

Abstract
Ghrelin is a gastrointestinal polypeptide that acts through the ghrelin receptor (GHSR) to promote food intake and increase adiposity. Activation of GHSR requires the presence of a fatty-acid (FA) side chain on amino acid residue serine 3 of the ghrelin molecule. However, little is known about the role that the type of FA used for acylation plays in the biological action of ghrelin. We therefore evaluated a series of differentially acylated peptides to determine whether alterations in length or stability of the FA side chain have an impact on the ability of ghrelin to activate GHSR in vitro or to differentially alter food intake, body weight, and body composition in vivo. Fatty acids principally available in the diet (such as palmitate C16) and therefore representing potential substrates for the ghrelin-activating enzyme ghrelin O-acyltransferase (GOAT) were used for dose-, time-, and administration/route-dependent effects of ghrelin on food intake, body weight, and body composition in rats and mice. Our data demonstrate that altering the length of the FA side chain of ghrelin results in the differential activation of GHSR. Additionally, we found that acylation of ghrelin with a long-chain FA (C16) delays the acute central stimulation of food intake. Lastly, we found that, depending on acylation length, systemic and central chronic actions of ghrelin on adiposity can be enhanced or reduced. Together our data suggest that modification of the FA side-chain length can be a novel approach to modulate the efficacy of pharmacologically administered ghrelin.
Ghrelin is a 28-amino acid peptide secreted mainly from the X/A-like cells of the stomach (1–3). A unique feature of ghrelin is the esterification with an 8-n acyl group at the serine in position 3. Acylation of ghrelin occurs during posttranslational modification in which an n-octanoic acid is esterified to the serine 3 residue of the peptide (4, 5). The enzyme that catalyzes this reaction is ghrelin O-acyltransferase (GOAT), which is a membrane-bound protein highly expressed in the stomach (4, 5). Ghrelin is found in circulation in both the acyl and des-acyl forms (1, 6). However, the presence of the fatty acid (FA) side chain is required for the binding of ghrelin and activation of its G protein-coupled receptor known as the GH-secretagogue receptor (GHSR) (1). This receptor retains significant baseline constitutive activity (7–9) and is highly expressed in the hypothalamus (10, 11), which is the nexus for the regulation of energy balance. The agouti-related peptide (AgRP)/neuropeptide Y (NPY) neurons of the hypothalamus express GHSR (12, 13) and are a primary target of ghrelin (14–17). Ghrelin stimulation of these neurons (18) results in an increase in NPY/AgRP peptide release in the paraventricular nucleus (PVN). NPY activates Y1 and Y5 (19) receptors, whereas AgRP antagonizes melanocortin 4 receptor in the PVN (20). Additionally, ghrelin activation of GHSR on NPY/AgRP neurons promotes the release of γ-aminobutyric acid (14), which acts to inhibit the anorectic effect of proopiomelanocortin-expressing neurons in the arcuate nucleus. The collective ghrelin-induced activation of GHSR on NPY/AgRP-expressing neurons in the hypothalamus is increased food intake (16, 21–23) and adiposity (24, 25).
Although the acylated form of ghrelin that was initially identified was octanoyl ghrelin (1), GOAT can actually acylate ghrelin with FA of different lengths (4, 5, 26). Furthermore, these FA can have dietary origin since mice were found to have circulating ghrelin acylated with a C7-FA side chain after being fed a diet rich in nonendogenously produced C7-chain FA (27, 28). In addition, ghrelin esterified by n-decanoic acid was isolated from human stomach and plasma (6). This form of ghrelin has the same potency to increase [Ca2+] in GHSR-expressing cells and to stimulate GH release in anesthetized rats as octanoyl ghrelin (6). One report demonstrated that altering the structure of the fatty acid side chain alters ghrelin's ability to activate GHSR in human embryonic kidney-293 cells (29), and another group found that replacing the ester bond linking the octanoic acid to the Ser3 residue of the ghrelin peptide with a more chemically stable ether or thioether bond resulted in similar GHSR activation in Chinese hamster ovary-GHSR62 cells as native ghrelin (30). Interestingly, administering ghrelin stabilized with a thioether bond iv to rats delayed the onset of GH secretion, although total GH secretion of the stable vs. native ghrelin was similar. Despite these findings exploring GHSR activation by ghrelin isoforms in cell-based assays and on GH secretion, it is unclear whether these effects will translate to the in vivo action of ghrelin on energy metabolism.
Circulating ghrelin levels change in pathophysiological conditions involving perturbed energy balance, such as obesity (31–35) and diabetes (36), and there are inconsistent reports that surgical procedures intended to reduce obesity and diabetes, such as Roux-en-Y gastric bypass, also result in changes in plasma ghrelin levels (37, 38). However, it is unknown whether different metabolic conditions have an impact on the levels of different acyl ghrelin isoforms, and the accurate measurement of different acyl ghrelin levels is still unfeasible due to the lack of specific high-throughput assays. Taken together, the changes in ghrelin associated with the metabolic disease, and the endogenous existence of ghrelin acylated with different fatty acids, warrants further investigation of the possible alternative physiological actions of these isoforms.
The aim of this study was to systematically dissect the in vitro and in vivo actions of synthetic ghrelin compounds acylated with FA of variable length (C2-C16) and stability to determine whether these alterations affect the action of ghrelin on GHSR activation, food intake, body weight, and body composition.
Materials and Methods
Peptide synthesis
Rat ghrelin was synthesized using in situ neutralization for Boc chemistry, purified by preparative chromatography, and characterized by HPLC and mass spectral analysis, as described previously (24). The native ghrelin used for these experiments contained an octanoic acid (C8) forming an ester bond at the Ser3 hydroxyl side chain. Ghrelin isoforms were synthesized using the 28-amino acid sequence of rat ghrelin, but the number of carbons in the FA side chain was altered (C2-C16). For the stabilized analog, the ester bond at the Ser3 was replaced with an isostere that has an amino group on the side chain. The amino group was acylated with octanoic acid to form an amide bond, which is less prone to hydrolysis, resulting in enhanced stability of bond of the octanoyl group to the peptide molecule. These peptide acyl amides are denoted as SC8 and SC16 to signify the absence of the labile acyl ester of endogenous ghrelin isoforms.
Transfections and tissue culture
COS-7 cells were grown in DMEM 1885 supplemented with 10% fetal calf serum, 2 mm glutamine, and 0.01 mg/ml gentamicin. The cells were transfected using the calcium phosphate precipitation method with chloroquine addition as previously described (39).
Phosphatidylinositol turnover
One day after transfection, COS-7 cells were incubated for 24 h with 5 μCi of [3H]-myo-inositol (Amersham Bioscience, Piscataway, NJ; PT6-271) in 1 ml medium supplemented with 10% fetal calf serum, 2 mm glutamine,180 U/ml penicillin, and 45 μg/ml streptomycin. Cells were washed twice in buffer, 20 mm HEPES (pH 7.4), supplemented with 140 mm NaCl, 5 mm KCl, 1 mm MgSO4, 1 mm CaCl2, 10 mm glucose, and 0.05% (wt/vol) bovine serum and were incubated in 0.5 ml buffer supplemented with 10 mm LiCl at 37 C for 30 min. After stimulation with various concentrations of peptide for 45 min at 37 C, cells were extracted with 10% ice-cold perchloric acid followed by incubation on ice for 30 min. The resulting supernatants were neutralized with KOH in HEPES buffer, and the generated [3H]-inositol phosphate was purified on Bio-Rad Laboratories (Hercules, CA) AG 1-X8 anion-exchange resin as described. Determinations were made in duplicate.
Animals
Male C57BL/6 mice (8 wk old; Jackson Labs, Bar Harbor, ME) and male Long-Evans rats (300–350 g; Harlan, Indianapolis, IN) were maintained on a standard chow diet (Teklad; Harlan). After receiving intracerebroventricular (icv) surgery, animals were singly housed on a 12-h light, 12-h dark cycle at 22 C with free access to food and water unless noted otherwise. Mice that received C6 or C16 acylated ghrelin were housed in an indirect calorimetry system (TSE LabMaster, Bad Homburg v.d.H., Germany) for the study period. Mice infused with C2, C12, or C14 were housed in conventional cages. Each study had a saline and C8 control group that was housed in the same manner as their corresponding treatment groups. Animals that received chronic sc injections were group housed (four mice per cage) in conventional cages. GHSR-knockout (KO) mice (8–12 wk) were received from Regeneron Pharmaceuticals (Tarrytown, NY) (40–42) and bred in our facilities as described previously (41). All studies were approved by and performed according to the guidelines of the Institutional Animal Care and Use Committee of the University of Cincinnati.
Acute icv cannula implantation in rats
A 22-gauge stainless steel cannula (Plastics One, Roanoke, VA) was stereotaxically implanted into the third ventricle of rats according to the following coordinates: −2.2 mm posterior to bregma and to a depth of −7.5 mm from the surface of the brain, with bregma and lambda being horizontal. Dental acrylic secured the cannula to the skull and an obturator extending 1 mm below the guide cannula was inserted. After a week of recovery, correct placement of the cannula was verified by icv administration of angiotensin II (1 μg/μl of 0.9% saline). Rats that failed to drink a minimum of 5 ml of water within 30 min were removed from the studies.
Determination of the acute effect of icv ghrelin on food intake
Food was weighed before icv injections which occurred at the beginning of the light phase. Briefly, an internal cannula (Plastics One) was connected to polyethylene-50 tubing, and the tubing was connected to a 25-μl Hamilton syringe (Hamilton, Reno, NV). The peptide was drawn into the tubing, the internal cannula was inserted, and a 1-μl volume of the peptide or saline was infused. Food intake was recorded 2, 4, and 24 h after injection. Doses were used as indicated and they included 0.3, 0.9, and 3 nmol (equivalent to 1, 3, and 10 μg of ghrelin isoforms).
Intracerebroventricular infusions in mice
Ghrelin was dissolved in isotonic saline and infused icv at a dose of 5 nmol/d per mouse (5 nmol = 15 μg of ghrelin isoforms) using osmotic minipumps (1007D; Alzet, Cupertino, CA), prepared following the manufacturer's instructions. Male C57BL/6 mice were anesthetized using 5% isoflurane in oxygen in an induction chamber and then maintained on 2.5% isoflurane delivered by a nose cone. Mice were stereotaxically implanted (David Kopf Instruments, Tujunga, CA) with a cannula (brain infusion kit no. 3; Alzet) placed in the lateral cerebral ventricle using the coordinates −0.7 mm posterior to bregma, −1.2 mm lateral to the midsagittal suture, and to a depth of −2.5 mm from the surface of the brain, with bregma and lambda being horizontal. A polyethylene catheter attached the cannula to an osmotic minipump that was sc implanted in the interscapular region. The pumps infused either ghrelin or saline at a rate of 0.5 μl/h for 7 d. After surgery, the animals received a single sc dose of 0.28 mg/kg buprenorphin (Buprenex; Reckitt Benckiser Healthcare, Richmond, VA) and were maintained on a heated surface until full recovery. Food intake and body weight were measured daily. Body composition was analyzed immediately before surgery and again on the final day of infusion.
Body composition measurements
Whole-body composition (fat and lean mass) was measured using nuclear magnetic resonance technology (43) (EchoMRI-100; Echo Medical Systems, Houston, TX).
Subcutaneous injections in mice
Mice were given a daily sc injection of saline, C8, C16, SC8, or SC16-acylated ghrelin. Ghrelin compounds were dissolved in saline and administered at a dose of 60 μmol/d per mouse (60 μmol = 200 μg of ghrelin isoforms). Food intake and body weight were measured daily. Body composition was measured before the initial injection and again after the final injection.
Calculations and statistical analysis
For inositol phosphate (IP) accumulation analysis in COS-7 cells, EC50 values were determined by nonlinear regression using Prism version 4.0 software (GraphPad Software, San Diego, CA). Statistical analysis for animal studies was performed using GraphPad Prism version 5.0 (GraphPad Software). The differences among treatments were assessed by using a one-way ANOVA and a Tukey's post hoc test. For analysis of time-dependent differences in body weight and food intake, two-way ANOVA were used with the Bonferroni post hoc. All results are given as means ± sem. Results were considered statistically significant when P < 0.05.
Results
Altering the fatty acid side-chain length of ghrelin results in variable activation of GHSR in COS-7 cells
We assessed synthetic ghrelin compounds acylated with different side-chain lengths (C2-C16) for their ability to activate GHSR by determining dose-response curves of phosphatidylinositol turnover (Fig. 1). The receptor activation of the isoforms was compared with that of native C8-acylated ghrelin induced receptor activation. Ghrelin acylated with a C8-FA stimulated IP turnover with a potency (EC50 = 0.43 nm) similar to what has previously been reported (7, 44). C2-acylated ghrelin did not stimulate IP accumulation in these cells (Fig. 1A), indicating that a FA side chain greater than 2 carbons is necessary to induce GHSR activation. Ghrelin acylated with a C6 medium chain FA (Fig. 1B) induced similar IP accumulation as C8-acylated ghrelin. Ghrelin acylated with longer FA side chains (C12 and C14, Fig. 1, C and D) shifted the dose response curves to the right with 80- and 37-fold decrease in potencies, respectively, compared with native C8-acylated ghrelin. Unlike ghrelin acylated with long-chain fatty acids of C12 and C14, ghrelin acylated with a C16 FA (Fig. 1E) caused a similar induction of IP accumulation as C8-acylated ghrelin.
Fig. 1.

In vitro receptor activation in COS-7 cells transiently expressing GHSR. Specificity of the receptor activation was established by the level of IP accumulation induced by C8-acylated ghrelin. A, In similar conditions, C2-acylated ghrelin has negligible activity. C6 and C16 have similar potency to activate GHSR as that of C8-acylated ghrelin (B and E), whereas C12 and C14 have a decreased potency (C and D).
Effect of acute icv injection of the different acyl ghrelin isoforms on food intake in rats
To determine whether altering the length of the FA side chain of ghrelin influences acute food intake, we conducted a preliminary screening of C2-, C6-, C8-, C12-, C14-, and C16-acylated ghrelin by administering these compounds icv (0.9 nmol) to rats at the beginning of the light phase (Fig. 2). Comparable with previous reports (16, 21, 23), rats treated with C8-acylated ghrelin had increased food intake at 2 and 4 h after injection compared with saline-treated controls (P < 0.05, C8 vs. saline, Student's t test). Ghrelin acylated with C2, C6, C12, C14, and C16 FA had no effect on food intake 2 and 4 h after injection compared with saline controls. Unexpectedly, rats treated with C16-acylated ghrelin, which had normal food intake at 2 and 4 h, had a significant increase in food intake after 24 h compared with saline controls (P < 0.05,C16 vs. saline, Student's t test), whereas food intake in the C2-, C6-, C8-, and C14-treated groups was similar compared with saline controls. No change in body weight was noted 24 h after injection in any group (data not shown). Taken together, these data suggest that modification of ghrelin with a C16 FA delays the induction of food intake in response to centrally administered ghrelin.
Fig. 2.
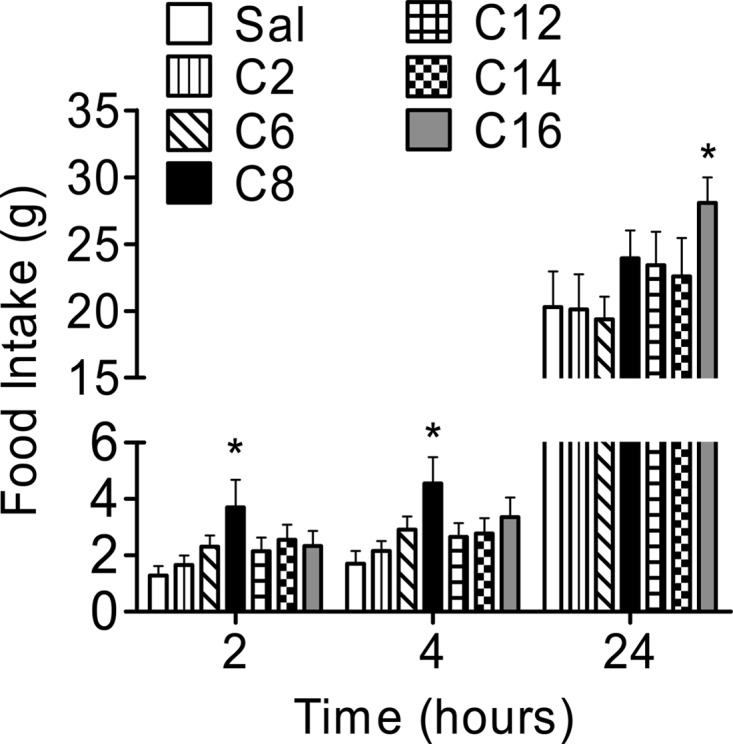
Preliminary evaluation of ghrelin isoforms on acute food intake in rats. Rats were administered icv saline or 0.9 nmol of C2-, C6-, C8-, C12-, C14-, or C16-acylated ghrelin at the beginning of the light phase. Food intake 2 and 4 h after injection was increased in animals treated with C8-acylated ghrelin compared with saline controls. *, P < 0.05, C8 vs. saline, Student's t test. Food intake 24 h after the injection was significantly increased in rats receiving C16-acylated ghrelin compared with saline-treated rats. *, P < 0.05, C16 vs. saline, Student's t test (n = 9–10 rats per group).
Effect of chronic icv infusion of different acyl-ghrelin isoforms on food intake, body weight, and adiposity in mice
To assess the chronic central effects of C2-, C6-, C8-, C12-, C14-, or C16-acylated ghrelin, mice were infused with each of the ghrelin analogs (5 nmol/d per mouse) via a sc implanted minipump that was attached to a cannula placed in the lateral ventricle of the brain. The effects on food intake and body weight are summarized in Table 1. Cumulative food intake was increased in the C12-acylated ghrelin group compared with saline-treated controls after 6 d of infusion (Table 1). Body weight was increased 6 d after treatment in the C8-, C12-, and C14-treated mice. Animals administered with C2-acylated ghrelin had no change in fat mass after the infusion period (Fig. 3A), whereas mice receiving C6-, C8-, C12-, C14-, and C16-acylated ghrelin all had increased fat mass (Fig. 3, B–E) compared with saline-treated controls. Although animals treated with C14-acylated ghrelin experienced an increase in fat mass, this was significantly less than animals infused with C8-acylated ghrelin. No change in lean mass was observed in any group (data not shown). Mice infused with C6- and C16-acylated ghrelin were placed in an indirect calorimetry system for the duration of the study for further analysis. There were no differences in energy expenditure, respiratory quotient, or locomotor activity compared with C8-acylated ghrelin (data not shown).
Table 1.
Chronic central effects of ghrelin isoforms on food intake and body weight in mice
| Cumulative FI (g) | ΔBody weight (g) | |
|---|---|---|
| C2 | ||
| Saline | 31.49 ± 1.24 | 1.29 ± 0.27 |
| C8 | 35.57 ± 1.72 | 2.75 ± 0.43a |
| C2 | 31.78 ± 1.60 | 1.70 ± 0.28 |
| C6 | ||
| Saline | 22.72 ± 1.22 | 0.09 ± 0.42 |
| C8 | 25.04 ± 1.26 | 1.06 ± 0.31 |
| C6 | 23.46 ± 1.35 | 0.64 ± 0.5 |
| C12 | ||
| Saline | 31.49 ± 1.24 | 1.29 ± 0.27 |
| C8 | 35.57 ± 1.72 | 2.75 ± 0.43a |
| C12 | 36.91 ± 0.97a | 2.85 ± 0.36a |
| C14 | ||
| Saline | 26.98 ± 1.0 | 0.24 ± 0.155 |
| C8 | 30.71 ± 1.65 | 1.79 ± 0.3275b |
| C14 | 27.69 ± 1.36 | 1.32 ± 0.23a |
| C16 | ||
| Saline | 22.72 ± 1.22 | 0.09 ± 0.42 |
| C8 | 25.04 ± 1.26 | 1.06 ± 0.31 |
| C16 | 22.88 ± 1.85 | 0.87 ± 0.32 |
Cumulative food intake and change in body weight after 6 d of chronic icv infusion of C2-, C6-, C8-, C12-, C14-, or C16-acylated ghrelin (5 nmol/d per mouse) in C57/BL6 mice.
P < 0.05 vs. saline (one way ANOVA; n = 8–10 mice per group).
P < 0.001 vs. saline (one way ANOVA; n = 8–10 mice per group).
Fig. 3.

Chronic central effects of ghrelin isoforms on adiposity. C57/BL6 mice were implanted with icv minipumps that infused C2-, C6-, C8-, C12-, C14-, or C16-acylated ghrelin (5 nmol/d per mouse). C2-acylated ghrelin did not have an effect on fat mass after a 7-d infusion period (A), whereas C6- (B), C12- (C), C41- (D), and C16-acylated ghrelin (E) all increased fat mass compared with saline treated controls. *, P < 0.05, **, P < 0.01, ***, P < 0.001 vs. saline, one-way ANOVA. D, C14-acylated ghrelin was significantly less effective at increasing fat mass as C8-treated animal. #, P < 0.05 C14 vs. C8 (one-way ANOVA with Tukey post hoc; n = 8–10 animals per group). Sal, Saline.
The lack of effect on fat mass observed in the C2-treated group as well as the decreased effect of C14-acylated ghrelin compared with C8-treated animals is consistent with our GHSR activation assay in which we found that C2-acylated ghrelin lacked the ability to induce GHSR activation and that C14-acylated ghrelin had decreased potency compared with C8-acylated ghrelin. Interestingly, we found that C12-acylated ghrelin had a similar potency to increase fat mass as C8-acylated ghrelin; however, C12-acylated ghrelin had a less potent effect on IP accumulation compared with C8-acylated ghrelin (Fig. 1C). A possible explanation for this is that C12-acylated ghrelin may have GHSR-independent actions.
C12-acylated ghrelin requires GHSR to increase food intake and fat mass in mice
To explore the possibility that C12-acylated ghrelin has GHSR-independent action, we infused GHSR-KO mice icv with C8- or C12-acylated ghrelin (5 nmol/d per mouse). Neither C8-nor C12-acylated ghrelin had an effect on food intake (Fig. 4A) during the infusion period. We also found that body weight (Fig. 4B), fat mass (Fig. 4C), and lean mass (data not shown) were not altered in either of the ghrelin treatment groups after the 7-d central treatment. From these data we concluded that C12-acylated ghrelin requires GHSR to mediate its effects in mice.
Fig. 4.

Effects of C12-acylated ghrelin on food intake, body weight, and body composition in GHSR-KO mice. GHSR-KO mice were infused icv for 7 d with C8- or C12-acylated ghrelin (5 nmol/d per mouse). A, Cumulative food intake in mice treated with C8- or C12-acylated ghrelin was similar to that of saline-treated controls 7 d after treatment. B, Change in body weight on d 7 of treatment was similar in both experimental groups compared with saline-treated controls. C, Change in fat mass on d 7 of treatment was also similar among all groups (n = 8 animals per group).
Effect of icv C16-acylated ghrelin on acute feeding in rats
To confirm the delayed effect of C16-acylated ghrelin on acute feeding, we administered doses of 0.3, 0.9, and 3 nmol of C16- and C8-acylated ghrelin and compared them with saline-treated controls (Fig. 5). Consistent with our findings in Fig. 2 as well as previous reports (16, 21, 23), icv C8-acylated ghrelin induced a significant increase in food intake at early time points (2 and 4 h) with doses of 0.9 and 3 nmol, whereas C16-acylated ghrelin had no effect at these time points (Fig. 5, A and B). C8-acylated ghrelin had no effect on 24-h food intake, whereas doses of 0.9 and 3 nmol of C16-acylated ghrelin significantly increased feeding at 24 h compared with saline-treated controls (Fig. 5C). The reason that C16-acylated ghrelin has a delayed effect on food intake is not clear, but a possible explanation is due to increased stability of the FA side chain to the ghrelin peptide or enhanced association with plasma proteins, such as albumin.
Fig. 5.

Central administration of C16-acylated ghrelin delays the onset of acute food intake in rats. Rats received icv saline or C8- or C16-acylated ghrelin at doses of 0.3, 0.9, and 3 nmol. A, At 2 h after the injection, rats treated with C8-acylated ghrelin experienced an increase in food intake with doses of 0.9 and 3 nmol. **, P < 0.01 C8 vs. saline (two way ANOVA). B, At 4 h after the injection, rats treated with C8-acylated ghrelin experienced an increase in cumulative food intake with doses of 0.9 and 3 nmol. *, P < 0.05, **, P < 0.01 C8 vs. saline (two way ANOVA). C, Rats receiving C16-acylated ghrelin had a delayed effect on food intake, and cumulative 24 h food intake was increased in rats treated with 0.9 and 3 nmol. *, P < 0.05, ***, P < 0.001 C16 vs. saline (two way ANOVA; n = 9–15 animals per group).
Effect of chronic peripheral administration of C16-acylated ghrelin and stabilized ghrelin compounds in mice
Ghrelin receptor activity is dependent on Ser3 acylation, and the ester bond is highly prone to hydrolysis by circulating esterases, rendering the duration of active ghrelin action relatively short (45). Thus, stabilizing this ester bond should increase the duration of action and enhance the efficacy of ghrelin. We hypothesized that one explanation for the delayed action of C16-acylated ghrelin on acute food intake is due to enhanced resistance to enzymatic cleavage of the ester bond. If so, chronic peripheral treatment with C16-acylated ghrelin or C8-acylated ghrelin with a stabilized bond (SC8) between the FA and ghrelin peptide should prolong the action of ghrelin and enable it to have a more potent effect on food intake, body weight, and adiposity. We therefore treated mice with daily sc injections of C8, C16, SC8, or SC16 ghrelin (Fig. 6). Mice were housed four animals per group (n = 2 cages per treatment). Food intake was similar among groups but tended to be increased in the cages that received the C16 compared with saline-treated controls after 6 d of injections (cumulative average per mouse: saline, 24.07 ± 2.41 g; C8-acylated ghrelin, 27.40 ± 0.12 g; SC8-acylated ghrelin, 27.23 ± 1.73 g; C16-acylated ghrelin, 33.34 ± 1.21 g; SC16-acylated ghrelin, 28.90 ± 0.38 g). Body weight was significantly increased in all ghrelin treatment groups compared with saline-treated controls (Fig. 6A). Body weight was significantly increased in the C16- and SC16-acylated ghrelin treatment groups relative to the C8 and SC8 treatment groups. Consistent with the effects on body weight, all ghrelin treatment groups experienced a significant increase in fat mass (Fig. 6B). Again, both C16-acylated compounds more effectively increased fat mass compared with both C8-acylated compounds (Fig. 6B). No change in lean mass was observed in any of the treatment groups (data not shown). These data suggest that acylation of ghrelin with a C16 FA enhances the efficacy of pharmacological doses of ghrelin.
Fig. 6.

Effect of chronic peripheral treatment of C16-acylated ghrelin and stabilized ghrelin compounds in mice. C57/BL6 mice were given daily sc injections of native C8-acylated ghrelin (C8), C16-acylated ghrelin (C16), stabilized C8-acylated ghrelin (SC8), or stabilized C16-acylated ghrelin (SC16) at a dose of 60 nmol/d per mouse. A, All ghrelin-treated mice had a significant increase in body weight compared with saline-treated animals. $, P < 0.05 vs. saline (two way ANOVA). Both C16- and SC16-treated groups had a greater increase in body weight compared with C8. #, P < 0.05 C16- and SC16- vs. C8 (two way ANOVA) and SC8-treated animals. $, P < 0.05 C16 and SC16 vs. SC8 (two way ANOVA). B, All ghrelin-treated mice had increased fat mass after 6 d of injections. **, P < 0.01, ***, P < 0.001 vs. saline (one way ANOVA). Mice treated with C16 and SC16 had a greater increase of fat mass compared with C8 and SC8. #, P < 0.05, ##, P < 0.01, ###, P < 0.001 C16 or SC16 vs. C8 or SC18 (one way ANOVA with Tukey post hoc test; n = 8 mice per group).
Discussion
Previous reports using cell-based assays demonstrate that altering the fatty acid side-chain length of the ghrelin molecule alters its ability to bind and activate GHSR (29). The ability of the enzyme GOAT to use dietary lipids as a substrate to acylate ghrelin, coupled with studies documenting the existence of ghrelin acylated with different fatty acids, led us to investigate whether altering the fatty-acid side-chain length of the ghrelin molecule will cause the peptide to have differential biological action on food intake, body weight, and/or adiposity. In addition to finding differential GHSR activation in cell-based assays, we report novel findings on energy metabolism of ghrelin acylated with FA of varying length (C2-C16) and chemical stabilities in rodents. Specifically, the presence of a FA side chain longer than C2 is required for ghrelin to activate the GHSR receptor and to exert its biological effects on food intake, body weight, and adiposity. Our chronic studies are in accordance with previous findings that demonstrate ghrelin's effects on adiposity can be independent of changes in food intake (23, 24, 46, 47) and body weight gain (46). As a rule, we find that GHSR activation in cells provides a reliable prediction of how ghrelin analogs perform on in vivo adiposity, but interestingly, this is not always the case. Ghrelin acylated with a C12 or a C14 FA exhibits a decreased potency to activate GHSR when compared with native C8-acylated ghrelin. As expected, chronic central treatment of C14-acylated ghrelin to mice has a weak effect on increasing adiposity. Unexpectedly, C12-acylated ghrelin increases adiposity in mice with a similar potency as C8-acylated ghrelin. We hypothesized that this could be a result of a GHSR-independent action of C12-acylated ghrelin. To test this, we icv infused C12-acylated ghrelin to GHSR-KO mice and found that C12-acylated ghrelin loses its effects on adiposity in GHSR-KO mice. Thus, GHSR is essential for the action of C12-acylated ghrelin.
These studies highlight the complexity of the in vivo ghrelin system. Many reports demonstrate that GHSR is able to heterodimerize with other G protein-coupled receptors (48, 49). Acylation of ghrelin with a C12 rather than a C8 FA may differentially affect the heterodimerization of these receptors. This could explain the strong effect of C12-acylated ghrelin on adiposity in rodents that we found as well as weak GHSR activation in COS-7 cells that are not transfected with these additional G protein-coupled receptor. Our data suggest that a combination of both in vitro screening and in vivo testing of drug candidates may be an important determinant for the development of effective therapies based on targeting GHSR activity.
The most striking findings of the present report are the acute and chronic actions of C16-acylated ghrelin in rodents. Despite having a similar activation of GHSR in COS-7 cells, centrally administered C16-acylated ghrelin in rats reveals a unique delayed time course of action on food intake. Comparable prolonged effects can be observed in many pharmacological agents that are commonly acylated with a C16 FA. We explored the possibility that the prolonged onset of increased food intake could be due to the increased stability of the C16 FA to the ghrelin peptide and therefore might increase the efficacy of chronic peripheral ghrelin administration. We found that chronic peripheral treatment with ghrelin acylated with a C16 FA side chain had a greater ability to increase body weight and fat mass than ghrelin acylated with a C8-FA side chain.
We tested a stabilized form of both C8- and C16-acylated ghrelin, in which the ester bond linking the FA to the Ser3 residue was replaced with a nonhydrolysable amide bond. The stabilized ghrelin compounds did not increase the efficacy of ghrelin on body weight or fat mass compared with their nonstabilized counterparts. However, acylation of ghrelin with a C16 FA, regardless of whether the ester bond is stabilized, enhances ghrelin's effects on body weight and adiposity. This demonstrates that C16 acylation increases the potency of ghrelin through a mechanism other than enhanced stability of the FA to the peptide molecule.
The mechanisms responsible for the enhancement of C16-acylated ghrelin on energy metabolism require further clarification. It has been reported that the octonyl side chain of ghrelin provides an anchor to bind the peptide to the cell membrane (50). It is possible that the C16-FA side chain acts as a more effective anchor and therefore has a decreased amount of clearance from the circulation than C8-acylated ghrelin. In a related fashion, it is known that serum albumin binds long-chain FA (51) and acylated peptides, such as liraglutide and detemir. Consequently, it is possible that the C16 FA ghrelin binds to albumin, allowing the peptide to remain in circulation for an extended period of time. This might also account for the diminished early action in acute central administration of the C16-acylated ghrelin because albumin binding could suppress such activity. The rates of clearance of C8- vs. C16-acylated ghrelin have yet to be determined, and whether a decreased rate of clearance contributes to the increased potency of chronic treatment with C16-acylated ghrelin requires further investigation. In addition to examining the rates of clearance, future studies investigating intracellular signaling pathways could illuminate possible mechanisms mediating the delayed effect on acute food intake found with icv administration of C16-acylated ghrelin. For example, AMP-activated protein kinase (AMPK) has previously been suggested to act as an intracellular energy sensor, and pharmacological activation of hypothalamic AMPK has been shown to increases food intake (52). Interestingly, the orexigenic action of icv C8-acylated ghrelin administration correlates with a time-dependent increase in AMPK phosphorylation that peaks acutely at 2 h and returns to normal levels at 6 h after treatment (53). It is possible that a delay in AMPK phosphorylation after icv administration of C16-acylated ghrelin mediates the delayed effect on food intake reported here. However, whether ghrelin's orexigenic action can exclusively be attributed to activation of AMPK remains to be determined. Additionally, central C8-acylated ghrelin administration causes neuronal activation in hypothalamic nuclei including the arcuate nucleus and PVN (54). Follow-up studies investigating induction of c-fos as a marker of neuronal activation in these hypothalamic nuclei after icv administration of the various ghrelin isoforms are underway to help clarify whether the differential effects on acute food intake are a result of alterations in neuronal activation.
Future studies examining the endogenous existence of these ghrelin isoforms and whether these isoforms are altered in metabolic diseases including obesity, cancer cachexia, and anorexia nervosa could help us understand the pathogenesis of these disorders. A study in which mice were fed diets enriched in FA of various lengths used mass spectroscopy to demonstrate that stomach-derived ghrelin can be acylated with FA of different lengths including C6, C10, and C10:1 (28). Furthermore, this study demonstrated that these various ghrelin isoforms can be secreted into the circulation. Although it seems that stomach derived ghrelin has a preference for medium chain FA, it would be of interest to explore whether other tissues expressing both ghrelin and GOAT (e.g. brain, kidney, adipose tissue, small intestine, and large intestine) are able to synthesize additional ghrelin isoforms when animals are exposed to diets enriched in short- or long-chain FA.
Collectively this report provides a novel investigation of the comparison of in vitro vs. in vivo effects of synthetic ghrelin peptides of varied acyl-character. Our data demonstrate that acyl chains of differing length possess unique pharmacology and imply the existence of differential biological action profiles for naturally occurring acyl-ghrelin isoforms. These effects are specific to GHSR activation but may involve other receptor signaling systems as well. Importantly, our data demonstrate that C16-acylation is a novel approach to enhance and prolong the efficacy of pharmacologically administered ghrelin.
Acknowledgments
We thank E. Bartley, J. Hembree, J. Holland, and C. Raver for their excellent technical assistance.
This work was supported by grant support from the Hilda and Preston Davis Foundation (Davis Foundation Postdoctoral Fellowship Program for Eating Disorders) (to T.D.M). M.H.T. received NIH funding for this manuscript from the following grants: R01 DK077975 and R01 DK069987.
Disclosure Summary: The authors have nothing to declare.
Footnotes
- AgRP
- Agouti-related peptide
- AMPK
- AMP-activated protein kinase
- FA
- fatty acid
- GHSR
- GH-secretagogue receptor
- GOAT
- ghrelin O-acyltransferase
- icv
- intracerebroventricular
- IP
- inositol phosphate
- KO
- knockout
- NPY
- neuropeptide Y
- PVN
- paraventricular nucleus.
References
- 1. Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. 1999. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402:656–660 [DOI] [PubMed] [Google Scholar]
- 2. Date Y, Kojima M, Hosoda H, Sawaguchi A, Mondal MS, Suganuma T, Matsukura S, Kangawa K, Nakazato M. 2000. Ghrelin, a novel growth hormone-releasing acylated peptide, is synthesized in a distinct endocrine cell type in the gastrointestinal tracts of rats and humans. Endocrinology 141:4255–4261 [DOI] [PubMed] [Google Scholar]
- 3. Ariyasu H, Takaya K, Tagami T, Ogawa Y, Hosoda K, Akamizu T, Suda M, Koh T, Natsui K, Toyooka S, Shirakami G, Usui T, Shimatsu A, Doi K, Hosoda H, Kojima M, Kangawa K, Nakao K. 2001. Stomach is a major source of circulating ghrelin, and feeding state determines plasma ghrelin-like immunoreactivity levels in humans. J Clin Endocrinol Metab 86:4753–4758 [DOI] [PubMed] [Google Scholar]
- 4. Gutierrez JA, Solenberg PJ, Perkins DR, Willency JA, Knierman MD, Jin Z, Witcher DR, Luo S, Onyia JE, Hale JE. 2008. Ghrelin octanoylation mediated by an orphan lipid transferase. Proc Natl Acad Sci USA 105:6320–6325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang J, Brown MS, Liang G, Grishin NV, Goldstein JL. 2008. Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 132:387–396 [DOI] [PubMed] [Google Scholar]
- 6. Hosoda H, Kojima M, Mizushima T, Shimizu S, Kangawa K. 2003. Structural divergence of human ghrelin. Identification of multiple ghrelin-derived molecules produced by post-translational processing. J Biol Chem 278:64–70 [DOI] [PubMed] [Google Scholar]
- 7. Holst B, Cygankiewicz A, Jensen TH, Ankersen M, Schwartz TW. 2003. High constitutive signaling of the ghrelin receptor—identification of a potent inverse agonist. Mol Endocrinol 17:2201–2210 [DOI] [PubMed] [Google Scholar]
- 8. Holst B, Holliday ND, Bach A, Elling CE, Cox HM, Schwartz TW. 2004. Common structural basis for constitutive activity of the ghrelin receptor family. J Biol Chem 279:53806–53817 [DOI] [PubMed] [Google Scholar]
- 9. Petersen PS, Woldbye DP, Madsen AN, Egerod KL, Jin C, Lang M, Rasmussen M, Beck-Sickinger AG, Holst B. 2009. In vivo characterization of high Basal signaling from the ghrelin receptor. Endocrinology 150:4920–4930 [DOI] [PubMed] [Google Scholar]
- 10. Howard AD, Feighner SD, Cully DF, Arena JP, Liberator PA, Rosenblum CI, Hamelin M, Hreniuk DL, Palyha OC, Anderson J, Paress PS, Diaz C, Chou M, Liu KK, McKee KK, Pong SS, Chaung LY, Elbrecht A, Dashkevicz M, Heavens R, Rigby M, Sirinathsinghji DJ, Dean DC, Melillo DG, Patchett AA, Nargund R, Griffin PR, DeMartino JA, Gupta SK, Schaeffer JM, Smith RG, Van der Ploeg LH. 1996. A receptor in pituitary and hypothalamus that functions in growth hormone release. Science 273:974–977 [DOI] [PubMed] [Google Scholar]
- 11. Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK. 2006. Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol 494:528–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Castañeda TR, Tong J, Datta R, Culler M, Tschöp MH. 2010. Ghrelin in the regulation of body weight and metabolism. Front Neuroendocrinol 31:44–60 [DOI] [PubMed] [Google Scholar]
- 13. Nogueiras R, Tschöp MH, Zigman JM. 2008. Central nervous system regulation of energy metabolism: ghrelin versus leptin. Ann NY Acad Sci 1126:14–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cowley MA, Smith RG, Diano S, Tschöp M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML, Garcia-Segura LM, Nillni EA, Mendez P, Low MJ, Sotonyi P, Friedman JM, Liu H, Pinto S, Colmers WF, Cone RD, Horvath TL. 2003. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37:649–661 [DOI] [PubMed] [Google Scholar]
- 15. Briggs DI, Andrews ZB. 2011. Metabolic status regulates ghrelin function on energy homeostasis. Neuroendocrinology 93:48–57 [DOI] [PubMed] [Google Scholar]
- 16. Nakazato M, Murakami N, Date Y, Kojima M, Matsuo H, Kangawa K, Matsukura S. 2001. A role for ghrelin in the central regulation of feeding. Nature 409:194–198 [DOI] [PubMed] [Google Scholar]
- 17. Wang L, Saint-Pierre DH, Taché Y. 2002. Peripheral ghrelin selectively increases Fos expression in neuropeptide Y-synthesizing neurons in mouse hypothalamic arcuate nucleus. Neurosci Lett 325:47–51 [DOI] [PubMed] [Google Scholar]
- 18. Willesen MG, Kristensen P, Romer J. 1999. Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology 70:306–316 [DOI] [PubMed] [Google Scholar]
- 19. Fekete C, Sarkar S, Rand WM, Harney JW, Emerson CH, Bianco AC, Beck-Sickinger A, Lechan RM. 2002. Neuropeptide Y1 and Y5 receptors mediate the effects of neuropeptide Y on the hypothalamic-pituitary-thyroid axis. Endocrinology 143:4513–4519 [DOI] [PubMed] [Google Scholar]
- 20. Cone RD, Lu D, Koppula S, Vage DI, Klungland H, Boston B, Chen W, Orth DN, Pouton C, Kesterson RA. 1996. The melanocortin receptors: agonists, antagonists, and the hormonal control of pigmentation. Recent Prog Horm Res 51:287–317; discussion 318 [PubMed] [Google Scholar]
- 21. Wren AM, Small CJ, Ward HL, Murphy KG, Dakin CL, Taheri S, Kennedy AR, Roberts GH, Morgan DG, Ghatei MA, Bloom SR. 2000. The novel hypothalamic peptide ghrelin stimulates food intake and growth hormone secretion. Endocrinology 141:4325–4328 [DOI] [PubMed] [Google Scholar]
- 22. Wren AM, Seal LJ, Cohen MA, Brynes AE, Frost GS, Murphy KG, Dhillo WS, Ghatei MA, Bloom SR. 2001. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab 86:5992. [DOI] [PubMed] [Google Scholar]
- 23. Perez-Tilve D, Heppner K, Kirchner H, Lockie SH, Woods SC, Smiley DL, Tschöp M, Pfluger P. 2011. Ghrelin-induced adiposity is independent of orexigenic effects. FASEB J 25:2814–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tschöp M, Smiley DL, Heiman ML. 2000. Ghrelin induces adiposity in rodents. Nature 407:908–913 [DOI] [PubMed] [Google Scholar]
- 25. van der Lely AJ, Tschöp M, Heiman ML, Ghigo E. 2004. Biological, physiological, pathophysiological, and pharmacological aspects of ghrelin. Endocr Rev 25:426–457 [DOI] [PubMed] [Google Scholar]
- 26. Ohgusu H, Shirouzu K, Nakamura Y, Nakashima Y, Ida T, Sato T, Kojima M. 2009. Ghrelin O-acyltransferase (GOAT) has a preference for n-hexanoyl-CoA over n-octanoyl-CoA as an acyl donor. Biochem Biophys Res Commun 386:153–158 [DOI] [PubMed] [Google Scholar]
- 27. Kirchner H, Gutierrez JA, Solenberg PJ, Pfluger PT, Czyzyk TA, Willency JA, Schürmann A, Joost HG, Jandacek RJ, Hale JE, Heiman ML, Tschöp MH. 2009. GOAT links dietary lipids with the endocrine control of energy balance. Nat Med 15:741–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nishi Y, Hiejima H, Hosoda H, Kaiya H, Mori K, Fukue Y, Yanase T, Nawata H, Kangawa K, Kojima M. 2005. Ingested medium-chain fatty acids are directly utilized for the acyl modification of ghrelin. Endocrinology 146:2255–2264 [DOI] [PubMed] [Google Scholar]
- 29. Bednarek MA, Feighner SD, Pong SS, McKee KK, Hreniuk DL, Silva MV, Warren VA, Howard AD, Van Der Ploeg LH, Heck JV. 2000. Structure-function studies on the new growth hormone-releasing peptide, ghrelin: minimal sequence of ghrelin necessary for activation of growth hormone secretagogue receptor 1a. J Med Chem 43:4370–4376 [DOI] [PubMed] [Google Scholar]
- 30. Matsumoto M, Hosoda H, Kitajima Y, Morozumi N, Minamitake Y, Tanaka S, Matsuo H, Kojima M, Hayashi Y, Kangawa K. 2001. Structure-activity relationship of ghrelin: pharmacological study of ghrelin peptides. Biochem Biophys Res Commun 287:142–146 [DOI] [PubMed] [Google Scholar]
- 31. Tschöp M, Weyer C, Tataranni PA, Devanarayan V, Ravussin E, Heiman ML. 2001. Circulating ghrelin levels are decreased in human obesity. Diabetes 50:707–709 [DOI] [PubMed] [Google Scholar]
- 32. le Roux CW, Patterson M, Vincent RP, Hunt C, Ghatei MA, Bloom SR. 2005. Postprandial plasma ghrelin is suppressed proportional to meal calorie content in normal-weight but not obese subjects. J Clin Endocrinol Metab 90:1068–1071 [DOI] [PubMed] [Google Scholar]
- 33. Shiiya T, Nakazato M, Mizuta M, Date Y, Mondal MS, Tanaka M, Nozoe S, Hosoda H, Kangawa K, Matsukura S. 2002. Plasma ghrelin levels in lean and obese humans and the effect of glucose on ghrelin secretion. J Clin Endocrinol Metab 87:240–244 [DOI] [PubMed] [Google Scholar]
- 34. English PJ, Ghatei MA, Malik IA, Bloom SR, Wilding JP. 2002. Food fails to suppress ghrelin levels in obese humans. J Clin Endocrinol Metab 87:2984. [DOI] [PubMed] [Google Scholar]
- 35. Hansen TK, Dall R, Hosoda H, Kojima M, Kangawa K, Christiansen JS, Jorgensen JO. 2002. Weight loss increases circulating levels of ghrelin in human obesity. Clin Endocrinol (Oxf) 56:203–206 [DOI] [PubMed] [Google Scholar]
- 36. Pöykkö SM, Kellokoski E, Hörkkö S, Kauma H, Kesaniemi YA, Ukkola O. 2003. Low plasma ghrelin is associated with insulin resistance, hypertension, and the prevalence of type 2 diabetes. Diabetes 52:2546–2553 [DOI] [PubMed] [Google Scholar]
- 37. Cummings DE, Weigle DS, Frayo RS, Breen PA, Ma MK, Dellinger EP, Purnell JQ. 2002. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N Engl J Med 346:1623–1630 [DOI] [PubMed] [Google Scholar]
- 38. Pournaras DJ, le Roux CW. 2010. Ghrelin and metabolic surgery. Int J Pept 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Holst B, Zoffmann S, Elling CE, Hjorth SA, Schwartz TW. 1998. Steric hindrance mutagenesis versus alanine scan in mapping of ligand binding sites in the tachykinin NK1 receptor. Mol Pharmacol 53:166–175 [DOI] [PubMed] [Google Scholar]
- 40. Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, Roth RH, Sleeman MW, Picciotto MR, Tschöp MH, Gao XB, Horvath TL. 2006. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest 116:3229–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pfluger PT, Kirchner H, Günnel S, Schrott B, Perez-Tilve D, Fu S, Benoit SC, Horvath T, Joost HG, Wortley KE, Sleeman MW, Tschöp MH. 2008. Simultaneous deletion of ghrelin and its receptor increases motor activity and energy expenditure. Am J Physiol Gastrointest Liver Physiol 294:G610–G618 [DOI] [PubMed] [Google Scholar]
- 42. Wortley KE, Anderson KD, Garcia K, Murray JD, Malinova L, Liu R, Moncrieffe M, Thabet K, Cox HJ, Yancopoulos GD, Wiegand SJ, Sleeman MW. 2004. Genetic deletion of ghrelin does not decrease food intake but influences metabolic fuel preference. Proc Natl Acad Sci USA 101:8227–8232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tinsley FC, Taicher GZ, Heiman ML. 2004. Evaluation of a quantitative magnetic resonance method for mouse whole body composition analysis. Obes Res 12:150–160 [DOI] [PubMed] [Google Scholar]
- 44. Holst B, Brandt E, Bach A, Heding A, Schwartz TW. 2005. Nonpeptide and peptide growth hormone secretagogues act both as ghrelin receptor agonist and as positive or negative allosteric modulators of ghrelin signaling. Mol Endocrinol 19:2400–2411 [DOI] [PubMed] [Google Scholar]
- 45. De Vriese C, Gregoire F, Lema-Kisoka R, Waelbroeck M, Robberecht P, Delporte C. 2004. Ghrelin degradation by serum and tissue homogenates: identification of the cleavage sites. Endocrinology 145:4997–5005 [DOI] [PubMed] [Google Scholar]
- 46. Theander-Carrillo C, Wiedmer P, Cettour-Rose P, Nogueiras R, Perez-Tilve D, Pfluger P, Castaneda TR, Muzzin P, Schürmann A, Szanto I, Tschöp MH, Rohner-Jeanrenaud F. 2006. Ghrelin action in the brain controls adipocyte metabolism. J Clin Invest 116:1983–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pfluger PT, Castaneda TR, Heppner KM, Strassburg S, Kruthaupt T, Chaudhary N, Halem H, Culler MD, Datta R, Burget L, Tschöp MH, Nogueiras R, Perez-Tilve D. 2011. Ghrelin, peptide YY and their hypothalamic targets differentially regulate spontaneous physical activity. Physiol Behav 105:52–61 [DOI] [PubMed] [Google Scholar]
- 48. Rediger A, Piechowski CL, Yi CX, Tarnow P, Strotmann R, Grüters A, Krude H, Schöneberg T, Tschöp MH, Kleinau G, Biebermann H. 2011. Mutually opposite signal modulation by hypothalamic heterodimerization of ghrelin and melanocortin-3 receptors. J Biol Chem 286:39623–39631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kern A, Albarran-Zeckler R, Walsh HE, Smith RG. 2012. Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron 73:317–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Grossauer J, Kosol S, Schrank E, Zangger K. 2010. The peptide hormone ghrelin binds to membrane-mimetics via its octanoyl chain and an adjacent phenylalanine. Bioorg Med Chem 18:5483–5488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hamilton JA, Era S, Bhamidipati SP, Reed RG. 1991. Locations of the three primary binding sites for long-chain fatty acids on bovine serum albumin. Proc Natl Acad Sci USA 88:2051–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, Carling D, Small CJ. 2004. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem 279:12005–12008 [DOI] [PubMed] [Google Scholar]
- 53. López M, Lage R, Saha AK, Pérez-Tilve D, Vázquez MJ, Varela L, Sangiao-Alvarellos S, Tovar S, Raghay K, Rodríguez-Cuenca S, Deoliveira RM, Castañeda T, Datta R, Dong JZ, Culler M, Sleeman MW, Alvarez CV, Gallego R, Lelliott CJ, Carling D, Tschöp MH, Diéguez C, Vidal-Puig A. 2008. Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab 7:389–399 [DOI] [PubMed] [Google Scholar]
- 54. Lawrence CB, Snape AC, Baudoin FM, Luckman SM. 2002. Acute central ghrelin and GH secretagogues induce feeding and activate brain appetite centers. Endocrinology 143:155–162 [DOI] [PubMed] [Google Scholar]