

Abstract
Lipoxygenase enzymes initiate diverse signaling pathways by specifically directing oxygen to different carbons of arachidonate and other polyunsaturated acyl chains, but structural origins of this specificity have remained unclear. We therefore determined the nature of the lipoxygenase interaction with the polar-end of a paramagnetic lipid by electron paramagnetic resonance spectroscopy. Distances between selected grid points on soybean seed lipoxygenase-1 (SBL1) and a lysolecithin spin-labeled on choline were measured by pulsed (electron) dipolar spectroscopy. The protein grid was designed by structure-based modeling so that five natural side chains were replaced with spin labels. Pairwise distances in 10 doubly spin-labeled mutants were examined by pulsed dipolar spectroscopy, and a fit to the model was optimized. Finally, experimental distances between the lysolecithin spin and each single spin site on SBL1 were also obtained. With these 15 distances, distance geometry localized the polar-end and the spin of the lysolecithin to the region between the two domains in the SBL1 structure, nearest to E236, K260, Q264, and Q544. Mutation of a nearby residue, E256A, relieved the high pH requirement for enzyme activity of SBL1 and allowed lipid binding at pH 7.2. This general approach could be used to locate other flexible molecules in macromolecular complexes.
Introduction
Lipoxygenases are a family of enzymes of importance in development (1) and in exerting exquisite control over the first oxidative step in many unsaturated-lipid signaling pathways (2). Lipoxygenase protein structures, and sequences, are highly conserved in eukaryotes and some bacteria, yet how different family members direct oxygen stereospecifically to carbons 5, 8, 9, 11, 12, or 15 when, for instance, arachidonate is substrate, or to C-9 or -13 on linoleate, remains, to our knowledge, poorly understood. The site of oxidation is specific to the pathway that will subsequently be activated.
Much of the lipoxygenase structure is devoted to shaping a curved substrate channel that approaches the centrally located, active-site iron ion, with ends of the curve near different surface locations (3–10). Ideas about how similar lipoxygenase structures might result in different sites of polyunsaturated lipid oxidation include reversed placement in the cavity of the polar and methyl ends of the substrate, depth of substrate penetration into the cavity (11,12), and the nature of the entrance to the cavity (8). Progress has been made in identifying substrate and product electron density internally, near the nonheme iron site of catalysis, but the carboxyl and methyl ends of the fatty-acid chains are disordered and hard to distinguish in lipoxygenase crystals prepared at pH values where the fatty-acid carboxyl is protonated (9,13). Esters and lipids with polyunsaturated chains are also lipoxygenase substrates, suggesting that the substrate polar-end marks a surface region proximal to the cavity entrance(s) (14). The spectroscopic approach taken here, with spin labels, permits examination of affinity, dynamics, and location of the polar-end of a lipid tethered to lipoxygenase in solution, all at the pH optimum for catalysis. The major isoform of lipoxygenase in soybean seeds, SBL1 (optimum pH range 8–9), was chosen for the study. Many of the seminal ideas about substrate orientation in the cavity (11) and on lipoxygenase mechanism (15) are based on studies of SBL1. SBL1 is highly water-soluble and functions in solution, but some other lipoxygenases have membrane affinity.
We determined the location of a lipid spin in the protein structure by triangulation, or distance geometry, using distances measured between the lipid spin and multiple spin-labeled side chains introduced into the protein (16–18). Doing so required an accurate model of spin-labeled protein side chains in the protein structure. Construction of the model was facilitated by well-defined x-ray structures of four different isoforms of soybean lipoxygenase (3–5,19) and a program for spin-label modeling (20). Sequestered sites were selected for placement of spin-label side chains so that side-chain dihedrals closest to the backbone, χ1 and χ2, were modeled as those of the x-ray structures, and the ranges of χ3–χ5 conformers were constrained by neighboring residues.
Experimental distances between spins, placed by site-directed spin labeling on the protein, and the lipid spin were determined by pulsed electron paramagnetic resonance (EPR). Distances characteristic of the dimensions of macromolecules and assemblies (the ∼10–>80 Å range) are available from pulsed (electron) dipolar spectroscopy (PDS), a technique that includes double electron-electron resonance (DEER) (21–25) and double-quantum coherence (DQC) EPR (16,22,26). Diverse biomolecular structure problems have been solved by selected PDS distance measurements (see, for example, the literature (17,18,27–31)). Fewer examples of flexible ligand docking, used in our study, exist among PDS studies, but these include peptide antigen binding to MHC class I proteins (32), inhibitor binding to monoamine oxidase (33), and fatty-acid binding to HSA (34). Reversible binding of a paramagnetic ligand to a macromolecule is well suited to DEER and DQC because distances are measured with frozen solutions, and the fraction of ligand bound is therefore trapped in a unique location, whereas the unbound, or nonspecifically bound, fraction has no defined spatial relation to macromolecular sites and only contributes to background. Some disorder in the position of the bound ligand spin can be evaluated as long as the partner sites on the protein are well ordered.
Lipoxygenases accept variations in substrate, including acyl-chain length and number of double bonds as well as variations at the polar-end of the acyl chain (35–37). Saturated and monounsaturated acyl chains have been characterized as inhibitors (36–38). Doxyl stearate spin labels have previously been used to examine motion of the acyl chain of these substrate analogs when they bind to ferrous SBL1 in solution (39). The new substrate analog, lysooleoylphosphatidylTEMPOcholine (LOPTC, see below), is a lysooleoyllecithin with a spin label replacing one choline methyl. Lysolecithin with a linoleoyl chain is a known lipoxygenase substrate (35), so LOPTC satisfies the requirements of being an unreactive (monounsaturated chain) and spin-labeled substrate analog.
Materials and Methods
Chemicals
LOPTC (1-oleoyl-2-hydroxy-sn-glycero-3-phospho(TEMPO)choline) was prepared by custom synthesis by Avanti Polar Lipids (Alabaster, AL). The spin-label reagent ((1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl) methanethiosulfonate) was purchased from Toronto Research Chemicals (North York, Ontario, Canada).
Preparation of spin-labeled SBL1 proteins
A SBL1 expression construct was reported previously (14). Four mutations changing native Cys to Ser gave a cysteine-free construct (NoCys) for our study. Five new single Cys mutations, F270C, L480C, A569C, A619C, and F782C, were introduced. Plasmids were sequenced with a model No. 3130×l Genetic Analyzer (Applied Biosystems, Foster City, CA). Ten double mutants, all possible combinations of the introduced single sites, were also expressed in either BL21(DE3) or CodonPlus (Stratagene, La Jolla, CA) Escherichia coli cells. Mutant proteins were labeled in elution buffer (pH 7.2, with 1 mM sodium azide) from Ni-NTA Superflow (Qiagen, Valencia, CA) for 16 h at 4°C with excess spin-label reagent. The previous protein-purification scheme (14) was modified in that the HPLC size-exclusion step was replaced by a second DE-53 anion-exchange chromatography step, at pH 7.2. Chromatography materials, Nickel-NTA Superflow and DE-53 (Whatman International, Maidstone, England), were discarded after a single use. SBL1 has limited stability at pH ∼9, so samples for EPR were prepared in 0.02 M TRIS, pH 7.2, and diluted with 0.2 M buffer (±60% sucrose) at the appropriate pH just before use. (See Section S1 in the Supporting Material.)
Continuous-wave EPR spectroscopy
Solution samples were examined at 0–22°C and at 60 K on a model No. E600 X-band spectrometer (Bruker, Billerica, MA) equipped with an SHQE_R2 high sensitivity resonator, an ESR9/10 cryostat (Oxford Instruments, Concord, MA) with a CERNOX sensor (Lakeshore Cryotronics, Westerville, OH), and an ITC503 controller (Oxford Instruments).
Pulsed-dipolar EPR spectroscopy
DEER and DQC experiments were performed on a home-built pulse EPR spectrometer operating at 17.3 GHz and modified for PDS (40). Generally, four-pulse DEER experiments used a standard setup (24,41). The detection pulse sequence was applied at the lower-field edge of the nitroxide EPR spectrum, and the pump pulse was applied 70 MHz higher, in the center region (mI = 0) of the spectrum. The pump π-pulse width was 16 ns (20 or 32 ns in several cases), and the detection π/2−π−π pulse sequence used 16 ns π/2 and 32 ns π pulses. (See Section S2 in the Supporting Material.)
DQC was used to measure distances in some cases where higher resolution was required (42). Our spectrometer configuration permits microwave pulses with B1 of 45 Gauss. The six-pulse DQC sequence π/2−π−π/2−π−π/2−π (16,22,43), with π/2-pulses of 2 ns and π-pulses of 4 ns, was applied at ∼5 G below the central maximum of the nitroxide spectrum. The data-averaging time for the same signal/noise ratio was approximately an order-of-magnitude shorter for DQC than for optimized DEER.
All pulsed EPR measurements were conducted at 60 K with 10–15-μL samples with protein concentrations typically in the range 20–220 μM (2–22 mg/mL) overall. LOPTC concentrations were in the range 15–100 μM. Samples for pulsed EPR were, in most cases, in 0.1 M TRICINE, pH 8.4 (20°C; ∼8.9 at −2°C). The glassing agent chosen was 30% sucrose because glycerol alters EPR spectra of ferric lipoxygenase.
Distance geometry modeling
Distance geometry used in distance analysis was based on MATLAB R2011A Statistical Toolbox functions (The MathWorks, Natick, MA). Fifteen distances (see Results) were used to solve the distance geometry. Probability-density isosurfaces, used to visualize the volume occupied by the LOPTC spin, were constructed by application of the Monte Carlo method. The final densities were normalized to unity integral, and the isosurfaces corresponding to 1σ and 2σ (in one dimension for each spin-pair) were determined with MATLAB 3D-plotting tools. The centers, radii, and tilts of the resulting envelopes (nearly ellipsoids) that represent the results were used to render solutions into the PDB:1YGE structure.
Protein models and figure rendering
A Protein DataBank (PDB) file was prepared as input for modeling spin labels in the PDB:1YGE structure by addition of hydrogens (GROMACS 4.5.4) (44) and deletion of the first 146 residues at the N-terminus. The program PRONOX (20) removes waters and heteroatoms from the PDB file and was used for spin-label placement at mutation sites under selected van der Waals cutoffs. Figures were rendered with the softwares PyMOL (http://www.pymol.org), KaleidaGraph 4.1.3 (http://www.synergy.com), and MATLAB (The MathWorks).
Results
Characterization of the lipoxygenase-LOPTC interaction in solution
LOPTC is a lysolecithin with an oleoyl acyl chain. It forms micelles in solution, as is evident from a broad EPR spectrum at ambient temperature (Fig. 1, A and B, lower traces). In the presence of native SBL1 (from soybeans), NoCys, or R1 mutants, sharper LOPTC signals increase as protein concentration increases (Fig. 1 A for NoCys). The fraction of LOPTC bound to NoCys also increases with pH (Fig. 1 B) from none in phosphate (pH 6.8, not shown) to a modest amount at pH 7.2 (TRIS) and to near maximum at pH 9 (TRIS), consistent with what is known about the pH optimum of catalysis by SBL1 (pH ∼ 9) and specific buffer effects (11,15). The EPR line shapes of LOPTC bound to the protein at 22°C suggest motional averaging of the polar-end bearing the spin label, even though LOPTC is evidently tethered to the protein. An immobilized component also becomes more pronounced at 2°C (see the top inset in Fig. 1 B of derivative presentations of EPR spectra). No changes in bound LOPTC EPR line-shape were found when the N-terminal, 46-amino-acid histidine tag was removed from NoCys or when the buffer contained 1 mM calcium. The EPR spectrum of a frozen solution (60 K) of LOPTC bound to NoCys is readily simulated (45) with parameters characteristic of a magnetically dilute, immobilized nitroxide (gx,y,z = [2.0102, 2.0057, 2.0029], Ax,y,z = [6.5, 6.7, 36.5 G], and line-width (full width half-maximum) = 8.5 G). Also, LOPTC at 10–40 μM is an effective inhibitor of NoCys kinetics when linoleate substrate is 4–23 μM (see Fig. S1 in the Supporting Material, lower right). In this limited range of substrate concentration, a competitive-inhibition model fits the data reasonably well with Ki ∼ 20 μM.
Figure 1.
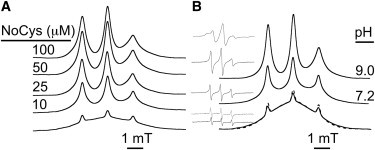
Solution electron paramagnetic resonance (EPR) spectra of 1-oleoyl-2-hydroxy-sn-glycero-3-phospho(TEMPO)choline (LOPTC) bound to cysteine-free construct (NoCys) lipoxygenase. Figures are shown as first integrals to emphasize differences between pure LOPTC micelles (lower traces) and varied amounts of LOPTC bound to protein (upper). (A) Constant LOPTC (30 μM) was titrated with increasing amounts of NoCys protein (in 0.1 M TRICINE, pH 9.0, 30% sucrose). (B) The fraction of LOPTC bound to NoCys was a function of pH. Total [LOPTC] was 60 μM, and NoCys was 100 μM (0.1 M TRIS, pH 9.0 or 7.2 as indicated). Micelle spectra at both pH values are shown at the bottom in panel B; the one with slightly increased monomer signal is at pH 9.0. (Insets) Derivative EPR spectra of each sample. The temperature in panels A and B was 295 K (except for the upper inset, for which the temperature was 275 K).
Establishing suitable spin-label sites in the lipoxygenase structure
The structure of SBL1 is known to high resolution (4), so by inspection and computer modeling, sequestered pockets can be chosen where a natural side chain could be replaced by spin-labeled cysteine. The strategy we took involves five sites for spin labeling SBL1 (Fig. 2 A). A convoluted cavity leading to the catalytic iron extends approximately from under the helix including spin-label site F270R1 (specifically from residue T259) to labeled site L480R1. The cavity is larger than one substrate molecule and it contains ordered water in the apoenzyme. The sites chosen for spin labeling are generally near residues conserved throughout the lipoxygenase family and could be studied in other variants. The one soybean isoform crystallized with a product fatty-acid bound (L-3) had a structure generally only 0.4 Å RMSD from the apo-form (13), so a reasonable expectation is that the chosen spin-label sites in SBL1 will vary little upon ligand binding, unless they are directly in contact with LOPTC.
Figure 2.
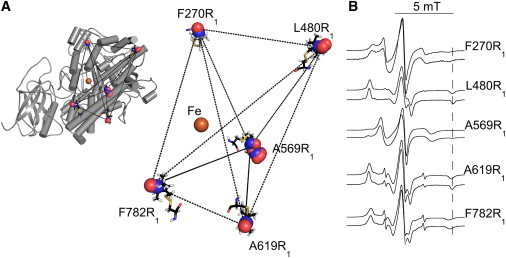
Experimental strategy. (A) Placement of the spin-labeled R1 side chain, replacing natural side chains, in the structure of soybean seed lipoxygenase-1 (SBL1) (PDB:1YGE). The coordinates of spin-labeled residues F270R1, L480R1, A519R1, A619R1, and F782R1 were generated with the software PRONOX. The allowed solutions for nitroxide oxygen (red) and nitrogen (blue) are illustrated (spheres), and one full nitroxide at each site is rendered (sticks). The catalytic iron ion is indicated (orange). Representative distances (lines) between spin labels to be measured by PDS. (Left) Placement of the spin-label sites in the overall SBL1 structure. Helices that contain <5 amino acids are simplified (loops), modifying PDB:1YGE from 46 to 26 helices. (Right) The calculated spin locations are enlarged. (B) Solution EPR spectra of SBL1 spin labeled at single sites. The maximum hyperfine separations, qualitative measures of R1 side-chain vary (verticle line drawn on the right side of the figure). Samples were at pH 8.4 (0.1 M TRICINE-HCl, 22°C), without (upper) and with (lower) 30% sucrose (w/v).
The four cysteines of native SBL1 are weakly reactive (3), and each was replaced with serine by mutagenesis (yielding NoCys). Single and double cysteine replacements were introduced, and each introduced cysteine was converted to a spin-labeled amino acid, named R1 by convention (46).
The large, catalytic domain of SBL1 (approximately residues 200–839) is predominantly α-helical. Selection criteria for mutants included placement of spin labels more than two residues from the ends of helices; near, but not on, the surface of the protein, natural side chains facing away from the protein center; and a distance ≥20 Å between the natural side-chain Cβ and the catalytic iron ion (to minimize relaxation). Spin-label mutations (and corresponding helix numbers) selected for this study were F270R1 (helix 2), L480R1 (helix 9), A569R1 (helix 13), A619R1 (helix 15), and F782R1 (helix 23 or, in some structures, helix 22, the last helix before the C-terminus). Alignment of residues in these helices in the four different isoforms of soybean lipoxygenase that have been crystallized is shown in Table S1 in the Supporting Material.
To examine the fit of spin-label mutations into the structure of SBL1 (with hydrogens added), we created models using the program PRONOX (20) and the x-ray structure of SBL1 (PDB:1YGE) (4), with additional input on torsion angles from the structures of other soybean lipoxygenase isoforms SBL3 (PDB:1RRH) (13), VLXB (PDB:2IUJ) (5), and VLXD (PDB:2IUK) (5). Initial torsion-angle inputs in PRONOX for χ1 and χ2, those closest to the backbone, were taken from PDB:1YGE for F270R1 and F782R1. For the two Ala mutation sites (569 and 619), values of χ1 (and χ2 for A619R1) were taken from structures of other isoforms having different amino-acid substitutions at corresponding positions (see Table S1). The disulfide-bond torsion angle, χ3, was set to either 90° or 270°, and increments of 30° or 15° in χ4 and χ5 were examined with each χ1−χ3 combination in a search for solutions having no clashes with protein atoms. Defining the clash criterion at 75% of interatomic van der Waals distances, with values of χ1−χ2 from soybean isoform x-ray structures, gave fits at all sites except F270R1. Two conformers of L480R1 resulted at this cutoff.
A fine search around χ1 and more relaxed clash criteria (64%) were required for a F270R1 model and resulted in a single fit with χ1 of 314°, compared to the PDB:1YGE value of 276.5°. A more stringent clash cutoff of 85% reduced the number of solutions to one set of χ1−χ3 for A619R1 and F782R1. In the case of A569R1, none of the isoforms had a side chain with a χ2, so gauche± and trans conformers were examined, and 10 solutions for A569R1 were obtained with the 85% cutoff. Fig. 2 A illustrates these PRONOX solutions for all five R1 sites in SBL1, and the calculated R1–R1 distances appear in Table 1. Two and 10 solutions for L480R1 and A569R1 respectively, and only one for the other sites, resulted from PRONOX modeling. Similar calculations for the residues that are Cys in native SBL1 revealed that only C357 could accommodate the R1 side chain, and then with the cutoff reduced to 60%, thus explaining the low reactivity of the native cysteines (3).
Table 1.
Experimental pulsed (electron) dipolar spectroscopy (PDS) and calculated PRONOX distances in doubly spin-labeled soybean seed lipoxygenase-1 mutants
| SDSL pair | PDS distance (Å)∗ | PRONOX distances (Å)† |
|---|---|---|
| 270-480 | 32.5 | 30.1 ± 0.5 |
| 270-569 | 37 | 37.6, 39.5 ± 0.2, 40.3 ± 0.9 |
| 270-619 | 47 | 48.5 |
| 270-782 | 38 | 41.8 |
| 480-569 | 37.5 | 37.4 ± 0.1, 39.6 ± 0.4, 39.9 ± 0.5 |
| 480-619 | 46.5 | 46.0 ± 0.5 |
| 480-782 | 52.5 | 52.0 ± 0.7 |
| 569-619 | 44 | 39.7, 41.3 ± 1.4, 49.9 ± 0.8 |
| 569-782 | 44/52 | 47.3, 48.7 ± 0.7, 48.9 ± 1.1 |
| 619-782 | 20/22 | 22.0 |
SDSL, site-directed spin label.
Experimental distances are the maxima in the distance distributions shown in Fig. 3. The corresponding maxima in Fig. 5 are LOPTC spin to F270R1 (23 and 30 Å), L480R1 (43 Å), A569R1 (38 Å), A619R1 (47 Å), and F782R1 (37.5 Å). The distance between residues A619R1 and F782R1 showed two close peaks, but a single value of 22 Å was used in calculations of the LOPTC spin location.
Calculated interspin distances are the average of the distance between N-N and O-O. Multiple acceptable PRONOX fits of the spin label at L480R1 and A569R1 were found, of 2 and 10, respectively. Average distances and their standard deviations are given for pairs involving these sites.
The extent of R1 side-chain motion, evident in continuous-wave EPR spectra recorded at 22°C (Fig. 2 B), ranged from immobile (L480R1) to very mobile (A569R1). The freedom of motion of the A569R1 side chain probably reflects the multiple conformers PRONOX was able to fit in this site. Because buffers for PDS measurements contained 30% w/v sucrose, Fig. 2 B also compares spectra for samples in the absence (upper) and presence (lower) of sucrose. The EPR spectra of spin-labeled mutants at pH 7.2, 8.4, and 9.0 were compared also (not shown). Only the F270R1 mutant had significant pH dependence of side chain mobility, the more mobile component being favored at higher pH.
The five singly spin-labeled constructs were characterized biochemically by enzyme kinetics and iron assay (see Table S2 and Fig. S1). Proteins from soybean, NoCys, and L480R1 had similar values of kcat (232–268 s−1), whereas kcat values were slightly lower for 569R1 and 782R1 mutants. Kinetic constants take into account iron occupancy, which was 78–87% of total protein (see Table S2). The F270R1 and A619R1 mutants exhibited the most altered kinetic properties; kcat and Km values were about half those of SBL1 from soybeans (see Table S2). Chiral HPLC analysis (14) of the product from F270R1 established that the reaction course at pH 9.0 was normal and gave ≥89% 13-S-hydroperoxyoctadecadienoic acid (and 11% racemic product).
Spin-labeled SBL1 as a five-point three-dimensional grid object
To establish experimental coordinates of spin-labeled side chains, we determined 10 pairwise distances in SBL1 double cysteine mutants, spin-labeled with two R1s, using four-pulse DEER and six-pulse DQC (see Section S2, Fig. S2, and Fig. S3 in the Supporting Material). Although the pH values at which x-ray structures of SBL1 were determined were low (7.0 and 5.6) (Boyington et al. (3) and Minor et al. (4), respectively), the pH optimum for substrate turnover is 8–9 (11). A compromise pH value of 8.4 (0.1 M TRICINE, 22°C) was therefore selected for most of the PDS measurements. The dependence of the primary echo amplitude on pulse repetition rate showed that R1 spins in SBL1 had spin-lattice relaxation time, T1, up to a factor of 2 shorter than those in various other spin-labeled proteins, indicating modest relaxation enhancement by the high-spin ferrous ion in SBL1. A higher repetition rate than usual was therefore possible, shortening signal-averaging time.
Background decays were measured for singly labeled SBL1 constructs under sample and instrumental conditions identical to those of the doubly labeled samples. In many cases, the background decay was not a simple exponential, as was also observed in related cases (23), partly because of spectral diffusion caused by ferrous iron. In these cases, low-order polynomial fits to the background were calculated. Distance distributions, P(r), were obtained by the Tikhonov regularization method followed by MEM refinement (47,48) (see Section S3 in the Supporting Material). The normalized, and background-subtracted, time-domain decays are shown for all 10 double mutants in Fig. 3. Corresponding distance distributions are given as inserts. Normalized amplitudes of dipolar signals by DEER from double mutants, after background subtraction, were compared to calculated theoretical values (verified in a large number of DEER measurements) and found to correspond to near 100% spin-label occupancy in most cases, but somewhat less for F270R1 paired with A569R1 or A619R1. To facilitate comparisons, we scaled DQC data in Fig. 3 to have the appearance of DEER data with typical modulation depth, but note that spin-labeling efficiencies are more difficult to determine by DQC because of the lack of convenient reference (see Section S3 in the Supporting Material).
Figure 3.
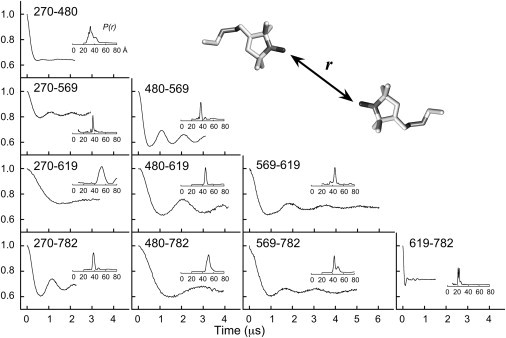
Time-domain double electron-electron resonance (DEER)/double-quantum coherence (DQC) data and reconstructed distance distributions, P(r), for doubly spin-labeled SBL1s. The residues involved in each measurement are specified simply by residue numbers in each subpanel. Most decays were obtained with DEER experiments, but those for A569R1–A619R1, A569R1–F782R1, and A619R1–F782R1 are from DQC experiments (all at 60 K). Decaying backgrounds in DEER data have been subtracted from the raw data, and the result normalized and shifted up to place the initial point at unity. DQC data, for better comparison with DEER data, were normalized to 1.0 at zero time and scaled down to nominal modulation depth, as modulation depth is not a relevant parameter for DQC (16,42). A 16-ns pump pulse was used to obtain all DEER data, except in the cases of 270/569 (32 ns) and 270/619 (20 ns). For 100% spin labeling efficiency, modulation depths will be 0.22, 0.31, and 0.36 when pump pulses are 32, 20, and 16 ns, respectively. Based on modulation depths, spin-labeling efficiency is 82–89% for the cases shown. Protein samples were 20–100 μM ± 5% in 0.1 M TRICINE-HCl (pH 8.4 at 22°C) with 30% sucrose (w/v).
Maxima of experimentally determined distance distributions were in very good agreement with the distances predicted from the PRONOX fits (Table 1), but some minor peaks, shoulders, and clearly bimodal distributions were seen, particularly for distances including 569R1 where distinct rotamers were predicted. Given the large set of 10 distances, these small features have negligible impact on the distance analyses discussed below in Distance Geometry to Locate the Polar-End of Lysolecithin Bound to SBL1.
Specificity of the lipoxygenase-LOPTC interaction
The modulation depth of calibrated DEER decays is a quantitative measure of the percentage of spin pairs. Various titrations of LOPTC with F270R1, A569R1, and A619R1 were examined, and generally, modulation depths were only slightly smaller than predicted from the total protein concentration, indicating that the majority of the protein does participate in binding. For example, at the modulation depth of the DEER signal at 60 K, a titration was made of F270R1 (50 μM total protein) with varying LOPTC (15.4–80 μM) (Fig. 4 A). The fraction, [LP]/[PTOT], of spin-labeled SBL1 [P] in complex [LP] with LOPTC was calculated with Eq. 1, obtained by considering the fractions of molecules with two spins that contribute to DEER modulation and those with a single spin that do not.
| (1) |
In this case, p′ is the normalized modulation depth, p′ = p/p0, where p is measured modulation depth and p0 is the maximal computed value (0.36) for the instrument and pulse sequence used. Other parameters in Eq. 1 are the spin-labeling efficiency f (∼0.95 in this example), and the ratio k of echo amplitudes of SBL1 to LOPTC in the DEER experiment, due to difference in their relaxation rates (k ∼0.7). Contributions to baselines from free LOPTC monomer are small and constant because the critical micelle concentration of LOPTC is low, and those from free micelles are very small in the range measured because only a small fraction of the broad micelle spectrum is excited and the molarity of micelles is also very low. Incorporation of iron in expressed SBL1 mutants was incomplete (78 ± 20% of total protein for F270R1; see Table S2). The results, shown as a Hill plot (Fig. 4 B), indicated single-site binding with Kd in the 40–80 μM range. The parameters f and k had little effect on the slope in the Hill plot, but they did affect the determination of Kd, which was also influenced to an unknown extent by the presence of ironless protein. In Fig. 4 B, the modulation-depth data are compared to a calculation for an effective concentration of F270R1 of 40 μM (80% of total protein) and Kd of 60 μM. The enzymatic Ki (∼20 μM) is not expected to be the same as a Kd measured in solution because the enzymatic reaction involves intermediates, including both ferric and ferrous states of the enzyme (38). These EPR experiments were all made with ferrous, resting enzyme. Clearly, the majority of F270R1 present contributed to the binding event measured by DEER, regardless of the uncertainties mentioned.
Figure 4.
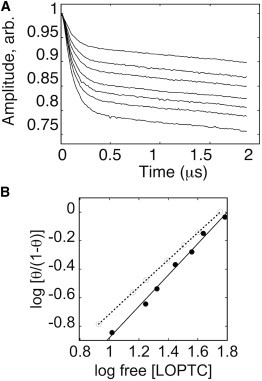
LOPTC binds to a unique site in SBL1. (A) DEER data were recorded at 60 K for F270R1 (50 μM) and varied amounts of LOPTC (15.4–80 μM, top to bottom). (B) The modulation depth of decays in panel A was compared to a calibrated standard for determination of the fraction of the spins giving a defined dipolar interaction between F270R1 and bound LOPTC. Experimental results are presented as a Hill plot (solid circles, linear fit of slope 1.1, solid line), with LOPTC concentrations in μM. These data are compared to a plot calculated with Kd of 60 μM, on the assumption that 80% of the protein (40 μM) contributed to the dipolar interaction.
Distance geometry to locate the polar-end of lysolecithin bound to SBL1
To determine, by PDS, the protein-docked location of the polar-end of the lipoxygenase substrate analog, LOPTC, we determined the five distances between the spin on the lipid and each of the five protein R1 sites. Distance distributions, P(r), between the LOPTC spin and each of the single protein R1 sites were determined from DEER measurements (Fig. 5 A). The narrowness of the distributions, and different distances for each pair, indicated uniqueness of the LOPTC site. The distance between F782R1 and the spin of 8-doxylstearic acid (8DSA) was also determined (Fig. 5 B). The affinity of this spin-labeled fatty acid for native SBL1, measured previously in Wu and Gaffney (39), is 31 μM at room temperature. The 8DSA spin was closer to F782R1 (30 Å) than was the LOPTC spin (37.5 Å). This distance is consistent with, but not proof that, the acyl chain of LOPTC and the 8DSA chain enter the SBL1 cavity at a similar location. One distance pair, L480R1–A619R1, had the same distribution with and without excess unlabeled lysooleoyllecithin, indicating that ligand binding did not induce a large conformational change, at least for this pair of sites. For another pair, F270R1–L480R1, some small deviations could be seen, but they were too small to have substantial effect on the distance analyses described below.
Figure 5.
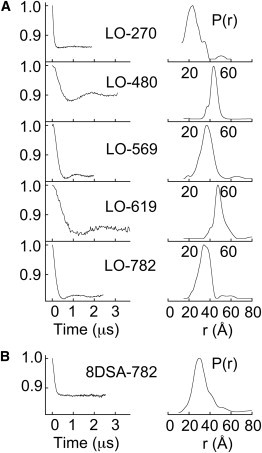
Distance determinations from the LOPTC spin to spins on SBL1. (A) Five single R1 SBL1 mutants (200 μM) with 0.8 equivalents of LOPTC, in TRICINE (0.1 M, pH 8.4 at 22°C), sucrose 30% (w/v), were examined by DEER, at 60 K. The maxima in the P(r) distributions are given in the footnote of Table 1. (B) Similar experiments measuring the distance between F782R1 and 8DSA, as the substrate analog, yielded a P(r) maximum at ∼30 Å. The 8-doxylstearic acid (8DSA) was added from an ethanol solution giving 1% ethanol in the final sample.
The coordinates of the LOPTC spin, relative to those of the R1 side chains, were determined from the complete set of 10 R1–R1 protein distances (Table 1) and the five LOPTC-R1 distances (Table 1 footnote). The peaks of the probability distributions (Figs. 3 and 5) were chosen as the experimental spin-spin distances. The P(r) distributions have significant second peaks for F270R1-LOPTC and A569R1-F782R1, and these were included in a general approach to determining locations of all spins that takes widths of the P(r) values into account. The steps of the approach are elaborated in Section S4 in the Supporting Material, and representative equations, vectors, and matrices resulting from the analysis are given in Eqs. S4–S12 in the Supporting Material. Briefly, all 15 experimental distances were embedded in a Euclidian space, R3, by implementation of a classic, metric-matrix distance geometry (DG) (because the distance set was complete) (49–51). The solution by DG was a polyhedron with five vertices corresponding to reference R1 sites and the sixth occupied by the NO moiety of LOPTC. Subsequent Procrustes analysis (PA) found the affinity transformation (rotation and translation only were allowed) that superimposed reference sites from DG onto those from the PRONOX model, thereby bringing the LOPTC spin to its location in the crystal structure of SBL1. The dissimilarity (see Eq. S10 in the Supporting Material) between the model and experimental grids was small (d = 0.0093), as required to locate the space occupied by the LOPTC spin. The widths of the R1–R1 distributions, in one dimension along the interspin distance, were incorporated into 106 Monte Carlo trials to define this space at 1σ and 2σ (see Section S4 in the Supporting Material).
The solutions place the spin of LOPTC just external to helices 2 and 11 (Fig. 6, cyan shape). The solution volume is close in shape to an ellipsoid. The ellipsoid shown in Fig. 6 is a smoothed representation of what in reality is a more intricate shape. The solution has limits imposed by the reconstruction of P(r) values, side-chain modeling, distance geometry, and PA, as well as any small perturbations to SBL1 structure due to substrate binding or changes in pH. How individual P(r) values are correlated is unknown; we addressed this problem partially by bounding the stress in DG and the disparity in PA. More importantly, the side-chain modeling was accurate enough (Table 1) and distributions were narrow enough (Fig. 3) to introduce only a very small error between the PRONOX model and DG/PA solution. The main contribution to location volumes is therefore the conformational range of the polar-end of LOPTC (which was tested by substitution of narrow distributions in distances to the spin of LOPTC). In the absence of such flexibility, the 2σ envelope of the LOPTC spin location was estimated to be <2 Å.
Figure 6.
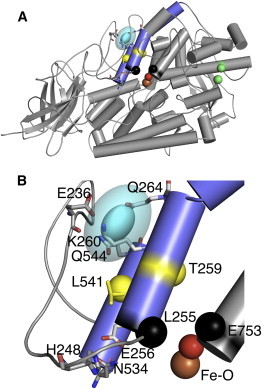
The LOPTC spin placed in the SBL1 structure (PDB:1YGE). (A) The experimental PDS solution for the location of the LOPTC spin is superimposed on the overall structure of SBL1(helices 2 and 11, slate gray). Helix 2 is separated into two portions by P268. The experimental LOPTC spin location (cyan ellipsoid) of radii 1.2, 2.3, and 3.0 Å, corresponding to the 1σ confidence level (distance distributions in one dimension), and an outer ellipsoid with radii twice those as an approximation of the 2σ confidence level. The coordinates of the center of the ellipsoid are 30.4, 62.2, and 12.2 Å. The catalytic Fe-water are colored by atom. The α-carbon atoms of side-chain pairs blocking possible entrances (3,4) into the cavity are show as colored spheres (yellow for T259 and L541, black for L255 and E753, and green for M341 and L480). (B) A portion of Fig. 6A (residues 213–276, 534–546, 750–756, and 840-841 (Fe-water)) is enlarged to provide details of the pocket in which the LOPTC spin resides (same orientation as in panel A). The loop region, composed of amino acids 213–254 (smooth loop) shows only the E236 side chain. Residues closest to the ellipsoid (cyan) are labeled (E236, K260, Q264, and Q544). (Lower region) E256 oxygens (helix 2) form a salt bridge with H248 (loop) and a hydrogen bond with N534 (helix 11). The orientation of SBL1 in this figure is related to that in Fig. 2 by rotations of the viewing direction by ∼40° about x and 30° about z.
Fig. 6 B is an enlargement showing features of a pocket surrounding the LOPTC spin position, a pocket that is formed largely by the loop preceding helix 2. Protein residues closest to the spin of docked LOPTC include E236 (loop), K260, Q264 (helix 2), and Q544 (helix 11). Suitable entrances to the large internal cavity of SBL1 have been carefully scrutinized (4). The α-carbons of side-chain pairs blocking entry are illustrated in Fig. 6 as colored spheres, and residues T259 and L541 (yellow) are closest to our solution for the LOPTC spin. Models of linoleic acid extending from this entrance to the catalytic iron have been published (14,39). Another entrance between L255 and E753 (black alpha carbon atoms (CA)) was also considered (4), but is less consistent with our data.
The possibilities that a fraction of bound LOPTC adopts a head-first orientation with the polar-end inside the cavity, or docks at the opposite side of the structure, between M341 and L480 (green CA atoms in Fig. 6 A), were found unreasonable because these possibilities would result in a short distance from the LOPTC spin to L480R1. No evidence indicated a short distance in the LOPTC/L480R1 P(r), even in the high signal/noise data of the early part of the DEER signal (see Fig. S4 and Fig. S5).
Mutation of E256 enhanced pH 7 binding of LOPTC to SBL1
Binding of LOPTC to SBL1 (NoCys) at pH 9.0 was greater than twofold higher than binding at pH 7.2 (Fig. 1 B), implying that the substrate cavity is less open at the lower pH. Because a triad of interacting side chains unique to the SBL1 isoform (19) forms the base of the pocket surrounding the location found for the LOPTC spin, the SBL1 mutant E256A in this triad was characterized. Glu256 (helix 2) forms a salt bridge to H248 (loop) and a hydrogen bond to N534 (helix 11) in the native SBL1 structure. Alanine is the natural residue in the equivalent position of the VLXB isoform (see Table S1) (5). Binding of LOPTC to the E256A mutant, as determined by continuous-wave EPR, increased 2.4-fold at pH 7.2 over that to NoCys. The enzyme-specific activity of E256A at pH 7.2 was also twice that of NoCys at the same pH, implying that the mutation rendered the cavity more open.
Discussion
These pulsed dipolar EPR distance measurements placed the polar-end of a paramagnetic lysolipid, LOPTC, in a surface pocket formed by a loop preceding helix 2 and adjacent helices 2 and 11 in lipoxygenase (SBL1). The 15 distances yielded, at one standard deviation, a convex envelope resembling an ellipsoid with radii (at 1 σ) 1.2, 2.3, and 3.0 Å (Fig. 6). The LOPTC spin is nearest a possible entrance to the internal substrate cavity that is blocked by T259 and L541 (yellow CA in Fig. 6 A) and is too far from the pair M341-L480 (green CA in Fig. 6 A) for that entry to be considered. A pair of side chains, L255 and E753 (black CA), discussed earlier (4) as a point where the cavity might reach the surface, is a less likely entrance for LOPTC. The location we find for the spin of LOPTC speaks to years of discussion of how substrates enter a lipoxygenase cavity (3–5); recent focus has been on the importance of localized changes in helix 2 of another 12/15 (arachidonate substrate) lipoxygenase (6). The possible significance of the pre-helix 2 loop was suggested when structures of the isoforms SBL1 and SBL3 were first compared (19). A triad of side chains, H248, E256, and N534, interact to connect the loop and the N-termini of helices 2 and 11 in SBL1 (Fig. 6). Indeed, mutant E256A converts SBL1 to the residue found in the VLXB isoform, and leads to increased enzyme activity of SBL1 at pH 7.2, similar to that of VLXB. The increase in enzyme activity at pH 7.2, of E256A, explains the origin of the uniquely high pH optimum for activity of SBL1.
An advantage of using a spin-labeled lysolecithin in this study was that transfer from micelle to protein was more facile than if a phospholipid spin label were used. EPR spectra of LOPTC in solution with SBL1 made immediately evident that the motion of the polar-end of this lysolecithin spin label is only modestly restricted when it is bound to the protein at ambient temperature (Fig. 1). The affinity of LOPTC for the protein, measured by modulation depth or by enzyme kinetics, is consistent with known affinities in tens of micromolar for substrate (15) and a variety of fatty-acid weak inhibitors (36,37). An earlier study using fatty acids with spin labels (doxyl stearates, DSAs) showed more restricted mobility of the acyl chain when the spin was on carbon 8 than when it was on C-5 (39). Together, these spin-label studies present a picture of the acyl chains being motionally restricted by binding to the lipoxygenase cavity while the more polar-ends sample multiple conformations and environments. During catalysis, the cis, cis pentadiene of the substrate rearranges to a cis, trans tetradiene moiety of the product. This change and oxygenation of the substrate must result in rearrangement or twist of the chain in the cavity, in preparation for product release. These motions probably propagate to the polar-end and would be facilitated by mobile headgroup locations.
A number of proposals for gated access to other lipoxygenase substrate cavities have been discussed, on the basis of x-ray structures of these proteins. In three cases a cavity entrance is wide open: reticulocyte lipoxygenase without inhibitor bound (6), leukocyte mini 12-lipoxygenase (lacking the N-terminal domain) with an inhibitor (52), and a stable human 5-lipoxygenase phosphomimetic mutant with arachidonic acid bound in reversed orientation (9). Structure in the helices (11,12) protecting the active site was lost upon substrate binding in the lattermost study. All of these cases point to a role for helices 2 and 11–12 in gating the active site cavity. Other lipoxygenase structures, including those of soybean isoforms (5), appear to have more closed cavities. Our finding, that a salt bridge/H-bond network involving the loop before helix 2 and the N-termini of helices 2 and 11 is responsible for the high pH optimum of SBL1, adds pH to the forms of gating. In this case, deprotonating H248 apparently switches the cavity access area from closed to more open.
Other soybean lipoxygenase isoforms lack the glutamate (E256) component of the network, and these isoforms have wider openings between helices 2 and 11. For instance, the separations of the backbones of these helices differ by ∼2 Å in an overlay of the structures of VLXB (5) and SBL1 (4), but the network stabilizing SBL1 may simply be one way of maintaining the closed state, which is accomplished by bulky hydrophobic side chains instead in other soybean isoforms (5). The recent analysis of the 11R-lipoxygenase from a coral, Gersemia fruticosa (10), led to discovery of a highly conserved π-cation pair that is positioned so that it might regulate possible cavity entrances in that, and other, enzyme(s). The corresponding residues in SBL1 are W130 and R242. This bridge from the N-terminal domain to the pre-helix 2 loop might be destabilized by a substrate with a bulky headgroup docking in the location we have found for LOPTC. These considerations, and the spectroscopic results for the LOPTC location, help pinpoint regions of the structures where future mutational/spectroscopic studies would be useful in identifying conformational changes associated with substrate binding.
The sites chosen as grid points for the SBL1-pulsed dipolar EPR structure representation are also strategically placed to facilitate future studies. Residue 270 is near the C-terminal end of helix 2, the helix that has two side chains in close proximity to the LOPTC location. Three sites are in conserved regions (see Table S1) of known lipoxygenase structures: L480R1, A619R1, and F782R1. Residues 619 and 782 are within the two long helices running antiparallel to each other on one side of lipoxygenase structures. Helix 15, containing residue A619 in SBL1, has five conserved residues, and the positional equivalent of F782 in helix 23 is followed by highly conserved equivalents of F785 and L789 in diverse members of the lipoxygenase family. Residue 480 follows W in the characteristic WXXAK sequence of lipoxygenases. Residue 480 is toward the beginning of long helix 9, which runs through the center, and iron-binding region, of lipoxygenase structures. The remaining residue, 569, is in a triad of short helices, α-11 to α-13, containing sites influencing product stereochemistry (14) and oxygen access to the active site (53). This group of helices is completely altered by substrate binding to a 5-lipoxygenase mutant (9). Residues 270 and 569 are therefore well positioned for future experiments examining functional changes in lipoxygenase. The SBL1 triangulation grid, constructed with these five sites, could be extended, as needed, by the addition of other sites likely to be sensitive to protein structural changes that may occur on substrate binding or other states of the enzyme.
Applications of PDS to animal lipoxygenases may depend on further development of site-directed spin labeling based on genetically encoded unnatural amino acids (54) because these lipoxygenases have 10–30 natural Cys. Of crystallized lipoxygenases, those from coral and bacterial sources have five or fewer cysteines, as do the soybean isoforms.
Conclusions
A spin-labeled lysolecithin (LOPTC) docked on SBL1 in a defined region, even though EPR spectra of the docked lipid demonstrated that the polar-end had considerable conformational flexibility at 22°C. The distances between five spin-label sites on the protein were determined with high accuracy by PDS, so that, after additional distances from these sites to the spin of LOPTC were measured, the uncertainty in the location and volume obtained for the LOPTC spin was attributed largely to conformers of the polar-end of LOPTC that were trapped by freezing. The location found for the LOPTC spin, external to helices 2 and 11, provides experimental support for suggestions that lipoxygenase substrates for SBL1 dock with a polar-end external to these helices and eliminates the opposite side of the enzyme as an entry site for this lipid.
The proximity of E236, in the loop preceding helix 2, to the LOPTC spin focuses attention on a pocket formed by this pre-helix 2 loop, covering the external surfaces of helices 2 and 11. Within the pocket, four polar residues were nearest neighbors of the LOPTC spin. Their proximity to the LOPTC headgroup implies that residues E236, K260, Q264, and Q544 must move when a substrate is bound to SBL1. Lower in the pocket, the hydrophobic residues I257 and L541 (Fig. 6) are positioned to direct a fatty-acid chain to a suggested (4) entrance between the helices. At the bottom of the pocket, the SBL1 isoform has three residues linking the loop and both N-terminal ends of helices 2 and 11 by a salt bridge and H-bond (19). Although not an original aim of this study, examination of the spectroscopic results also led to the finding that mutant E256A abolished the high pH optimum for activity in SBL1.
A distance-geometry approach was adapted to assign the location of the polar-end of LOPTC on the protein surface. The approaches developed here could be applied similarly to pinpoint the location and volume occupied by other flexible small molecules, for instance a peptide, cofactor, or inhibitor, in a macromolecular complex.
Acknowledgments
Avanti Polar Lipids kindly prepared LOPTC for this study. We also are grateful to A. Ozarowska for technical help with protein preparations and to Elka R. Georgieva for help with sample preparations for pulse EPR.
This study was supported by the National Institutes of Health under grant No. GM065268 and the American Recovery and Reinvestment Act supplement to B.J.G., and National Institutes of Health/National Institute of General Medical Sciences grant No. P41GM103521 and National Institutes of Health/National Center for Research Resources grant No. RR016292 to J.H.F.
Footnotes
Fayi Wu’s present address is Department of Molecular and Cellular Pharmacology, University of Miami Miller School of Medicine, 1600 NW 10th Ave., Miami, FL 33136.
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Haas U., Raschperger E., Haeggström J.Z. Targeted knock-down of a structurally atypical zebrafish 12S-lipoxygenase leads to severe impairment of embryonic development. Proc. Natl. Acad. Sci. USA. 2011;108:20479–20484. doi: 10.1073/pnas.1117094108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rådmark O., Werz O., Samuelsson B. 5-Lipoxygenase: regulation of expression and enzyme activity. Trends Biochem. Sci. 2007;32:332–341. doi: 10.1016/j.tibs.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 3.Boyington J.C., Gaffney B.J., Amzel L.M. The three-dimensional structure of an arachidonic acid 15-lipoxygenase. Science. 1993;260:1482–1486. doi: 10.1126/science.8502991. [DOI] [PubMed] [Google Scholar]
- 4.Minor W., Steczko J., Axelrod B. Crystal structure of soybean lipoxygenase L-1 at 1.4 Å resolution. Biochemistry. 1996;35:10687–10701. doi: 10.1021/bi960576u. [DOI] [PubMed] [Google Scholar]
- 5.Youn B., Sellhorn G.E., Kang C. Crystal structures of vegetative soybean lipoxygenase VLX-B and VLX-D, and comparisons with seed isoforms LOX-1 and LOX-3. Proteins. 2006;65:1008–1020. doi: 10.1002/prot.21182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choi J., Chon J.K., Shin W. Conformational flexibility in mammalian 15S-lipoxygenase: reinterpretation of the crystallographic data. Proteins. 2008;70:1023–1032. doi: 10.1002/prot.21590. [DOI] [PubMed] [Google Scholar]
- 7.Neau D.B., Gilbert N.C., Newcomer M.E. The 1.85 Å structure of an 8R-lipoxygenase suggests a general model for lipoxygenase product specificity. Biochemistry. 2009;48:7906–7915. doi: 10.1021/bi900084m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gilbert N.C., Bartlett S.G., Newcomer M.E. The structure of human 5-lipoxygenase. Science. 2011;331:217–219. doi: 10.1126/science.1197203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gilbert N.C., Rui Z., Newcomer M.E. Conversion of human 5-lipoxygenase to a 15-lipoxygenase by a point mutation to mimic phosphorylation at Serine-663. FASEB J. 2012;26:3222–3229. doi: 10.1096/fj.12-205286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eek P., Järving R., Samel N. Structure of a calcium-dependent 11R-lipoxygenase suggests a mechanism for Ca2+ regulation. J. Biol. Chem. 2012;287:22377–22386. doi: 10.1074/jbc.M112.343285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gardner H.W. Soybean lipoxygenase-1 enzymically forms both (9S)- and (13S)-hydroperoxides from linoleic acid by a pH-dependent mechanism. Biochim. Biophys. Acta. 1989;1001:274–281. doi: 10.1016/0005-2760(89)90111-2. [DOI] [PubMed] [Google Scholar]
- 12.Coffa G., Schneider C., Brash A.R. A comprehensive model of positional and stereo control in lipoxygenases. Biochem. Biophys. Res. Commun. 2005;338:87–92. doi: 10.1016/j.bbrc.2005.07.185. [DOI] [PubMed] [Google Scholar]
- 13.Skrzypczak-Jankun E., Bross R.A., Funk M.O., Jr. Three-dimensional structure of a purple lipoxygenase. J. Am. Chem. Soc. 2001;123:10814–10820. doi: 10.1021/ja011759t. [DOI] [PubMed] [Google Scholar]
- 14.Coffa G., Imber A.N., Brash A.R. On the relationships of substrate orientation, hydrogen abstraction, and product stereochemistry in single and double dioxygenations by soybean lipoxygenase-1 and its Ala542Gly mutant. J. Biol. Chem. 2005;280:38756–38766. doi: 10.1074/jbc.M504870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Glickman M.H., Klinman J.P. Nature of rate-limiting steps in the soybean lipoxygenase-1 reaction. Biochemistry. 1995;34:14077–14092. doi: 10.1021/bi00043a013. [DOI] [PubMed] [Google Scholar]
- 16.Borbat P.P., McHaourab H.S., Freed J.H. Protein structure determination using long-distance constraints from double-quantum coherence ESR: study of T4 lysozyme. J. Am. Chem. Soc. 2002;124:5304–5314. doi: 10.1021/ja020040y. [DOI] [PubMed] [Google Scholar]
- 17.Borbat P.P., Surendhran K., McHaourab H.S. Conformational motion of the ABC transporter MsbA induced by ATP hydrolysis. PLoS Biol. 2007;5:e271. doi: 10.1371/journal.pbio.0050271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park S.Y., Borbat P.P., Crane B.R. Reconstruction of the chemotaxis receptor-kinase assembly. Nat. Struct. Mol. Biol. 2006;13:400–407. doi: 10.1038/nsmb1085. [DOI] [PubMed] [Google Scholar]
- 19.Skrzypczak-Jankun E., Amzel L.M., Funk M.O., Jr. Structure of soybean lipoxygenase L3 and a comparison with its L1 isoenzyme. Proteins. 1997;29:15–31. [PubMed] [Google Scholar]
- 20.Hatmal M.M., Li Y., Haworth I.S. Computer modeling of nitroxide spin labels on proteins. Biopolymers. 2012;97:35–44. doi: 10.1002/bip.21699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milov A.D., Ponomarev A.B., Tsvetkov Y.D. Electron-electron double resonance in electron spin echo: model biradical systems and the sensitized photolysis of decalin. Chem. Phys. Lett. 1984;110:67–72. [Google Scholar]
- 22.Borbat P.P., Freed J.H. Measuring distances by pulsed dipolar ESR spectroscopy: spin-labeled histidine kinases. Methods Enzymol. 2007;423:52–116. doi: 10.1016/S0076-6879(07)23003-4. [DOI] [PubMed] [Google Scholar]
- 23.Schiemann O., Prisner T.F. Long-range distance determinations in biomacromolecules by EPR spectroscopy. Q. Rev. Biophys. 2007;40:1–53. doi: 10.1017/S003358350700460X. [DOI] [PubMed] [Google Scholar]
- 24.Jeschke G., Polyhach Y. Distance measurements on spin-labeled biomacromolecules by pulsed electron paramagnetic resonance. Phys. Chem. Chem. Phys. 2007;9:1895–1910. doi: 10.1039/b614920k. [DOI] [PubMed] [Google Scholar]
- 25.Pannier M., Veit S., Spiess H.W. Dead-time free measurement of dipole-dipole interactions between electron spins. J. Magn. Reson. 2000;142:331–340. doi: 10.1006/jmre.1999.1944. [DOI] [PubMed] [Google Scholar]
- 26.Borbat P.P., Freed J.H. Double-quantum ESR and distance measurements. In: Berliner L.J., Eaton G.R., Eaton S.S., editors. Vol. 19. Kluwer Academics/Plenum Publishers; New York: 2000. pp. 383–459. (Biological Magnetic Resonance). [Google Scholar]
- 27.Bhatnagar J., Borbat P.P., Crane B.R. Structure of the ternary complex formed by a chemotaxis receptor signaling domain, the CheA histidine kinase, and the coupling protein CheW as determined by pulsed dipolar ESR spectroscopy. Biochemistry. 2010;49:3824–3841. doi: 10.1021/bi100055m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schultz K.M., Merten J.A., Klug C.S. Characterization of the E506Q and H537A dysfunctional mutants in the E. coli ABC transporter MsbA. Biochemistry. 2011;50:3599–3608. doi: 10.1021/bi101666p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vishnivetskiy S.A., Francis D., Gurevich V.V. The role of arrestin α-helix I in receptor binding. J. Mol. Biol. 2010;395:42–54. doi: 10.1016/j.jmb.2009.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim S., Brandon S., Beth A.H. Determination of structural models of the complex between the cytoplasmic domain of erythrocyte band 3 and ankyrin-R repeats 13-24. J. Biol. Chem. 2011;286:20746–20757. doi: 10.1074/jbc.M111.230326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roessler M.M., King M.S., Hirst J. Direct assignment of EPR spectra to structurally defined iron-sulfur clusters in complex I by double electron-electron resonance. Proc. Natl. Acad. Sci. USA. 2010;107:1930–1935. doi: 10.1073/pnas.0908050107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Herget M., Baldauf C., Bordignon E. Conformation of peptides bound to the transporter associated with antigen processing (TAP) Proc. Natl. Acad. Sci. USA. 2011;108:1349–1354. doi: 10.1073/pnas.1012355108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Upadhyay A.K., Borbat P.P., Edmondson D.E. Determination of the oligomeric states of human and rat monoamine oxidases in the outer mitochondrial membrane and octyl β-D-glucopyranoside micelles using pulsed dipolar electron spin resonance spectroscopy. Biochemistry. 2008;47:1554–1566. doi: 10.1021/bi7021377. [DOI] [PubMed] [Google Scholar]
- 34.Junk M.J., Spiess H.W., Hinderberger D. Characterization of the solution structure of human serum albumin loaded with a metal porphyrin and fatty acids. Biophys. J. 2011;100:2293–2301. doi: 10.1016/j.bpj.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang L.S., Kim M.R., Sok D.E. Linoleoyl lysophosphatidylcholine is an efficient substrate for soybean lipoxygenase-1. Arch. Biochem. Biophys. 2006;455:119–126. doi: 10.1016/j.abb.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 36.Zhu Z.Y., Funk M.O. Lipoxygenase-1 inhibition with a series of half-product analogs. Bioorg. Chem. 1996;24:95–109. [Google Scholar]
- 37.Mogul R., Johansen E., Holman T.R. Oleyl sulfate reveals allosteric inhibition of soybean lipoxygenase-1 and human 15-lipoxygenase. Biochemistry. 2000;39:4801–4807. doi: 10.1021/bi992805t. [DOI] [PubMed] [Google Scholar]
- 38.Moody J.S., Marnett L.J. Kinetics of inhibition of leukocyte 12-lipoxygenase by the isoform-specific inhibitor 4-(2-oxapentadeca-4-yne)phenylpropanoic acid. Biochemistry. 2002;41:10297–10303. doi: 10.1021/bi020320q. [DOI] [PubMed] [Google Scholar]
- 39.Wu F., Gaffney B.J. Dynamic behavior of fatty acid spin labels within a binding site of soybean lipoxygenase-1. Biochemistry. 2006;45:12510–12518. doi: 10.1021/bi061415l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borbat P.P., Crepeau R.H., Freed J.H. Multifrequency two-dimensional Fourier transform ESR: an X/Ku-band spectrometer. J. Magn. Reson. 1997;127:155–167. doi: 10.1006/jmre.1997.1201. [DOI] [PubMed] [Google Scholar]
- 41.Georgieva E.R., Ramlall T.F., Eliezer D. The lipid-binding domain of wild type and mutant α-synuclein: compactness and interconversion between the broken and extended helix forms. J. Biol. Chem. 2010;285:28261–28274. doi: 10.1074/jbc.M110.157214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borbat P.P., Freed J.H. Pulse dipolar ESR: distance measurements. In: Harmer J., Timmel C., editors. Structural Information from Spin-Labels and Intrinsic Paramagnetic Centres in the Biosciences. Structure and Bonding. Springer; Berlin, Heidelberg, Germany: 2012. Published online 2012. 101.1007/430_2012_82. [Google Scholar]
- 43.Borbat P.P., Freed J.H. Multiple-quantum ESR and distance measurements. Chem. Phys. Lett. 1999;313:145–154. [Google Scholar]
- 44.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 45.Stoll S., Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 46.Langen R., Oh K.J., Hubbell W.L. Crystal structures of spin labeled T4 lysozyme mutants: implications for the interpretation of EPR spectra in terms of structure. Biochemistry. 2000;39:8396–8405. doi: 10.1021/bi000604f. [DOI] [PubMed] [Google Scholar]
- 47.Chiang Y.W., Borbat P.P., Freed J.H. Maximum entropy: a complement to Tikhonov regularization for determination of pair distance distributions by pulsed ESR. J. Magn. Reson. 2005;177:184–196. doi: 10.1016/j.jmr.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 48.Chiang Y.W., Borbat P.P., Freed J.H. The determination of pair distance distributions by pulsed ESR using Tikhonov regularization. J. Magn. Reson. 2005;172:279–295. doi: 10.1016/j.jmr.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 49.Havel T., Wuthrich K. A distance geometry program for determining the structures of small proteins and other macromolecules from nuclear magnetic-resonance measurements of intramolecular 1H-1H proximities in solution. Bull. Math. Biol. 1984;46:673–698. [Google Scholar]
- 50.Havel T.F., Wüthrich K. An evaluation of the combined use of nuclear magnetic resonance and distance geometry for the determination of protein conformations in solution. J. Mol. Biol. 1985;182:281–294. doi: 10.1016/0022-2836(85)90346-8. [DOI] [PubMed] [Google Scholar]
- 51.Havel T.F., Kuntz I.D., Crippen G.M. The theory and practice of distance geometry. Bull. Math. Biol. 1983;45:665–720. [Google Scholar]
- 52.Xu S., Mueser T.C., Funk M.O., Jr. Crystal structure of 12-lipoxygenase catalytic-domain-inhibitor complex identifies a substrate-binding channel for catalysis. Structure. 2012;20:1490–1497. doi: 10.1016/j.str.2012.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knapp M.J., Klinman J.P. Kinetic studies of oxygen reactivity in soybean lipoxygenase-1. Biochemistry. 2003;42:11466–11475. doi: 10.1021/bi0300884. [DOI] [PubMed] [Google Scholar]
- 54.Fleissner M.R., Brustad E.M., Hubbell W.L. Site-directed spin labeling of a genetically encoded unnatural amino acid. Proc. Natl. Acad. Sci. USA. 2009;106:21637–21642. doi: 10.1073/pnas.0912009106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zabinski R., Münck E., Wood J.M. Kinetic and Mössbauer studies on the mechanism of protocatechuic acid 4,5-oxygenase. Biochemistry. 1972;11:3212–3219. doi: 10.1021/bi00767a012. [DOI] [PubMed] [Google Scholar]
- 56.Najfeld I., Havel T.F. Embedding with a rigid substructure. J. Math. Chem. 1997;21:223–260. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.