philadelphia and London: W. B. Saunders Co [Google Scholar]
- 34. Luque RM, Lin Q, Córdoba-Chacón J, Subbaiah PV, Buch T, Waisman A, Vankelecom H, Kineman RD. 2011. Metabolic impact of adult-onset, isolated, growth hormone deficiency (AOiGHD) due to destruction of pituitary somatotropes. PLoS One 6:e15767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fäldt J, Wernstedt I, Fitzgerald SM, Wallenius K, Bergström G, Jansson JO. 2004. Reduced exercise endurance in interleukin-6-deficient mice. Endocrinology 145:2680–2686 [DOI] [PubMed] [Google Scholar]
- 36. Seidell JC, Muller DC, Sorkin JD, Andres R. 1992. Fasting respiratory exchange ratio and resting metabolic rate as predictors of weight gain: the Baltimore Longitudinal Study on Aging. Int J Obes Relat Metab Disord 16:667–674 [PubMed] [Google Scholar]
- 37. Zurlo F, Lillioja S, Esposito-Del Puente A, Nyomba BL, Raz I, Saad MF, Swinburn BA, Knowler WC, Bogardus C, Ravussin E. 1990. Low ratio of fat to carbohydrate oxidation as predictor of weight gain: study of 24-h RQ. Am J Physiol 259:E650–E657 [DOI] [PubMed] [Google Scholar]
- 38. Tschöp M, Smiley DL, Heiman ML. 2000. Ghrelin induces adiposity in rodents. Nature 407:908–913 [DOI] [PubMed] [Google Scholar]
- 39. Short KR, Moller N, Bigelow ML, Coenen-Schimke J, Nair KS. 2008. Enhancement of muscle mitochondrial function by growth hormone. J Clin Endocrinol Metab 93:597–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mauras N, O'Brien KO, Welch S, Rini A, Helgeson K, Vieira NE, Yergey AL. 2000. Insulin-like growth factor I and growth hormone (GH) treatment in GH-deficient humans: differential effects on protein, glucose, lipid, and calcium metabolism. J Clin Endocrinol Metab 85:1686–1694 [DOI] [PubMed] [Google Scholar]
- 41. Moustaïd N, Jones BH, Taylor JW. 1996. Insulin increases lipogenic enzyme activity in human adipocytes in primary culture. J Nutr 126:865–870 [DOI] [PubMed] [Google Scholar]
- 42. Kersten S. 2001. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep 2:282–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Carroll PV, Christ ER, Bengtsson BA, Carlsson L, Christiansen JS, Clemmons D, Hintz R, Ho K, Laron Z, Sizonenko P, Sönksen PH, Tanaka T, Thorne M. 1998. Growth hormone deficiency in adulthood and the effects of growth hormone replacement: a review. Growth Hormone Research Society Scientific Committee. J Clin Endocrinol Metab 83:382–395 [DOI] [PubMed] [Google Scholar]
- 44. Götherström G, Elbornsson M, Stibrant-Sunnerhagen K, Bengtsson BA, Johannsson G, Svensson J. 2009. Ten years of growth hormone (GH) replacement normalizes muscle strength in GH-deficient adults. J Clin Endocrinol Metab 94:809–816 [DOI] [PubMed] [Google Scholar]
- 45. Götherström G, Elbornsson M, Stibrant-Sunnerhagen K, Bengtsson BA, Johannsson G, Svensson J. 2010. Muscle strength in elderly adults with GH deficiency after 10 years of GH replacement. Eur J Endocrinol 163:207–215 [DOI] [PubMed] [Google Scholar]
- 46. Pfluger PT, Castañeda TR, Heppner KM, Strassburg S, Kruthaupt T, Chaudhary N, Halem H, Culler MD, Datta R, Burget L, Tschöp MH, Nogueiras R, Perez-Tilve D. 2011. Ghrelin, peptide YY and their hypothalamic targets differentially regulate spontaneous physical activity. Physiol Behav 105:52–61 [DOI] [PubMed] [Google Scholar]
- 47. Bruun JM, Lihn AS, Verdich C, Pedersen SB, Toubro S, Astrup A, Richelsen B. 2003. Regulation of adiponectin by adipose tissue-derived cytokines: in vivo and in vitro investigations in humans. Am J Physiol Endocrinol Metab 285:E527–E533 [DOI] [PubMed] [Google Scholar]
- 48. Van Cauter E, Copinschi G. 2000. Interrelationships between growth hormone and sleep. Growth Horm IGF Res 10(Suppl B):S57–S62 [DOI] [PubMed] [Google Scholar]
- 49. Van Cauter E, Plat L. 1996. Physiology of growth hormone secretion during sleep. J Pediatr 128:S32–S37 [DOI] [PubMed] [Google Scholar]
- 50. Van Cauter E, Plat L, Copinschi G. 1998. Interrelations between sleep and the somatotropic axis. Sleep 21:553–566 [PubMed] [Google Scholar]
- 51. van der Lely AJ, Tschöp M, Heiman ML, Ghigo E. 2004. Biological, physiological, pathophysiological, and pharmacological aspects of ghrelin. Endocr Rev 25:426–457 [DOI] [PubMed] [Google Scholar]
- 52. Obal F, Jr, Alt J, Taishi P, Gardi J, Krueger JM. 2003. Sleep in mice with nonfunctional growth hormone-releasing hormone receptors. Am J Physiol Regul Integr Comp Physiol 284:R131–R139 [DOI] [PubMed] [Google Scholar]
- 53. Obál F, Jr, Fang J, Taishi P, Kacsóh B, Gardi J, Krueger JM. 2001. Deficiency of growth hormone-releasing hormone signaling is associated with sleep alterations in the dwarf rat. J Neurosci 21:2912–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Obal F, Jr, Krueger JM. 2004. GHRH and sleep. Sleep Med Rev 8:367–377 [DOI] [PubMed] [Google Scholar]
- 55. Kawakami N, Takatsuka N, Shimizu H. 2004. Sleep disturbance and onset of type 2 diabetes. Diabetes Care 27:282–283 [DOI] [PubMed] [Google Scholar]
- 56. Knutson KL, Van Cauter E. 2008. Associations between sleep loss and increased risk of obesity and diabetes. Ann NY Acad Sci 1129:287–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Taheri S, Lin L, Austin D, Young T, Mignot E. 2004. Short sleep duration is associated with reduced leptin, elevated ghrelin, and increased body mass index. PLoS Med 1:e62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tasali E, Leproult R, Spiegel K. 2009. Reduced sleep duration or quality: relationships with insulin resistance and type 2 diabetes. Prog Cardiovasc Dis 51:381–391 [DOI] [PubMed] [Google Scholar]
- 59. Trujillo ME, Scherer PE. 2005. Adiponectin—journey from an adipocyte secretory protein to biomarker of the metabolic syndrome. J Intern Med 257:167–175 [DOI] [PubMed] [Google Scholar]
- 60. Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, Kuriyama H, Nishida M, Yamashita S, Okubo K, Matsubara K, Muraguchi M, Ohmoto Y, Funahashi T, Matsuzawa Y. 1999. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun 257:79–83 [DOI] [PubMed] [Google Scholar]
- 61. Matsuzawa Y. 2010. Adiponectin: a key player in obesity related disorders. Curr Pharm Des 16:1896–1901 [DOI] [PubMed] [Google Scholar]
- 62. Buechler C, Wanninger J, Neumeier M. 2011. Adiponectin, a key adipokine in obesity related liver diseases. World J Gastroenterol 17:2801–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Okamoto Y, Folco EJ, Minami M, Wara AK, Feinberg MW, Sukhova GK, Colvin RA, Kihara S, Funahashi T, Luster AD, Libby P. 2008. Adiponectin inhibits the production of CXC receptor 3 chemokine ligands in macrophages and reduces T-lymphocyte recruitment in atherogenesis. Circ Res 102:218–225 [DOI] [PubMed] [Google Scholar]
- 64. Steffens S, Mach F. 2008. Adiponectin and adaptive immunity: linking the bridge from obesity to atherogenesis. Circ Res 102:140–142 [DOI] [PubMed] [Google Scholar]
- 65. Azuma K, Katsukawa F, Oguchi S, Murata M, Yamazaki H, Shimada A, Saruta T. 2003. Correlation between serum resistin level and adiposity in obese individuals. Obes Res 11:997–1001 [DOI] [PubMed] [Google Scholar]
- 66. Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. 2001. The hormone resistin links obesity to diabetes. Nature 409:307–312 [DOI] [PubMed] [Google Scholar]
- 67. Steppan CM, Lazar MA. 2002. Resistin and obesity-associated insulin resistance. Trends Endocrinol Metab 13:18–23 [DOI] [PubMed] [Google Scholar]
- 68. Christ ER, Simpson HL, Breen L, Sonksen PH, Russell-Jones DL, Kohner EM. 2003. The effect of growth hormone (GH) replacement therapy in adult patients with type 1 diabetes mellitus and GH deficiency. Clin Endocrinol (Oxf) 58:309–315 [DOI] [PubMed] [Google Scholar]
- 69. Menezes Oliveira JL, Marques-Santos C, Barreto-Filho JA, Ximenes Filho R, de Oliveira Britto AV, Oliveira Souza AH, Prado CM, Pereira Oliveira CR, Pereira RM, Ribeiro Vicente Tde A, Farias CT, Aguiar-Oliveira MH, Salvatori R. 2006. Lack of evidence of premature atherosclerosis in untreated severe isolated growth hormone (GH) deficiency due to a GH-releasing hormone receptor mutation. J Clin Endocrinol Metab 91:2093–2099 [DOI] [PubMed] [Google Scholar]
- 70. Oliveira CR, Salvatori R, Barreto-Filho JA, Rocha IE, Mari A, Pereira RM, Campos VC, Menezes M, Gomes E, Meneguz-Moreno RA, Araujo VP, Leite NT, Nascimento-Junior AC, Farias MI, Viscente TA, Araujo RD, Melo EV, Aguiar-Oliveira MH. 2011. Insulin sensitivity and β-cell function in adults with lifetime, untreated isolated growth hormone deficiency. J Clin Endocrinol Metab [DOI] [PubMed] [Google Scholar]
- 71. Yakar S, Setser J, Zhao H, Stannard B, Haluzik M, Glatt V, Bouxsein ML, Kopchick JJ, LeRoith D. 2004. Inhibition of growth hormone action improves insulin sensitivity in liver IGF-1-deficient mice. J Clin Invest 113:96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Getty-Kaushik L, Song DH, Boylan MO, Corkey BE, Wolfe MM. 2006. Glucose-dependent insulinotropic polypeptide modulates adipocyte lipolysis and reesterification. Obesity (Silver Spring) 14:1124–1131 [DOI] [PubMed] [Google Scholar]
- 73. Fulurija A, Lutz TA, Sladko K, Osto M, Wielinga PY, Bachmann MF, Saudan P. 2008. Vaccination against GIP for the treatment of obesity. PLoS One 3:e3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gault VA, O'Harte FP, Flatt PR. 2003. Glucose-dependent insulinotropic polypeptide (GIP): anti-diabetic and anti-obesity potential? Neuropeptides 37:253–263 [DOI] [PubMed] [Google Scholar]
- 75. Oben J, Morgan L, Fletcher J, Marks V. 1991. Effect of the entero-pancreatic hormones, gastric inhibitory polypeptide and glucagon-like polypeptide-1(7–36) amide, on fatty acid synthesis in explants of rat adipose tissue. J Endocrinol 130:267–272 [DOI] [PubMed] [Google Scholar]
- 76. Eckel RH, Fujimoto WY, Brunzell JD. 1979. Gastric inhibitory polypeptide enhanced lipoprotein lipase activity in cultured preadipocytes. Diabetes 28:1141–1142 [DOI] [PubMed] [Google Scholar]
- 77. Hauner H, Glatting G, Kaminska D, Pfeiffer EF. 1988. Effects of gastric inhibitory polypeptide on glucose and lipid metabolism of isolated rat adipocytes. Ann Nutr Metab 32:282–288 [DOI] [PubMed] [Google Scholar]
- 78. Singh SK, Bartoo AC, Krishnan S, Boylan MO, Schwartz JH, Michael Wolfe M. 2008. Glucose-dependent insulinotropic polypeptide (GIP) stimulates transepithelial glucose transport. Obesity (Silver Spring) 16:2412–2416 [DOI] [PubMed] [Google Scholar]
- 79. Wellen KE, Hotamisligil GS. 2003. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest 112:1785–1788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Beutler B, Greenwald D, Hulmes JD, Chang M, Pan YC, Mathison J, Ulevitch R, Cerami A. 1985. Identity of tumour necrosis factor and the macrophage-secreted factor cachectin. Nature 316:552–554 [DOI] [PubMed] [Google Scholar]
- 81. Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, Simsolo RB. 1995. The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest 95:2111–2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Syed M, Cozart M, Crane C, Haney A, Akhter N, Odle AK, Syed F, Allensworth-James M, Childs GV. 2012. Functional defects in mice somatotropes caused by abalation of leptin receptor is reversed, in vitro, by stimulation with ghrelin. FASEB J 26:Abstract 909.4 [Google Scholar]