

Abstract
The interplay between epigenetic modification and chromatin compaction is implicated in the regulation of gene expression, and it comprises one of the most fascinating frontiers in cell biology. Although a complete picture is still lacking, it is generally accepted that the differentiation of embryonic stem (ES) cells is accompanied by a selective condensation into heterochromatin with concomitant gene silencing, leaving access only to lineage-specific genes in the euchromatin. ES cells have been reported to have less condensed chromatin, as they are capable of differentiating into any cell type. However, pluripotency itself—even prior to differentiation—is a split state comprising a naïve state and a state in which ES cells prime for differentiation. Here, we show that naïve ES cells decondense their chromatin in the course of downregulating the pluripotency marker Nanog before they initiate lineage commitment. We used fluorescence recovery after photobleaching, and histone modification analysis paired with a novel, to our knowledge, optical stretching method, to show that ES cells in the naïve state have a significantly stiffer nucleus that is coupled to a globally more condensed chromatin state. We link this biophysical phenotype to coinciding epigenetic differences, including histone methylation, and show a strong correlation of chromatin condensation and nuclear stiffness with the expression of Nanog. Besides having implications for transcriptional regulation and embryonic cell sorting and suggesting a putative mechanosensing mechanism, the physical differences point to a system-level regulatory role of chromatin in maintaining pluripotency in embryonic development.
Introduction
Embryonic stem (ES) cells are derived from the preimplantation mammalian epiblast and can undergo indefinite symmetrical cell division while retaining the ability to differentiate into the three primary germ layers of the embryo. Understanding the hallmark of ES cells—the pluripotent state—has inspired a quest to discover the mechanisms that act as a gateway for the pluripotent state. Much of that quest has centered on the trio of transcription factors (TFs)—Oct4, Sox2, and Nanog (1)—that seem to be at the heart of pluripotency (2). Of these TFs, Nanog alone preserves pluripotency in the absence of pluripotency maintenance signals (3). Furthermore, loss-of-function studies have implicated the necessity of Nanog at seminal time points in the development of mouse embryos (reviewed in Theunissen and Silva (1)), indicating the vital role of Nanog in orchestrating embryogenesis. Given this leading role, it is, at first sight, surprising that Nanog expression is not essential for maintaining pluripotency. This apparent paradox was resolved by the discovery that Nanog acts as a global regulator of differentiation (4). We can therefore define two states of pluripotency—high-Nanog-expressing and low-Nanog-expressing, both of which express Sox2 and Oct4—with high Nanog expression representing a stable naïve state and low Nanog expression a more heterogeneous and unstable primed state (5). Importantly, low-Nanog-expressing cells cultured in ES cell conditions still self-renew indefinitely and can contribute to chimaeras (4).
To study Nanog function, a mouse ES cell line with a green fluorescent protein (GFP) insertion into one of the Nanog loci (TNGA) was developed (4). GFP expression in TNGA cells shows a bimodal distribution in which high GFP expression is well correlated with high Nanog (HN) expression, whereas low-GFP cells constitute a more heterogeneous population of cells with primarily low Nanog (LN) expression (4,6). The HN and LN states are transcriptionally similar, with a slight but discernible downregulation in Oct4 accompanying an upregulation of lineage-specific genes in the LN state (6); furthermore, ES cells do not directly differentiate from the HN state but must first downregulate Nanog (5). These experimental facts justify the designation of the HN state as a naïve state with a well-regulated pluripotent phenotype and the LN state as a primed state poised for lineage commitment.
There is a potential unification between the molecular underpinnings and the epigenetic basis of pluripotency. The Sox2-Oct4-Nanog (SON) transcriptional network is seemingly involved crucially in regulating covalent histone modifications and chromatin remodeling, both indirectly, via transcriptional control of remodeling-associated proteins, and directly, by protein-protein interactions with remodeling complexes (reviewed in Orkin and Hochedlinger (7)). Pluripotency is hypothesized to be regulated in part by bivalent chromatin domains, which constitute at least two counteracting epigenetic marks at specific gene sites, silencing them while keeping them poised for activation (8). The discovery of these domains, present at the site of many developmentally important TF genes, is one of many breakthroughs exemplifying the high importance of epigenetic states in regulating pluripotency and differentiation (9).
Importantly, changes in epigenome have a significant consequence: these changes in the epigenome lead to multiscale chromatin remodeling, as epitomized by global condensation and decondensation of chromatin. A number of studies have underlined the importance of global chromatin states in the transition from the pluripotent to the committed state (10). It is well-established that there is widespread chromatin condensation in the nucleus associated with lineage commitment (9,11). Furthermore, there are significant increases in cell stiffness (12) and nuclear stiffness in differentiating ES cells (13). The cell stiffening is due to an increasingly organized cytoskeleton, whereas the nuclear stiffening is due in large part to upregulation of lamin A/C intermediate filaments (14), but chromatin structure could also play a significant role (15,16). These findings implicate a relationship between nuclear mechanics/chromatin structure and differentiation.
Unlike many previous studies of ES cells discussed above, we are investigating not differentiation, but the regulation of pluripotency before differentiation on a single cell-level. We show here a connection between nuclear mechanics and chromatin condensation using a unique combination of well-established and, to our knowledge, novel methods. We use optical stretching, a technique that employs a dual-beam optical trap for single-cell mechanical phenotyping (17), to measure cell compliance, and we use optical stretching in a novel, to our knowledge, way to measure single-cell nuclear mechanics. We augment these results with more established methods to study covalent histone modifications, thereby integrating physical and biochemical cell properties that influence developmental processes. We find that LN cells are more compliant, i.e., more deformable, than HN cells and that this increased compliance is due to a softening of the nucleus correlated with a more decondensed global state of chromatin. With this, we demonstrate biophysical states of pluripotency that are potentially critical for mediating fate decisions in biological development.
Materials and Methods
Cell culture, FACS analysis, and sorting
The TNGA mouse embryonic stem (mES) cell line we used was a kind gift from the lab of Austin Smith and has been described previously (4). Briefly, an eGFP reporter gene was inserted at the AUG codon of one of the Nanog-alleles and eGFP expression is correlated with Nanog promotor activation (4). ES cells were cultured in GMEM- medium (G5154, Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS) (Biosera S1900-500, lot S0495251900), 2 mM GlutaMAX (35050), penicillin-streptomycin (100 U/ml and 100 μg/ml, respectively, 15140-148), 1×MEM nonessential amino acids (11140035), 100 μM 2-mercaptoethanol (31350010, all from Invitrogen, Carlsbad, CA), 1 mM sodium pyruvate (S8636-100ML, Sigma-Aldrich) and 1000 U/ml LIF (ESGRO, ESG1107, Millipore, Billerica, MA). This medium will be referred to as LIF+ medium.
To monitor the expression of eGFP and the correlated Nanog expression, the cells were regularly checked using a Cyan ADP FACS analyzer (Dako, Glostrup, Denmark). Cells from a T25 flask or a six-well plate were trypsinized and resuspended in LIF+ medium. A protocol developed by T. Kalmar (6) was used to analyze the GFP profile of a cell population. The calibration was done in initial experiments with the nonGFP-expressing parental cell line E14. The GFP-positive cells (HN) typically showed signal intensities 100-fold those of GFP-negative cells (LN) and could therefore easily be distinguished. Only if the profile showed the typical two-peaked shape, with 20–35% of the cells being GFP-negative and 50–65% being GFP-positive, were the cells used for experiments. To sort the cells into HN and LN populations, cells were trypsinized and resuspended in LIF+ medium at a concentration of 1.5–2 E7 cells/ml. The suspension was filtered and the cells were sorted using a Dako MoFlo high-speed cell sorter. Only viable single cells were selected according to their forward-scatter (FSC)/side-scatter (SSC)/pulse width characteristics. Cells from the LN and HN peaks were collected, and cells with intermediate fluorescence intensity were discarded. Sorted cells were either used directly in stretching experiments or recultured.
Optical stretching experiments
Optical stretcher setup
All optical cell stretching experiments were performed on a custom built optical stretcher (OS), and the same equipment and settings were used throughout all experiments of this study. The basics of setting up an OS have been described elsewhere (18,19). Specifically, the microscope used for imaging phase-contrast videos in operating-system (OS) experiments was an inverted Eclipse TE2000–U equipped with a Plan Fluor ELWD 40×/0.60 NA objective (both from Nikon, Tokyo, Japan). A 1.5× C-mount was used to attach either a Hamamatsu ORCA-05G or an AVT Marlin F146B video camera leading to an overall magnification of 60×. The stage of the microscope was modified so that a microfluidic flow chamber designed for OS experiments could be attached. The laser used in OS experiments was a single-mode, continuous-wave fiber laser at a wavelength of λ = 1064 nm (YLM-5-1070-LP, IPG Photonics, Oxford, MA). Both the laser and the cameras were controlled using a custom-made LabVIEW-software (National Instruments, Austin, TX).
Data processing and statistical evaluation
A custom-built edge-detection program (LabVIEW, National Instruments) (19) was run for each cell to quantify the deformation of cells or nuclei. An ellipse was fitted to the cells and the relative deformation along the laser axis (strain) was quantified for each time point. The peak strain at the time point with the highest deformation was used to compare different cell populations.
Experimental procedure
For OS experiments, cells were grown to ∼80–90% confluency, trypsinized, optionally sorted as described above, and resuspended in LIF+ medium at a concentration of 1–2 E6 cells/ml. After an incubation period of 10–20 min at room temperature, cells were injected into the microfluidic system. Cells were trapped in the optical trap and then stretched. In all OS experiments presented in this study, the relative deformation (strain) of cells or nuclei in response to the application of a step stress was measured. In creep-compliance experiments, each cell or nucleus was recorded over a period of trapping (1 s, 0.2 W), stretching (3 s, 1.1 W), and relaxation (3 s, 0.2 W).
Compliance calculation
For compliance calculations, exact cell sizes were determined (see Supporting Material) and the refractive index of cells was measured using digital holographic microscopy (20). This information, together with known properties of the optical trap (geometry and material properties) were used to calculate the stress distribution on the cell surface according to the methods of Boyde et al. (21) and a geometrical factor (22). Using these parameters, the cellular strain during stretching was converted into compliance values.
Drug and chemical treatments
To test the role of different structures of the cell on deformability, cells were treated with different drugs or chemicals. To depolymerize the actin cytoskeleton, cells were treated with 1 μM cytochalasin D (C8273, Sigma-Aldrich) for 10 min (16,23). To disrupt the microtubule (MT) cytoskeleton, nocodazole (M1404, Sigma-Aldrich) was used (30 min, 1 μM) (24). To influence chromatin condensation, the histone deacetylase (HDAC) inhibitor trichostatin A (TSA) (19-138, Millipore) was used (300 nM, 2 h) (25) as was 5-AZA-2′-deoxycytidine (189825, Millipore) (250 nM, 2 h). These dosages and time course were chosen to minimize cytotoxicity: with these treatments, TNGAs could be passaged at full viability for at least two passages beyond treatment. In addition, these treatment regimes did not cause any change in the Nanog expression profile. To reversibly condense chromatin, divalent cations (MgCl2 and CaCl2, 2 mM each, 30 min, both from Sigma-Aldrich) were used. Divalent cations were shown to enhance chromatin condensation both in vivo and in vitro (26).
Stretching of nuclei within intact cells
For the quantification of nuclear deformation within living cells, the cells were stained with the cell-permeable nucleic acid dye Hoechst 33342 (H3570, Invitrogen). The cells were incubated with 5–10 μg/ml for 15 min before trypsinization, and the dye was kept in the resuspension medium of the cells. Except for the Hoechst staining, cells were processed in exactly the same way as for whole-cell measurements. Data analysis was performed in a way similar to that used for whole cells. The LabVIEW edge detection program was used to track edges of the nuclei and data were further processed using Matlab software. Due to lack of knowledge of the refractive index of the nuclei inside the cells, only relative nuclear deformation, rather than nuclear compliance, is reported.
Quantitative analysis of HP1α- and H3K9-methylation distributions in the nucleus
Immunofluorescence staining
The HP1α-staining protocol we used was modified from Bártová et al. (27) and was identical to the staining protocol used for H3K9/H3K27 trimethylation, as well as for Oct4. To prepare for the staining, cells were sorted (optionally), seeded in gelatinized 35-mm dishes, and incubated. When the cells reached ∼70% confluency, typically 24–36 h after seeding, they were washed twice with 2 ml of prewarmed phosphate-buffered saline (PBS) and fixed for 10 min at room temperature (RT) in prewarmed 4% formaldehyde in PBS. The dishes were then washed twice with PBS and processed immediately or stored covered with 2 ml PBS at 4°C. For permeabilization, we incubated the cells first with 0.1% triton X-100 diluted in PBS for 8 min and then with 0.1% saponin for 12 min. The dishes were washed twice with PBS for 10 min, blocked for 1 h with 1% bovine serum albumin (BSA) dissolved in PBS, and then washed for 15 min in PBS. The anti-HP1α antibody (05-689, Millipore) and the H3MeK9 antibody (abcam ab8898) were diluted 1:200 in 1% BSA in PBS, and cells were incubated o/n at 4° with the primary antibody. The cells were washed twice for 5 min in PBS and then incubated with a 1:200 dilution (in 1% BSA in PBS) of an Alexa Fluor 633 (A633) goat anti-mouse (A21126, Invitrogen) for 1 h at RT. For H3triMeK9, Alexa Fluor 568 (A568) goat anti-rabbit was used (A-11011, Invitrogen). The stained cells were washed three times for 5 min with PBS. Cells were mounted in FluorSave reagent (Calbiochem 345789, Millipore) and covered with a 22-mm coverslip.
The stained cells were imaged using the SP5 II microscope (Leica, Wetzlar, Germany). The images were recorded with a 63×, 1.4 NA oil objective. In addition to the A633 stain, a green channel was recorded to ensure purity of sorted cells. Gain settings were kept the same for all samples of a given experiment. In each experiment, multiple replicates (8, 11, 21, and 27) of stained dishes were imaged, and five to seven different regions from each replicate were imaged.
Image processing and analysis
To quantify differences in the spatial distribution of HP1α, we analyzed the fractal dimension, DF, as previously demonstrated (25,28) in a similar way. DF is a measure for complexity and defines the relationship between N, the number of pieces, and ε, the scale used to get the new pieces, as given by
For digital image analysis of immunohistochemistry stains, we used the box-counting dimension, DB, which can be calculated from the relation between the box count, Nε, and box size, ε, in box counting as
The limit is found as the slope of the regression line (29). For the sake of clarity, the calculated box-counting dimension was termed fractal dimension, or DF, in text and figures. The average nucleus diameter was ∼15 μm and pixel size was 100–200 nm, so that nearly two decades of spatial sizes were used for fractal-dimension calculation. To account for differences in staining intensity, individual nuclei were thresholded so that only pixels above the median intensity were considered. To get the fractal dimension, a standard box count was run in the ImageJ plugin Fraclac (29).
Quantitative analysis of H3triMeK27 methylation and Oct4 stainings
The samples were prepared and imaged with protocols identical to those delineated above, but with H3triMeK27 antibody (ABE44, Millipore) and Oct3/4 (c-10) antibody (sc-5279, Santa Cruz Biotechnology, Santa Cruz, CA). Alexa 568 goat anti-mouse was used for the Oct4 staining and Alexa 568 goat anti-rabbit was used for the H3triMeK27 staining. The TNGAs were not sorted before fixation, because we were interested in comparing intensities. Both 488-nm excitation and 568-nm excitation were used sequentially to measure the Nanog and Oct4/H3triMeK27 signals to ensure that the 568-nm channel was not contaminated with the Nanog signal. Two replicates in two experiments were used to ensure reproducibility. For each image (no less than 10 nuclei were present in each image), the intensities of all nuclei in the region of interest were measured in gray scale and divided by the median intensity for the respective channel for purposes of normalization. Only the bottom 35% and top 40% of Nanog signals were retained to correspond with the percentages of LN and HN cells present in the flow cytometry profile.
Fluorescence recovery after photobleaching
Preparation
The plasmid vector for the expresson of an RFP-tagged histone H2B was purchased from Evrogen (pTag-RFP-H2B vector, cat. no. FP368, Evrogen, Moscow, Russia). The tag protein (TagRFP) was selected for its compatibility in microscopy with the eGFP transgene and its monomeric structure. For experiments using fluorescence recovery after photobleaching (FRAP), transient expression of H2B-RFP was achieved by transfecting cells using Lipofectamine LTX and PLUS reagent (15338-100, Invitrogen). Cells were cultured in microdishes (80136, Ibidi, Verona, WI) for FRAP experiments, which allowed live-cell imaging on an inverted microscope.
Experiments
All FRAP experiments were performed as reported in Meshorer et al. (10), here using a Leica SP5 II confocal microscope equipped with an environmental chamber to control for temperature and CO2. All experiments were performed at 37°C and with 5% CO2 concentration using a 63.0×, NA 1.4–0.6 oil objective. To ensure good bleaching depth and a high section thickness during the imaging, the NA of the objective was adjusted to 1. Only cells that were clearly GFP-positive or GFP-negative were included in the measurements.
To set up FRAP experiments, the FRAP wizard of the Leica LAS AF software was used. Three prebleach frames were taken; then a circular area in the middle of the nucleus was bleached and 20 postbleach frames were taken at 30-s intervals.
Data processing and analysis
For image analysis, the Fiji software (ImageJ) was used. For each individual cell, the StackReg plugin (30) using the Rigid Body option was run to correct for movements of the cell in the x and y axis and rotations. Measurements with strong movements or focus drift were excluded from the analysis. To quantitatively measure the intensities of the FRAP region, the whole cell, and a background region, the FRAP Norm plugin for ImageJ was used.
We used the double-normalization method, which is based on the method described by Phair et al. for dynamic nuclear proteins (31,32). Before normalization, the background signal, Bt, for each time point is subtracted from the measured values for each time point. The first normalization step normalizes the signal of the FRAP region to its prebleach value (It/I0). The fluorescence intensity of the whole nucleus for each time point is then compared to the averaged prebleach value to calculate a correction factor to account for signal intensity lost during the bleach pulse and also during the imaging process (N0/Nt). The relative (double-normalized) intensity of the FRAP region is then calculated as
The obtained FRAP curves were then plotted normalized to the first postbleach frame. The data were fitted with the following equation to extract the diffusion timescales:
Statistical analysis
For optical stretching, quantitative image analysis, and FRAP experiments, plotting and statistical analysis were performed using Origin 8.5 software (OriginLab) or Matlab (MathWorks, Natick, MA). For each experimental day, the null hypothesis that two populations were the same was tested using a two-sample, two-tailed Student’s t-test. The normality of the samples was checked using a Kolmogorov-Smirnov test. To compare replicates of the same experiment from different days, a two-way ANOVA test was performed. Throughout the figures, N refers to the number of experiments and the error bars indicate the mean ± SD of each experiment, whereas n refers to the total number of measurements. In figures in which n is reported, the error bars represent the mean ± SE.
Results
Low Nanog expression is coupled to higher deformability in ES cells
We used TNGA cells (4) to establish physical phenotypes of priming in ES cells. We first applied optical stretching, a laser-trap-based technique used to measure whole-cell mechanics (17). Optical stretching relies on the principle that changes in refractive index cause changes in photon momentum, which induces optical forces at the surface of dielectric objects such as biological cells (Fig. 1, A and B). The LN cells were significantly more compliant (p < 0.01) than the HN cells (Fig. 2, A and B, and Fig. S1 in the Supporting Material). The peak compliance of the LN cells was 0.0296 ± 0.0047 Pa−1 (mean ± SD), whereas the peak compliance of the HN cells was 0.0259 ± 0.0028 Pa−1. We ruled out the possibility of these differences being due to variations in refractive index by using digital holographic microscopy to deduce the refractive index of the cells ((33) and Fig. S2). We also ruled out the influence of cell cycle, cell size, or nucleus/cytoplasm ratio, as these parameters are identical in LN and HN cells (Fig. S3 and Fig. S6). We also showed that LN cells recovered from an HN population after several passages were significantly more compliant than HN cells (Fig. S1). Moreover, the relative differences in cell deformability remained unchanged after cells were treated with cytoskeleton-disrupting drugs like cytochalasin D (cytoD), which disrupts filamentous actin, and Nocodazole (Noco), which disrupts microtubules (Fig. S1), but all cells were rendered more compliant by such treatment (Fig. 2 D). This result, along with the observation that the organization of the microtubules and the actin cytoskeleton (Fig. S4 and Fig. S5) in LN cells is indistinguishable from that seen in HN cells, indicates that the differences between these cells are not due to changes in cytoskeletal structure. Finally, it is unlikely that these differences are due to contamination from differentiating cells, given that this subpopulation would be intermixed with LN cells, and that differentiating ES cells are significantly stiffer than ES cells (12). Also, the LN state is reversible (Fig. S3), and LN cells express Oct4 at nearly the same level as HN cells (Fig. 3 E), signifying they are not a differentiating phenotype.
Figure 1.
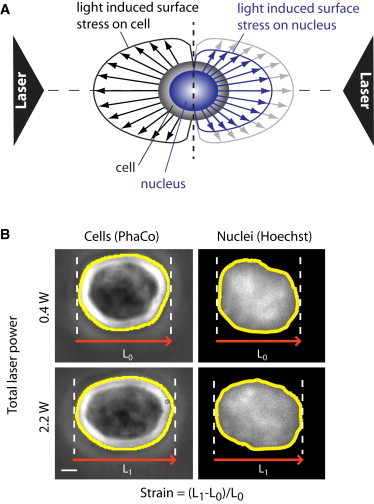
(A) Schematic representation of forces acting on cells and nuclei during optical stretching. Cells are trapped and stretched between two counterpropagating laser beams. Refractive index changes between the surrounding medium and the cell lead to a momentum transfer from light to the cell surface. The resulting forces are schematically represented by black arrows (left). In addition to forces acting on the cell surface, refractive index changes between the cytoplasm and the nucleus lead to a deformation of the nucleus (blue arrows, right). Optical stretching can be used to probe nuclear mechanics within living cells, where nuclei are visualized with DNA-specific dyes (Hoechst). (B) Representative examples of a cell and a nucleus that have been trapped (0.4 W total laser power) and stretched (2.2 W) in optical-stretching experiments. Scale bar (same for cell and nucleus), 1 μm. Edge detection was used to measure the relative deformation along the laser axis: Strain = (L1 − L0)/L0.
Figure 2.
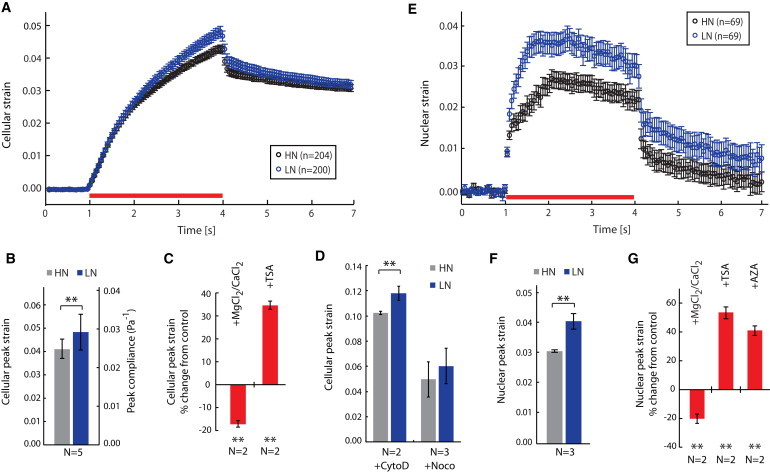
Optical stretching experiments show that LN cells, as well as LN nuclei, are more deformable than HN cells and HN nuclei, respectively. (A) Strain-versus-time plot for HN and LN cells. The stretching period is indicated by the red bar. (B) Cellular peak strains of HN and LN cells in OS experiments. The secondary y axis gives the values for cell compliance (see Methods). (C) TNGA cells were treated with MgCl2 + CaCl2 to condense, or TSA to decondense, chromatin y axis, and cellular peak strains were compared to an untreated control population. (D) Cells were treated with CytoD or Noco to disrupt actin or microtubule filaments, respectively. Note that the relative differences between HN and LN cells remain unchanged upon cytoskeleton disruption. (E) Strain-versus-time plot for the deformation of HN and LN nuclei stretched within intact cells. (F) Nuclear peak strains for HN and LN populations. (G) Treatment of TNGA cells with chromatin-modifying agents led to changes in nuclear deformability relative to an untreated control population (AZA decondenses chromatin). These changes correspond well to differences in whole-cell deformability (compare to C). Data in plots A and E are given as the mean ± SE from n individual measurements. Data in all bar diagrams represent the mean ± SD of N independent experiments unless stated otherwise. Statistical significance is indicated by asterisks: ∗p ≤ 0.05; ∗∗p ≤ 0.01; ∗∗∗p ≤ 0.001.
Figure 3.
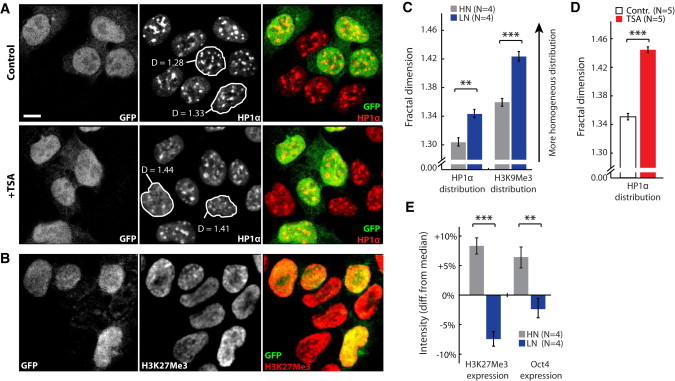
Confocal imaging and quantification of heterochromatin markers in TNGA cells. (A) TNGA cells (HN, GFP-positive; LN, GFP-negative) were treated with TSA to decondense chromatin and stained for HP1α, a marker for heterochromatic foci (middle column). The fractal dimension, D, was calculated for individual nuclei; examples are highlighted and D value is indicated. (B) TNGA cells stained for the heterochromatin marker H3K27Me3. (C) Fractal dimension was quantified for HP1α and H3K27Me3 distributions and compared between HN and LN cells. Note that a higher fractal dimension corresponds to a more homogeneous distribution pattern. (D) Quantitative results of the experiment described in A. (E) HN cells show a higher staining intensity for both H3K27Me3 and Oct4 than do LN cells, indicating a higher expression of both heterochromatin and pluripotency markers. Scale bar is 5 μM.
Nuclear deformability is modulated by chromatin condensation and is higher in low-Nanog-expressing ES cells
Given that the nuclei of ES cells comprise ∼50% of the total cell volume (Fig. S6), we tested whether the nucleus caused the observed differences in mechanical phenotype. For this, we used the optical stretching method—for the first time to our knowledge—to measure nuclear mechanics inside intact cells. We stained TNGAs with Hoechst, a live-cell, DNA-specific dye, to record the nuclear deformation during cell stretching. The mechanical stress optically induced at the cell surface, where the change in refractive index is largest, will be transmitted through the cytoskeleton to the nucleus, which will be deformed along with the entire cell. In turn, a stiffer nucleus at the core will also result in a reduced overall cell deformation. In addition, since nuclei likely have a higher refractive index than cytoplasm (34,35), there is an additional optical stress induced directly at the nuclear surface (Fig. 1 A), which leads to additional nuclear deformation. Modeling of the resultant nuclear deformation under various conditions (Fig. S7) demonstrates that nuclear deformation is insensitive to the mechanical properties of the cytoplasm, and in the absence of different optical properties (Fig. S2), this modeling can discern differently deformable nuclei. Significantly, this technique of assessing nuclear mechanics, besides being high-throughput and contact-free, leaves the cell intact, unlike many earlier nuclear-mechanics measurement approaches, which either require extraction of the nucleus from the cell or are relatively low-throughput (for instance, micropipette aspiration (13)).
Using our optical stretching method, we found LN nuclei to be significantly more deformable than HN nuclei (Fig. 2, E and F). We observed the same relative difference when we treated the cells with CytoD (Fig. S8), strengthening the case that the nucleus is being stretched independently of the cytoskeleton, and that the cytoskeleton cannot account for the overall cell deformability differences. We used Nanog knockout cells and Nanog overexpressing cells (generously provided by the laboratory of Austin Smith; prepared as in Silva et al. (36)) to see how nuclear mechanics responded to a direct perturbation in Nanog expression levels. We found that the nucleus became more deformable in the knockout cells, and less deformable in the Nanog overexpressing cell line (Fig. S1). Furthermore, we investigated whether GFP expression is itself affecting the nuclear mechanics, and found that there is no significant change in cellular and nuclear deformability upon insertion of GFP into one of the Nanog loci of the parental ES cell line (Fig. S1). We also note that when the cell deformability data and nuclear deformability data are fit to a standard liquid solid (SLS) model ((37), the cell data fit best with d = 0.62 (95% confidence bounds of (0.57,0.66)) and the nucleus data fit best with d = 6.48 (95% confidence bounds of (4.94,8.01)) for LN cells; the results are very similar for HN cells (Fig. S9). The number d is a dimensionless constant that amounts to the elasticity/viscosity ratio in the sample, and the SLS results indicate that the mechanical properties of the nucleus are dramatically different from those of the cell, with the lower value of d indicating that the nucleus is more of an elastic solid than a whole cell (a whole cell is more viscous).
Moreover, important structural proteins of the nucleus/nuclear membrane, such as lamin A/C, are only expressed in differentiated phenotypes, and not in ES cells (14). Due to this lack of nuclear scaffolding, we assume that chromatin compaction plays a more prominent role in determining the mechanical properties of nuclei (15). To test this, we artificially modified chromatin condensation by chemical treatment and measured the deformability of TNGA nuclei in three different conditions: 1), addition of 2 mM of MgCl2 and CaCl2, divalent cations that condense the highly charged chromatin; 2), addition of 300 nM Trichostatin A (TSA), a histone deacetylase inhibitor that decondenses chromatin; 3), addition of 250 nM 5-AZA-2′-deoxycytodine (AZA), a DNA methylation inhibitor that also decondenses chromatin. We verified the decondensation of chromatin with microscopy, as discussed in the next section. We saw decreased nuclear deformability with the addition of divalent cations, and increased nuclear deformability with the addition of TSA and AZA (Fig. 2 G). Our findings strongly suggest that the nuclear mechanics of ES cells can be modulated by interfering with chromatin structure. The data also indicate that decondensing chromatin makes the nuclei more compliant, whereas condensing it makes them stiffer. To test whether interfering with chromatin structure would modulate whole-cell mechanics, we used the above-mentioned conditions and measured cell mechanics. The results were in line with the nuclear mechanics measurements (Fig. 2 C), suggesting that alteration of chromatin structure may be responsible for the observed differences in mechanical phenotype of whole HN and LN cells.
Naïve ES cells have more condensed chromatin than primed ES cells
To establish the chromatin structure of ES cells, we first stained for HP1α and histone 3 trimethylated at lysine 9 (H3K9Me3), which are predominantly colocalized in chromatin and are associated with stability of polycomb repressive complexes (PRCs) and condensed heterochromatin (38) (Fig. 3 A). A more euchromatic state, such as that expected in undifferentiated ES cells, has a more diffuse distribution of HP1α/H3K9Me3. The diffuse distribution is consistent with a destabilization of local regions of condensed chromatin arbitrated by PRCs (39). Using a fractal analysis (40), which quantifies the uniformity of a distribution, we showed that even within the undifferentiated ES cells there is a clear modulation of this distribution, with HP1α/H3K9Me3 being more diffuse in LN cells than in HN cells (Fig. 3 C, Fig. S10, and Fig. S11). We verified the decondensation of chromatin with TSA treatment (Fig. 3 D) and the significant condensation of chromatin with the addition of divalent cations (data not shown). Significantly, we also found that there was a higher expression of H3K27Me3 (Fig. 3 C) in HN cells than in LN cells (Fig. 3 E). Given that H3K27Me3 is highly associated with transcriptional repression (8), this result indicates that there is less transcriptional accessibility in the naïve ES cells, which is reversed only when they are primed for differentiation. Hence, we propose that genome-wide changes in the chromatin condensation state occur when ES cells prime for differentiation.
Chromatin kinetics are faster in primed ES cells
We subsequently assessed the chromatin state of TNGA cells using FRAP of H2B-RFP. H2B is a histone protein that has a large fraction with fast kinetics in ES cells; the recovery dynamics of H2B have been positively correlated with a euchromatic state and pluripotency in similar experiments testing pluripotent versus differentiated cells (10). For FRAP measurements, we transiently transfected TNGA populations with H2B-RFP (Fig. 4 A). We found that the recovery kinetics of H2B was significantly faster in LN cells than in HN cells (Fig. 4 B). We fit an exponential decay function (1 – exp(−t/τ)) and found that the time constant, τ, for HN cells was 2.6 × 103 s, whereas the time constant for the LN cells was 1.6 × 103 s indicating a faster recovery time for LN cells. We also observed that a TSA-treated population of HN cells, with less condensed chromatin, exhibited a significantly faster recovery time of H2B fluorescence than an untreated HN cell population, signifying that recovery dynamics is strongly associated with a euchromatic, decondensed state (Fig. 4 C).
Figure 4.
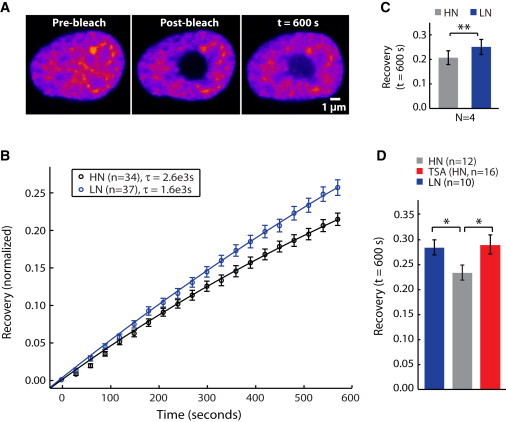
FRAP experiments for labeled histone proteins. (A) Histone H2B was fluorescently labeled with RFP and a bleach pulse was applied to a circular area. (B) Fluorescence recovery in the bleached area was recorded for 600 s for HN and LN cells. Data were fit to an exponential decay function, f(t) = 1 − exp(−t/τ), and time constant, τ, is reported in the figure key. (C) At the end of the observation period, LN cells show a significantly higher mean recovery than HN cells (inset; mean ± SD from four independent experiments). (D) HN cells treated with the histone deacetylase inhibitor TSA to decondense chromatin show fluorescence recovery kinetics similar to those observed for LN cells (mean ± SE from one representative experiment).
Altogether, our data on chromatin condensation and modifications point to a globally more heterochromatic state, and stiffer nucleus, in the naïve HN cells than in the differentiation-primed LN cells. This is highly provocative in light of research pointing to a more heterochromatic state, with a stiffer nucleus, in differentiated cells compared to pluripotent cells. Superficially, therefore, our results are a contradiction, but the contradiction is resolved when we consider pluripotency to have substates—naïve and primed—in which the primed state is possibly a global minimum of chromatin condensation. These findings have important implications for the nature of pluripotent cells and embryogenesis, as discussed below.
Discussion
The molecular mechanisms underpinning the passage from pluripotency to lineage commitment have recently been under intense scrutiny. Much of this research has focused on the biochemical interactions of the SON regulatory network of TFs. The SON network occupies a large number of transcriptionally inactive gene promoters in ES cells, demonstrating a possible regulatory role for these TFs in silencing genes that are important for lineage specification (2). Current research on the SON network is largely devoted to discovering its interface with the epigenome. For instance, various findings demonstrate that there is a significant overlap between the target genes of TFs that directly limit chromatin decondensation, such as UTF1, and the SON transcriptional network (7). Another hint of the overlap is that Nanog-dependent aspects of development are associated with extensive epigenetic reprogramming (41), such as the high degree of DNA demethylation at E3.5–E4.5, when the epiblast of DNA demethylation at the mouse developmental stages E3.5 is formed and between E11.5 and E12.5, where germ cells are formed (42).
Recent discoveries have strengthened the crucial role of Nanog in the establishment of ground-state pluripotency: Although Nanog was found to be expressed from both alleles at the transition to ground-state pluripotency when the epiblast is formed in the blastocyst, only one Nanog allele is expressed before and after epiblast maturation (43). Nanog dosage seems to be crucial for an efficient transition to pluripotency and is itself regulated by covalent histone modifications at the Nanog promotors. It has been proposed that dynamic allele-switching and fluctuations in Nanog expression offer the possibility to enter lineage commitment for otherwise stable pluripotent cells (6,43). It is important to note that the experiments we performed were on mouse ES cells, and many of the biochemical and biophysical observations we have made are not necessarily applicable to human ES cells. Moreover, it is not possible at this time to correlate cell-to-cell compliance with Nanog expression in the HN population, since if we look at all GFP-expressing cells (HN), the GFP within that high-expressing population correlates with cell cycle. So we would not be measuring anything like Nanog expression, but really how compliance is correlating with cell cycle. We did show, in Fig. S3, that the cell cycle is identical in HN and LN cells, so the differences should be averaged over in our measurements.
The interface between the SON network and the epigenome should be put into a larger context of models of pluripotency and differentiation (reviewed in Meshorer and Misteli (44)). These models include the early transcriptional competence marks model, in which genes specific to somatic cells are not actively transcribed in ES cells but epigenetically marked for later expression by, for instance, bivalent chromatin domains (45). Bivalent domains are specific chromatin-modification patterns consisting of, for instance, large regions of H3K27Me3 housing smaller regions of H3K4Me3 (8). H3K27Me3 negatively regulates transcription by condensing chromatin (46), whereas H3K4Me3 decondenses chromatin by recruiting histone acetylases (47). These bivalent domains have been found at the sites of many developmental TFs—silencing them while keeping them primed for activation (8). During the course of differentiation, most bivalent domains become resolved in either an active (H3K4Me3 only) or repressed (H3K27Me3 only) state. The second possible model is the promiscuous transcription model, in which lineage-specific genes are expressed at low levels in ES cells (48). Lineage-specific TFs then reinforce the expression of genes specific to a particular somatic fate.
Could both models be correct? It is clear that pluripotent cells can be separated into two distinct states, the HN and LN states, which are distinguished by a heterogeneous—bistable—expression of Nanog. LN cells bear some characteristics of the promiscuous transcription model, with low-level expression of lineage-specific genes (6), whereas HN cells, with the higher expression of repressive epigenetic marks also found in this work, bear hallmarks of the early transcriptional competence marks model. Here, we show a very strong correlation between Nanog expression and chromatin condensation, further buttressing the probability of the regulatory role of Nanog for epigenetic control, and for switching between the two models. Future work should focus on bringing into clear relief how the relationship between Nanog and the epigenome regulates pluripotency and lineage commitment. To move closer to a full understanding of this regulation, we must gain an appreciation for the emerging evidence that development is also driven by biophysics.
This evidence includes the role of cell/nuclear mechanics in embryogenesis. There are a number of well-established examples of the importance of mechanics in development, including the convergent extension in vertebrate gastrulation, where myosin-dependent cell intercalations lead to the elongation of the anterior-posterior axis in the embryo (49). Moreover, contractions and cortical tension apparently play a significant role in cell sorting of germ-layer organization in gastrulation (50). Also, in the early blastocyst stage, epiblast and primitive endoderm markers indicate a mixed population that uses active actin-dependent processes (51), among others, to sort into two distinct cell layers in the late blastocyst stage (52). Many of these studies focus on cell mechanics and the cytoskeleton, but given the large nucleus/cytoplasm ratio in pluripotent cells, nuclear mechanics should be considered a significant agent of whole-cell mechanics. Accepting that there is a link between cell/nuclear mechanics and embryonic cell positioning naturally leads to the question of whether or not there might also be a direct biophysical regulation of cell-fate decisions.
Supporting the hypothesis that pluripotency is biophysically regulated, there are widespread biophysical changes associated with the exit from pluripotency, including nuclear stiffening (13), as well as global condensation of chromatin structure (9). It is important to note that given the lack of lamin A/C expression in ES cells, it has been proposed that chromatin plays a larger role in the mechanical properties of nuclei within these cells (13), and we have justified that proposal in this study. The link between chromatin structure and nuclear mechanics is essential, because there are established connections between chromatin and the cytoskeleton (53), and externally applied forces are transmitted to the nucleus (54). It has also been shown that nuclei change shape due to external forces (55) and that mechanical signals affect chromatin assembly (56). Significantly, changes in ES cell state, including downregulation of pluripotency genes such as Oct4, have been observed with the application of physical forces (57). In addition, substrate stiffness and topography have been used to induce differentiation of stem cells (58,59). The overarching hypothesis is that the nucleus, acting as a mechanosensor, with its stiffness modulating its response, changes shape due to applied forces, leading to changes in gene expression. Much research effort is directed toward understanding how these modifications in gene expression can be predicted and controlled. Ultimately, there may be a central interplay between mechanics and mechanosensing on one hand and sorting and fate decisions in the embryo on the other.
Our work explores that interplay and supports two major points of emphasis. First, there is potential bootstrapping between chromatin condensation, nuclear mechanics, and cell mechanics, which together constitute a biophysical phenotype in ES cells. Second, this biophysical phenotype, along with epigenome monitoring, distinguishes between ES cells in a primed and naïve state of pluripotency. These focal points predict a systems approach toward investigating pluripotency. In the last few years, it has been demonstrated several times that epigenetic changes are highly dynamic throughout development, ensuring access at the proper time to specific locations of the genome (reviewed in Fisher and Fisher (9)). These results illuminate a clear pathway from the molecular aspects of pluripotency to the whole cell within the framework of a systematic and coherent biophysical signature (see summary figure in the Supporting Material). It is likely that these systemic changes significantly alter the manner in which ES cells interact with their physical and chemical environment. Ultimately, a full understanding of pluripotency and differentiation demands that we take a systems approach in examining ES cells, accounting for their environmental coupling as well as their physical properties across scales—from molecular chromatin substructure to whole-cell mechanics.
Acknowledgments
The authors acknowledge Tibor Kalmar, Penelope Hayward, Chea Lim, Graziano Martello, and Silvia Muñoz-Descalzo for help with biological methods and cell culture.
The authors acknowledge financial support from the University of Vienna (to M.H.), the Medical Research Council (to K.J.C. and J.G.), the Human Frontiers in Science Program (to J.G. and K.J.C.), and the Royal Society (to K.J.C.).
Supporting Material
References
- 1.Theunissen T.W., Silva J.C. Switching on pluripotency: a perspective on the biological requirement of Nanog. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2011;366:2222–2229. doi: 10.1098/rstb.2011.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boyer L.A., Lee T.I., Young R.A. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chambers I., Colby D., Smith A. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell. 2003;113:643–655. doi: 10.1016/s0092-8674(03)00392-1. [DOI] [PubMed] [Google Scholar]
- 4.Chambers I., Silva J., Smith A. Nanog safeguards pluripotency and mediates germline development. Nature. 2007;450:1230–1234. doi: 10.1038/nature06403. [DOI] [PubMed] [Google Scholar]
- 5.Nichols J., Smith A. Naive and primed pluripotent states. Cell Stem Cell. 2009;4:487–492. doi: 10.1016/j.stem.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 6.Kalmar T., Lim C., Martinez Arias A. Regulated fluctuations in nanog expression mediate cell fate decisions in embryonic stem cells. PLoS Biol. 2009;7:e1000149. doi: 10.1371/journal.pbio.1000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orkin S.H., Hochedlinger K. Chromatin connections to pluripotency and cellular reprogramming. Cell. 2011;145:835–850. doi: 10.1016/j.cell.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernstein B.E., Mikkelsen T.S., Lander E.S. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 9.Fisher C.L., Fisher A.G. Chromatin states in pluripotent, differentiated, and reprogrammed cells. Curr. Opin. Genet. Dev. 2011;21:140–146. doi: 10.1016/j.gde.2011.01.015. [DOI] [PubMed] [Google Scholar]
- 10.Meshorer E., Yellajoshula D., Misteli T. Hyperdynamic plasticity of chromatin proteins in pluripotent embryonic stem cells. Dev. Cell. 2006;10:105–116. doi: 10.1016/j.devcel.2005.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bártová E., Krejcí J., Kozubek S. Differentiation of human embryonic stem cells induces condensation of chromosome territories and formation of heterochromatin protein 1 foci. Differentiation. 2008;76:24–32. doi: 10.1111/j.1432-0436.2007.00192.x. [DOI] [PubMed] [Google Scholar]
- 12.Pillarisetti A., Desai J.P., Keefer C.L. Mechanical phenotyping of mouse embryonic stem cells: increase in stiffness with differentiation. Cell. Reprogram. 2011;13:371–380. doi: 10.1089/cell.2011.0028. [DOI] [PubMed] [Google Scholar]
- 13.Pajerowski J.D., Dahl K.N., Discher D.E. Physical plasticity of the nucleus in stem cell differentiation. Proc. Natl. Acad. Sci. USA. 2007;104:15619–15624. doi: 10.1073/pnas.0702576104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Constantinescu D., Gray H.L., Csoka A.B. Lamin A/C expression is a marker of mouse and human embryonic stem cell differentiation. Stem Cells. 2006;24:177–185. doi: 10.1634/stemcells.2004-0159. [DOI] [PubMed] [Google Scholar]
- 15.Dahl K.N., Engler A.J., Discher D.E. Power-law rheology of isolated nuclei with deformation mapping of nuclear substructures. Biophys. J. 2005;89:2855–2864. doi: 10.1529/biophysj.105.062554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mazumder A., Roopa T., Shivashankar G.V. Dynamics of chromatin decondensation reveals the structural integrity of a mechanically prestressed nucleus. Biophys. J. 2008;95:3028–3035. doi: 10.1529/biophysj.108.132274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guck J., Ananthakrishnan R., Käs J. The optical stretcher: a novel laser tool to micromanipulate cells. Biophys. J. 2001;81:767–784. doi: 10.1016/S0006-3495(01)75740-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lincoln B., Schinkinger S., Guck J. Reconfigurable microfluidic integration of a dual-beam laser trap with biomedical applications. Biomed. Microdevices. 2007;9:703–710. doi: 10.1007/s10544-007-9079-x. [DOI] [PubMed] [Google Scholar]
- 19.Lincoln B., Wottawah F., Guck J. High-throughput rheological measurements with an optical stretcher. Methods Cell Biol. 2007;83:397–423. doi: 10.1016/S0091-679X(07)83017-2. [DOI] [PubMed] [Google Scholar]
- 20.Chalut K.J., Ekpenyong A.E., Guck J. Quantifying cellular differentiation by physical phenotype using digital holographic microscopy. Integr Biol (Camb) 2012;4:280–284. doi: 10.1039/c2ib00129b. [DOI] [PubMed] [Google Scholar]
- 21.Boyde L., Chalut K.J., Guck J. Interaction of Gaussian beam with near-spherical particle: an analytic-numerical approach for assessing scattering and stresses. J. Opt. Soc. Am. A Opt. Image Sci. Vis. 2009;26:1814–1826. doi: 10.1364/josaa.26.001814. [DOI] [PubMed] [Google Scholar]
- 22.Ananthakrishnan R., Guck J., Käs J. Quantifying the contribution of actin networks to the elastic strength of fibroblasts. J. Theor. Biol. 2006;242:502–516. doi: 10.1016/j.jtbi.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 23.Petersen N.O., McConnaughey W.B., Elson E.L. Dependence of locally measured cellular deformability on position on the cell, temperature, and cytochalasin B. Proc. Natl. Acad. Sci. USA. 1982;79:5327–5331. doi: 10.1073/pnas.79.17.5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Head J., Lee L.L., Lee J.C. Equilibrium and rapid kinetic studies on nocodazole-tubulin interaction. J. Biol. Chem. 1985;260:11060–11066. [PubMed] [Google Scholar]
- 25.Tóth K.F., Knoch T.A., Rippe K. Trichostatin A-induced histone acetylation causes decondensation of interphase chromatin. J. Cell Sci. 2004;117:4277–4287. doi: 10.1242/jcs.01293. [DOI] [PubMed] [Google Scholar]
- 26.Widom J. Physicochemical studies of the folding of the 100 Å nucleosome filament into the 300 Å filament. Cation dependence. J. Mol. Biol. 1986;190:411–424. doi: 10.1016/0022-2836(86)90012-4. [DOI] [PubMed] [Google Scholar]
- 27.Bártová E., Pacherník J., Kozubek S. Differentiation-specific association of HP1α and HP1β with chromocentres is correlated with clustering of TIF1β at these sites. Histochem. Cell Biol. 2007;127:375–388. doi: 10.1007/s00418-006-0259-1. [DOI] [PubMed] [Google Scholar]
- 28.Chalut K.J., Kulangara K., Leong K.W. Stem cell differentiation indicated by noninvasive photonic characterization and fractal analysis of subcellular architecture. Integr Biol (Camb) 2011;3:863–867. doi: 10.1039/c1ib00003a. [DOI] [PubMed] [Google Scholar]
- 29.Karperien A. Charles Sturt University; Bathurst, Australia: 2007. FracLac for ImageJ. [Google Scholar]
- 30.Thévenaz P., Ruttimann U.E., Unser M. A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 1998;7:27–41. doi: 10.1109/83.650848. [DOI] [PubMed] [Google Scholar]
- 31.Phair R.D., Gorski S.A., Misteli T. Measurement of dynamic protein binding to chromatin in vivo, using photobleaching microscopy. Methods Enzymol. 2004;375:393–414. doi: 10.1016/s0076-6879(03)75025-3. [DOI] [PubMed] [Google Scholar]
- 32.Phair R.D., Misteli T. High mobility of proteins in the mammalian cell nucleus. Nature. 2000;404:604–609. doi: 10.1038/35007077. [DOI] [PubMed] [Google Scholar]
- 33.Ekpenyong A.E., Man S.M., Chalut K.J. Bacterial infection of macrophages induces decrease in refractive index. J. Biophotonics. 2012 doi: 10.1002/jbio.201200113. Published online. [DOI] [PubMed] [Google Scholar]
- 34.Chalut K.J., Kulangara K., Leong K.W. Deformation of stem cell nuclei by nanotopographical cues. Soft Matter. 2010;6:1675–1681. doi: 10.1039/B921206J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mourant J.R., Freyer J.P., Johnson T.M. Mechanisms of light scattering from biological cells relevant to noninvasive optical-tissue diagnostics. Appl. Opt. 1998;37:3586–3593. doi: 10.1364/ao.37.003586. [DOI] [PubMed] [Google Scholar]
- 36.Silva J., Chambers I., Smith A. Nanog promotes transfer of pluripotency after cell fusion. Nature. 2006;441:997–1001. doi: 10.1038/nature04914. [DOI] [PubMed] [Google Scholar]
- 37.Findley W.N., Lai J.S., Onaran K. Elsevier/North Holland; Amsterdam; New York: 1976. Creep and Relaxation of Nonlinear Viscoelastic Materials, with an Introduction to Linear Viscoelasticity. [Google Scholar]
- 38.Maison C., Almouzni G. HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 2004;5:296–304. doi: 10.1038/nrm1355. [DOI] [PubMed] [Google Scholar]
- 39.Eskeland R., Freyer E., Bickmore W.A. Histone acetylation and the maintenance of chromatin compaction by polycomb repressive complexes. Cold Spring Harb. Symp. Quant. Biol. 2010;75:71–78. doi: 10.1101/sqb.2010.75.053. [DOI] [PubMed] [Google Scholar]
- 40.Boyde L., Chalut K.J., Guck J. Near- and far-field scattering from arbitrary three-dimensional aggregates of coated spheres using parallel computing. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2011;83:026701. doi: 10.1103/PhysRevE.83.026701. [DOI] [PubMed] [Google Scholar]
- 41.Surani M.A., Hayashi K., Hajkova P. Genetic and epigenetic regulators of pluripotency. Cell. 2007;128:747–762. doi: 10.1016/j.cell.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 42.Reik W., Dean W., Walter J. Epigenetic reprogramming in mammalian development. Science. 2001;293:1089–1093. doi: 10.1126/science.1063443. [DOI] [PubMed] [Google Scholar]
- 43.Miyanari Y., Torres-Padilla M.E. Control of ground-state pluripotency by allelic regulation of Nanog. Nature. 2012;483:470–473. doi: 10.1038/nature10807. [DOI] [PubMed] [Google Scholar]
- 44.Meshorer E., Misteli T. Chromatin in pluripotent embryonic stem cells and differentiation. Nat. Rev. Mol. Cell Biol. 2006;7:540–546. doi: 10.1038/nrm1938. [DOI] [PubMed] [Google Scholar]
- 45.Mikkelsen T.S., Ku M., Bernstein B.E. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Francis N.J., Kingston R.E., Woodcock C.L. Chromatin compaction by a polycomb group protein complex. Science. 2004;306:1574–1577. doi: 10.1126/science.1100576. [DOI] [PubMed] [Google Scholar]
- 47.Pray-Grant M.G., Daniel J.A., Grant P.A. Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature. 2005;433:434–438. doi: 10.1038/nature03242. [DOI] [PubMed] [Google Scholar]
- 48.Efroni S., Duttagupta R., Meshorer E. Global transcription in pluripotent embryonic stem cells. Cell Stem Cell. 2008;2:437–447. doi: 10.1016/j.stem.2008.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Skoglund P., Rolo A., Keller R. Convergence and extension at gastrulation require a myosin IIB-dependent cortical actin network. Development. 2008;135:2435–2444. doi: 10.1242/dev.014704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krieg M., Arboleda-Estudillo Y., Heisenberg C.P. Tensile forces govern germ-layer organization in zebrafish. Nat. Cell Biol. 2008;10:429–436. doi: 10.1038/ncb1705. [DOI] [PubMed] [Google Scholar]
- 51.Meilhac S.M., Adams R.J., Zernicka-Goetz M. Active cell movements coupled to positional induction are involved in lineage segregation in the mouse blastocyst. Dev. Biol. 2009;331:210–221. doi: 10.1016/j.ydbio.2009.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Plusa B., Piliszek A., Hadjantonakis A.K. Distinct sequential cell behaviours direct primitive endoderm formation in the mouse blastocyst. Development. 2008;135:3081–3091. doi: 10.1242/dev.021519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dahl K.N., Ribeiro A.J., Lammerding J. Nuclear shape, mechanics, and mechanotransduction. Circ. Res. 2008;102:1307–1318. doi: 10.1161/CIRCRESAHA.108.173989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guilak F., Tedrow J.R., Burgkart R. Viscoelastic properties of the cell nucleus. Biochem. Biophys. Res. Commun. 2000;269:781–786. doi: 10.1006/bbrc.2000.2360. [DOI] [PubMed] [Google Scholar]
- 55.Deguchi S., Maeda K., Sato M. Flow-induced hardening of endothelial nucleus as an intracellular stress-bearing organelle. J. Biomech. 2005;38:1751–1759. doi: 10.1016/j.jbiomech.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 56.Wang N., Tytell J.D., Ingber D.E. Mechanotransduction at a distance: mechanically coupling the extracellular matrix with the nucleus. Nat. Rev. Mol. Cell Biol. 2009;10:75–82. doi: 10.1038/nrm2594. [DOI] [PubMed] [Google Scholar]
- 57.Chowdhury F., Na S., Wang N. Material properties of the cell dictate stress-induced spreading and differentiation in embryonic stem cells. Nat. Mater. 2010;9:82–88. doi: 10.1038/nmat2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dalby M.J., Gadegaard N., Oreffo R.O. The control of human mesenchymal cell differentiation using nanoscale symmetry and disorder. Nat. Mater. 2007;6:997–1003. doi: 10.1038/nmat2013. [DOI] [PubMed] [Google Scholar]
- 59.Engler A.J., Sen S., Discher D.E. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.