

Abstract
Neutron scattering and nuclear magnetic resonance relaxation experiments are combined with molecular dynamics (MD) simulations in a novel, to our knowledge, approach to investigate the change in internal dynamics on substrate (camphor) binding to a protein (cytochrome P450cam). The MD simulations agree well with both the neutron scattering, which furnishes information on global flexibility, and the nuclear magnetic resonance data, which provides residue-specific order parameters. Decreased fluctuations are seen in the camphor-bound form using all three techniques, dominated by changes in specific regions of the protein. The combined experimental and simulation results permit a detailed description of the dynamical change, which involves modifications in the coupling between the dominant regions and concomitant substrate access channel closing, via specific salt-bridge, hydrogen-bonding, and hydrophobic interactions. The work demonstrates how the combination of complementary experimental spectroscopies with MD simulation can provide an in-depth description of functional dynamical protein changes.
Introduction
Cytochrome P450s are essential hemoprotein monooxygenases that catalyze a large variety of biochemical reactions involved in carcinogenesis, drug metabolism, lipid and steroid biosynthesis, and degradation of pollutants in higher organisms (1). The chemical reactions catalyzed include hydroxylation, sulfoxidation, epoxidation, dehalogenation, deformylation, dealkylation, and C-C coupling (2).
Cytochrome P450cam (CYP101), an enzyme from Pseudomonas putida that catalyzes the regio- and stereo-specific hydroxylation of camphor, has long served as a model system for P450s (3). Early x-ray crystal structures of CYP101 exhibited only slight differences in the protein backbone of the substrate-free and camphor-bound forms, with a root-mean-square deviation of 0.5 Å (4,5). In both forms, the active site is buried with no channel open to the protein surface and this led to a long-standing question as to how the substrate accesses the buried protein active site. Numerous experimental and theoretical studies have been performed to address this problem (6), and camphor analog ligands with synthetic molecular wires have also been designed to probe the substrate access channel (7–9).
Very recent CYP101 structures, obtained by crystallizing the protein under conditions different from those used in Poulos et al. (4,5), showed that substrate-free CYP101 adopts an “open” conformation in which the F/G loop, F, and G helices are retracted from the B′ helix, leading to an open channel between them (Fig. 1 a) (10). In contrast, the camphor-bound form of the protein was found in a “closed” conformation as previously observed (10). X-ray structures of CYP101 bound by camphor analogs with molecular wires also revealed an intermediate “partially open” conformation (9). These crystallographic studies of CYP101 indicate that the channel can open and close to allow substrate entry and product egress.
Figure 1.
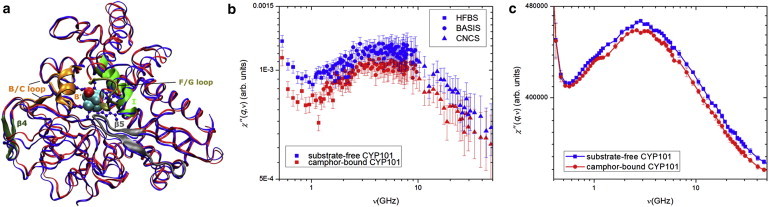
(a) Superposed x-ray crystal structures of substrate-free (dark-gray/blue) and camphor-bound (light-gray/red) CYP101, depicting the five regions listed in Table 1 for which significant dynamic changes are observed between the two protein forms. The five regions are labeled individually in the camphor-bound form. (b) Comparison of neutron dynamic susceptibility measured over a broad energy range for the two protein forms. The wings of the susceptibility peak observed in the BASIS instrumental energy window are supplemented by the data taken from HFBS in the low-energy range and CNCS in the high-energy range. (c) Comparison of the dynamic susceptibility for the two protein forms calculated from MD simulations over the same energy range.
Molecular dynamics (MD) simulations have been applied to characterize the location of substrate access, product exit, and water diffusion channels in several cytochrome P450 enzymes (6,11–15). Random expulsion and steered MD simulations, applying external forces on the substrate, were used to probe substrate exit pathways (11,12). The predicted pathways were found to correlate with regions of CYP101 that display high x-ray crystallographic temperature factors (13).
The crystallographic, biochemical, and computational studies performed so far have provided evidence of functional structural flexibility of CYP101, and it is likely that dynamic modulation of specific regions in CYP101 is required for substrate binding. However, direct experimental characterization of CYP101 flexibility and quantitative measurement of the relative contribution of these regions to the global dynamical changes is lacking.
Experimental methods such as neutron scattering and nuclear magnetic resonance (NMR) spectroscopy have proven to be useful tools for investigating the dynamics of biological molecules (16–18). Neutron-scattering probes atomic motions over timescales of ∼10−1−104 ps and ∼1–10 Å length-scales and has been widely used to characterize the global dynamics of proteins, hydration water, membranes, and even entire cells (19–22). The incoherent scattering cross-section of hydrogen atoms is ∼40 times larger than that of other atoms, and as hydrogen atoms are distributed nearly homogeneously within proteins, neutron scattering provides a global picture of the protein internal dynamics. Furthermore, the time and space domains probed by neutron scattering are well covered by contemporary MD simulation techniques. The neutron dynamic structure factor can be computed from atomic MD trajectories and compared with experimental data quantitatively (23). Contributions of the measured neutron scattering spectra can be decomposed, enabling a direct interpretation of the neutron scattering experimental data. NMR, in contrast, can be used to extract dynamic information in a residue-specific manner on the nanosecond timescale from a variety of NMR spin-relaxation experiments (17,24,25). NMR spin-relaxation measurements thus provide a good indication of the conformational mobility of specific regions in a protein and can be used to validate the potential of MD simulations on a residue level. Visualization of the corresponding dynamics at atomic detail is also possible using MD simulations (26) and is especially valuable when the experimental NMR data available are sparse. NMR and MD can be used in conjunction by comparing order parameters of NMR spin-relaxation measurements with those calculated from MD simulations.
In this study, we present a novel, to our knowledge, combined approach incorporating MD simulation with neutron scattering and NMR to obtain a multifaceted insight into the dynamic modulation of CYP101 upon substrate binding. The neutron scattering is used to measure the global flexibility and the NMR for local fluctuations, and then both are compared to MD simulation. The results from all three techniques are found to agree, and lead to a coherent, distinct picture of dynamical changes in the protein on substrate binding. This picture involves coupled dynamical changes of specific regions in the protein, potentially permitting adaptive modulation of the channel dynamics to accommodate a variety of substrates.
Materials and Methods
Neutron scattering and NMR experiments
Expression and purification of camphor-bound CYP101 followed methods described previously (27) and the substrate-free sample was prepared by extensive dialysis of camphor-bound protein with multiple buffer exchanges using substrate-free buffer, followed by gel filtration chromatography to ensure complete removal of the camphor. Quasielastic neutron scattering experiments were performed on hydrogenated CYP101 hydrated to h = 0.4 g D2O/g protein for both substrate-free and camphor-bound CYP101 using three neutron-scattering instruments, i.e., the NG2 high-flux backscattering spectrometer (HFBS) at the Center for Neutron Research at National Institute of Standards and Technology), and the Backscattering Spectrometer (BASIS) (28) and Cold Neutron Chopper Spectrometer (29) at the Spallation Neutron Source (SNS) at Oak Ridge National Laboratory. NMR relaxation measurements were carried out on substrate-free and camphor-bound CYP101 to determine 15N T1 and T2 relaxation times, which along with 15N-{1H} nuclear Overhauser effects were used to calculate order parameters of individual residues using a model-free formalism (24). Details of experimental procedures and data analysis are described in the Supporting Material.
Molecular dynamics simulations
MD simulations were performed with NAMD2 (30) by using the 3L61 x-ray crystal structure for substrate-free CYP101 and 3L63 for the camphor-bound form (10,31). A schematic representation of the two structures is shown in Fig. 1 a. The CHARMM22 force field (32) was used for the protein and the TIP3P model (33) for water molecules. Standard CHARMM force-field parameters (32) were used for the heme group including an explicit Fe-S bond to Cys357, except that the atomic partial charge of the SG atom in Cys357 was adjusted to −0.07e to account for the resting state of CYP101 in both substrate-free and camphor-bound forms (34). CHARMM parameters for camphor were obtained from Schöneboom et al. (35). MD simulations were carried out for both protein forms in aqueous solution as well as in hydrated powder. The simulations of the hydrated powder exhibit results similar to those of the aqueous solution in terms of the protein regions that show significant dynamic changes between the two protein forms. Hence, for consistent comparison with the experimental data, neutron scattering spectra and NMR backbone order parameters were both calculated from the hydrated powder simulations with 100-ns product MD trajectories. Details of the simulations are provided in the Supporting Material.
Calculations of neutron spectra and NMR order parameters from MD simulations
The neutron incoherent intermediate scattering function, Iinc(q,t), was calculated from 100-ns production MD trajectories by using the SASSENA program (36) as
| (1) |
where binc is the incoherent neutron scattering length of atom i, Ri(t) is the position of atom i at time t, and N is the number of atoms. Iinc(q,t) was Fourier-transformed to the experimentally measurable dynamic structure factors, Sinc(q,ω):
| (2) |
Sinc(q,ω) was used to calculate the dynamic susceptibility, χ″(q,ν) via
| (3) |
where nB(ω) = (eħω/kT −1)−1 is the Bose temperature factor and ω = 2πν. For decomposition of neutron scattering spectra in terms of protein regions (Table 1), residues corresponding to each selected region (e.g., 78–105 in the B/C loop) were chosen from the MD trajectories and used to calculate Iinc(q,t) using Eq. 1, which was then converted to Sinc(q,ω) and χ″(q,ν) for further analysis.
Table 1.
Change in the integral of quasielastic neutron dynamic susceptibility χ″(q,ν) and dynamic structure factor S(q,ω)
| Region | Residues | Number of H values | χ″(q,ν) Relative increase (%) | S(q,ω) Relative increase (%) |
|---|---|---|---|---|
| B/C loop | 78–105 | 161 | 8.8 ± 0.6 | 13.5 ± 1.1 |
| F/G loop | 186–193 | 43 | 6.1 ± 1.6 | 9.4 ± 2.3 |
| β5 sheet | 385–400 | 88 | 6.7 ± 0.4 | 7.5 ± 0.8 |
| β4 sheet | 305–313 | 59 | 5.0 ± 1.0 | 6.1 ± 1.4 |
| I-helix kink | 243–254 | 78 | 4.6 ± 0.3 | 5.4 ± 0.3 |
| Total of the above | — | 429 | 6.8 ± 0.7 | 9.4 ± 1.1 |
| Remainder protein | — | 2121 | 0.5 ± 0.03 | 0.9 ± 0.04 |
| Entire protein | 1–414 | 2550 | 3.3 ± 0.1 | 2.6 ± 0.1 |
These are for various regions in substrate-free CYP101, relative to the camphor-bound form over energy range 2–200 μeV (i.e., 0.5–50 GHz) calculated from MD simulations.
For NMR protein-backbone order parameters, the autocorrelation function Ci (t) of normalized N−H bond vectors were first calculated as
| (4) |
where P2(x) = 1/2(3x2 − 1) is the second-order Legendre polynomial and 〈⋅〉 denotes a time average over the MD trajectory. The order parameters were then obtained by fitting Ci (t) as its long-time limit (24,37) via
| (5) |
Because there is no overall translation/rotation for the protein simulated in hydrated powder, 100-ns product MD trajectories were divided into 10 equal time windows and each of them was used to calculate Si2 directly. Reported order parameters correspond to the mean of the ten Si2 values and the error bars correspond to their standard deviation.
Results
Quasielastic neutron scattering
Quasielastic neutron scattering experiments were performed on substrate-free and camphor-bound CYP101 hydrated to h ≈ 0.4 g D2O/g protein at 300 K by using three neutron instruments with different energy resolutions. These are HFBS at 1 μeV, BASIS (28) at the SNS at 3.5 μeV, and CNCS (29) at the SNS at 25 μeV (see experiment details in the Supporting Material). These instruments cover a broad energy range from 1 μeV to 1.6 meV, corresponding to 10−1−103 ps timescales. It is informative to present the neutron scattering spectra as the imaginary part of dynamic susceptibility χ″(q,ν). Because this allows direct identification of relaxation amplitudes in the protein from the distinct spectral peaks (νmax) and the associated relaxation times, τ are then given by (2πνmax)−1 (19). The value χ″(q,ν) can be obtained by dividing the experimentally measured dynamic structure factors, S(q,ω), by the Bose temperature factor (see Materials and Methods).
Fig. 1 b combines χ″(q,ν) from all three neutron scattering instruments. For both substrate-free and camphor-bound CYP101, a broad peak is seen at νmax ≈ 3.5 GHz (E = 14.5 μeV). The characteristic relaxation time is τ ≈ 45 ps for both protein forms. However, χ″(q,ν) of substrate-free CYP101 is of higher intensity at the peak position, and the integral of χ″(q,ν) over the quasielastic energy range (2–200 μeV) is 4.8 ± 0.5% greater than the camphor-bound form.
The room temperature measurements of S(q,ω) measured on the three instruments are shown in Fig. S2, a, b, and c, respectively, in the Supporting Material. Substrate-free CYP101 possesses higher quasielastic intensity than the camphor-bound form on all three instruments, consistent with the above results for χ″(q,ν). Quasielastic scattering arises from diffusive dynamics. Hence, the results indicate that, on the ∼10−1−103 ps time- and ∼1–10 Å length-scales, substrate-free CYP101 undergoes larger-amplitude diffusive dynamics than does the camphor-bound form. This conclusion was confirmed by an analysis of S(q,ω) as fitted by the sum of a δ-function and a Kohlrausch-Williams-Watts function plus background (38), from which the obtained elastic incoherent structure factor of substrate-free CYP101 is found 4.3 ± 0.4% lower than that of the camphor-bound form (see Fig. S2 e in the Supporting Material). Hence, both the elastic and quasielastic neutron scattering indicate that the diffusive dynamics of substrate-free CYP101 is distributed over similar relaxation times, but is larger in amplitude than the camphor-bound form.
The value χ″(q,ν) calculated from the MD simulations of the two protein forms is plotted in Fig. 1 c. The simulation-derived χ″(q,ν) exhibits a similar peak (albeit somewhat sharper) at a comparable frequency of νmax ≈ 3.2 GHz (τ ≈ 50 ps) as the experiment. Again, the substrate-free CYP101 susceptibility peak is of larger amplitude than that of the camphor-bound form. Integration of χ″(q,ν) over energy range 2–200 μeV yields that the quasielastic neutron scattering of substrate-free CYP101 is 3.3 ± 0.1% greater in amplitude than that of camphor-bound CYP101, in quantitative agreement with the experimental results.
NMR protein backbone order parameters
Examination of two-dimensional 15N-1H TROSY-HSQC NMR spectra of substrate-free and camphor-bound CYP101 revealed that the backbone amide resonances exhibit major chemical shift and line-width changes for several residues between the two protein forms (see Fig. S3). The most prominent spectral changes are for resonances corresponding to residues in the B′ helix, the F/G loop, the β5 sheet, and the I-helix kink near the active site, signifying conformational/dynamic changes in these regions. Apart from being shifted, resonances for most residues in the B′ helix (e.g., 90–96) are also markedly broadened, indicating corresponding increased fluctuations.
To characterize the local dynamic differences between substrate-free and camphor-bound CYP101, NMR 15N T1,T2 relaxation measurements were performed to obtain generalized squared-backbone-order parameters (S2) (for experiment details, see the Supporting Material). Although limited backbone assignments (39) and resonance overlap prevented NMR measurements for many residues in the protein, NMR backbone-amide order parameters were able to be determined from heteronuclear relaxation experiments for ∼100 residues in both the substrate-free and camphor-bound forms and these residues fortuitously correspond to most of the substrate-binding regions in CYP101. NMR order parameters were also calculated from MD simulations as the long time-limit of the autocorrelation function of normalized N−H bond vectors (Eq. 5), for which example fits of Tyr78 and Gln388 are shown in Fig. S5.
Fig. 2 shows a comparison of the NMR protein backbone-order parameters between substrate-free and camphor-bound CYP101 as obtained from both experiment and simulation. On average, substrate-free CYP101 exhibits lower values of order parameters compared to camphor-bound CYP101, indicating overall increased dynamics in the substrate-free form, similar to the observations from the neutron-scattering experiments. Further analysis of the order parameters permits us to identify key regions exhibiting significant differences in protein dynamics between the two protein forms. Indeed, in both experiment and simulation, significantly lower order parameters are found in substrate-free CYP101 for residues 78–105 in the B/C loop, 243–254 in the I-helix kink near the active site, and 385–400 in the β5 sheet. A small but significant decrease in the average order parameters is also seen in residues 186–193 in the F/G loop and 305–313 in the β4 sheet, although the agreement between experiment and simulation in these regions is imperfect, especially for the β4 sheet. Taken together, the MD and NMR results indicate that five key regions in CYP101 exhibit increased dynamic fluctuations between the two protein forms: the B/C loop containing the B′ helix, the F/G loop, the I-helix kink near the active site, and the β4 and β5 sheets.
Figure 2.
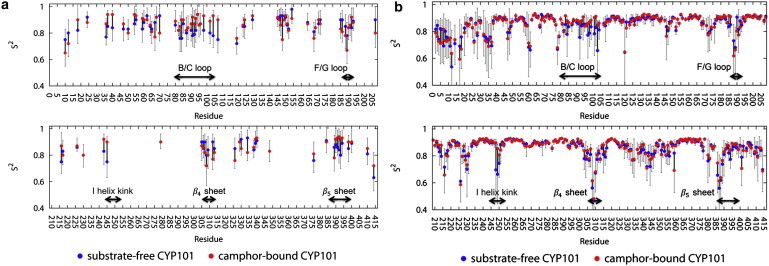
Comparison of backbone order parameters between substrate-free (dark-gray/blue) and camphor-bound CYP101 (light-gray/red) on a residue-by-residue basis. Order parameters were derived for the two protein forms using (a) experimental NMR data and (b) MD simulations.
Decomposition of neutron-scattering spectra
Contributions to the quasielastic neutron scattering spectra of the above five protein regions that exhibit largest differences between substrate-free and camphor-bound CYP101 in the NMR order parameters were calculated as described in Materials and Methods. As shown in Fig. 3, whereas the dynamic susceptibility χ″(q,ν) of substrate-free CYP101 is closely similar to that of the camphor-bound form for the remainder of the protein (Fig. 3 a), for the five regions themselves it is significantly higher in intensity (Fig. 3 b). For the remainder of the protein, the integral of χ″(q,ν) over the quasielastic energy range 2–200 μeV of substrate-free CYP101 is only 0.5% greater than the camphor-bound form, whereas it is ∼7% greater from the five high-flexibility regions (Table 1). Similar results were obtained for the integral of S(q,ω). Hence, the difference between the quasielastic neutron spectra also originates mainly from increased flexibility of these five regions in substrate-free CYP101.
Figure 3.
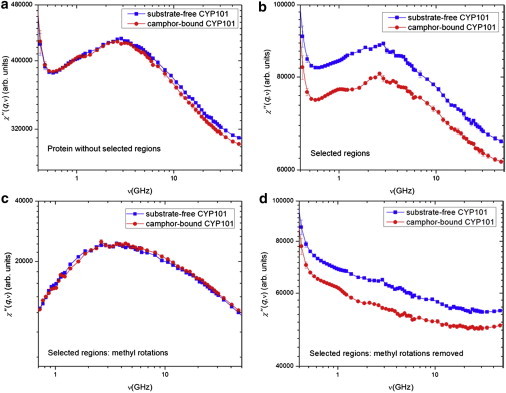
Decomposition of total dynamic susceptibility of substrate-free (dark-gray/blue) and camphor-bound (light-gray/red) CYP101 into contributions from: (a) protein without the five regions listed in Table 1, (b) the five regions only, (c) methyl rotations in the five regions, and (d) the five regions with methyl rotations removed.
The dynamical contributions to χ″(q,ν) from the five regions were examined more closely. It has been shown that χ″(q,ν) of a globular protein can be decomposed into three classes of motion: methyl rotations, localized diffusion, and nonmethyl jumps (40). The methyl rotations in substrate-free and camphor-bound CYP101 possess very similar dynamic susceptibility spectra (Fig. 3 c), peaked at νmax ≈ 3.2 GHz and yielding an average relaxation time τ ≈ 50 ps. Based on the fact that the rotation of methyl groups usually follows an Arrhenius temperature dependence, τ ∼ τ0 exp(ΔE/kT) with τ0 ∼ 0.18 ps (41), the above methyl relaxation time corresponds to an average rotational barrier of ΔE = 3.3 kcal/mol, in agreement with the ∼3 kcal/mol barrier obtained previously (41,42). In contrast to the similarity of χ″(q,ν) for methyl rotations, χ″(q,ν) from the localized diffusion and nonmethyl jumps is significantly larger in substrate-free CYP101 than the camphor-bound form over the entire energy range (see Fig. 3 d). Hence, the increased dynamics in substrate-free CYP101 in the five selected regions arises mainly from jump motions (conformational transitions) and localized diffusion, but not methyl rotations.
Dynamic differences between substrate-free and camphor-bound CYP101
The average structures and atomic root-mean-square fluctuations of camphor-bound and substrate-free CYP101 calculated from the MD simulations are illustrated in Fig. 4, a and b, respectively. In camphor-bound CYP101, flexibility is observed in only the F/G loop and the β4, β5 sheets (Fig. 4 a). In the substrate-free form, flexibility is increased in these three regions, and significantly higher fluctuations are also visible in the B/C loop that contains the B′ helix and the I-helix kink near the active site (Fig. 4 b).
Figure 4.
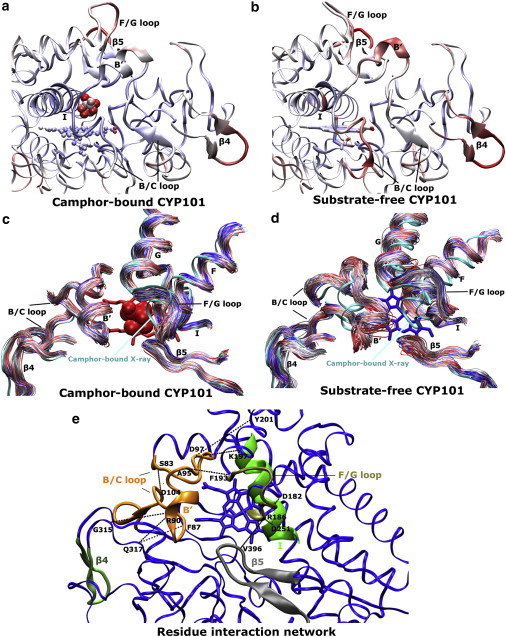
Average structures of (a) camphor-bound and (b) substrate-free CYP101 obtained from MD simulations colored by atomic root-mean-square fluctuations in a grayscale (color-scale online: 0 Å, blue; midpoint, white; and 1.2 Å, red). The structures are rotated from Fig. 1a so as to look down the I-helix for better viewing of the five regions. Time series of backbone traces of the five protein regions during 100-ns MD trajectories of (c) camphor-bound and (d) substrate-free CYP101 in a grayscale (color-scale online: 0 ns, blue; midpoint, white; and 100 ns, red). The closed 3L63 crystal structure is shown as reference. (e) Key salt-bridge, hydrogen-bonding, and hydrophobic interactions in CYP101, in which the five regions are shown similar to Fig. 1a.
Further analysis of MD simulations also revealed that the putative substrate access channel width (defined by the distance between Ser190 in the F/G loop and Pro89 in the B′ helix) retains a value of ∼15 Å in the camphor-bound form with relatively small fluctuations (∼0.9 Å), whereas in the substrate-free form the channel width is larger (∼19.5 Å on average), and exhibits significantly larger fluctuations, sampling the range ∼18–22 Å (see Fig. S6 a). Thus, the increased global amplitudes manifested in the quasielastic neutron scattering are translated into larger channel-width fluctuations in substrate-free CYP101 (see Fig. 4, c and d). Moreover, in the simulation of substrate-free CYP101, the F and G helices move closer to the B′ helix by ∼1 Å and ∼2 Å, respectively, relative to the initial configuration. However, on average these two helices are still ∼3.5 Å and ∼2.2 Å, respectively, away from their positions in the closed crystallographic conformation (see Fig. S6 b). We therefore surmise that in substrate-free CYP101, although the channel samples a wide variety of values between the fully open and partially open conformations, it has not reached the partially open state as defined in Lee et al. (9) (for analysis details, see section SIII.3 in the Supporting Material), and the closed conformation is not accessed by the substrate-free form.
Detailed analysis of the five flexible protein regions identified above allows us to pinpoint changes in specific interactions responsible for the dynamic differences between the two forms. There are a number of salt-bridge, hydrogen-bonding, and hydrophobic interactions in and around the putative substrate access channel of CYP101 that connect the five regions, maintaining a scaffold that dynamically couples the motion as shown in Fig. 4 e. These interactions have been previously suggested to play important roles in the stability and function of CYP101 (9,43,44). The key interactions involved comprise side-chain salt bridges and hydrogen bonds connecting the F-helix to the I-helix and the F/G loop (Arg186-Asp251 and Arg186-Asp182), the G-helix to the B′ helix (Asp97-Lys197 and Asp97-Tyr201), and the hydrogen bond between the side chain of Arg186 and the carbonyl oxygen of Val396 that connects the F-helix to the β5 sheet. In addition, there are several hydrophobic interactions that connect the B′ helix to surrounding structural elements including the G-helix (Ala95-Phe193) and β5 sheet (Phe87-Ile395). Distances and fluctuations of the above specific interactions are listed in Table 2 and time series of the distances are plotted in Fig. S8. In the substrate-free form, the interaction distances and the corresponding fluctuations are significantly higher. Hence, upon loss of camphor, a weakening is observed of these interactions connecting the five structural elements.
Table 2.
Characteristic distances of residue pairs in the x-ray crystal structures and MD simulations of substrate-free and camphor-bound CYP101 for key salt-bridge, hydrogen-bonding, and hydrophobic interactions
| Distance∗ (Å) | Camphor-bound |
Substrate-free |
Region(s) involved | ||
|---|---|---|---|---|---|
| 3L63 crystal | Simulation | 3L61 crystal† | Simulation | ||
| Arg186-Asp182‡ | 4.56 | 4.56 ± 0.22 | 4.66 | 5.97 ± 1.85 | F-helix, F/G loop |
| Arg186-Asp251‡ | 3.97 | 3.93 ± 0.07 | 5.63 | 4.44 ± 1.13 | F-helix, I-helix |
| Arg186-Val396§ | 2.83 | 2.82 ± 0.12 | 9.78 | 6.84 ± 1.89 | F-helix, β5 sheet |
| Asp97-Lys197‡ | 3.58 | 3.39 ± 0.55 | — | 4.18 ± 1.20 | B′ helix, G-helix |
| Asp97-Tyr201§ | 2.83 | 3.25 ± 0.36 | — | 4.68 ± 1.94 | B′ helix, G-helix |
| Ala95-Phe193¶ | 4.78 | 4.91 ± 0.30 | — | 5.86 ± 1.04 | B′ helix, G-helix |
| Ser83-Asp104§ | 3.10 | 3.32 ± 0.88 | — | 9.87 ± 2.64 | B/C loop |
| Phe87-Gln317§ | 2.87 | 2.88 ± 0.09 | 3.06 | 3.80 ± 1.76 | B/C loop, β3 sheet |
| Arg90-Gly315§ | 2.91 | 5.58 ± 0.97 | — | 8.07 ± 1.48 | B′ helix, β3 sheet |
| Arg90-Gln317§ | 3.37 | 3.60 ± 0.13 | — | 5.14 ± 1.12 | B′ helix, β3 sheet |
Distance fluctations are also given above in the simulations. Note that the individual hydrogen bonds are identified between Arg186 (side chain) and Val396 (carbonyl oxygen), Asp97 (side chain) and Tyr201 (-OH group), Ser83 (side chain) and Asp104 (side chain), Phe87 (carbonyl oxygen) and Gln317 (side chain), Arg90 (side chain) and Gly315 (carbonyl oxygen), and Arg90 (backbone nitrogen) and Gln317 (side chain).
The distance is calculated among the centers of the charged groups for ionic pairs (i.e., Cγ for Asp, Cδ for Glu, Nζ for Lys, Cζ for Arg, and Nδ or Nε for doubly protonated His, whichever is closer to the negatively charged residue in the ionic pair), centers of side chains for hydrophobic interactions (i.e., Cβ for Ala and Cγ for Phe), and donor-acceptor for hydrogen-bonding interactions.
Because residues 90–97 and 104 are missing in the 3L61 crystal structure, the corresponding distances are not given.
Salt-bridge interactions.
Hydrogen-bonding interactions.
Hydrophobic interactions.
The disorder in the B′ helix observed in the crystal structure of substrate-free CYP101 is also seen in the MD simulation, with several residues in the helix exhibiting increased fluctuations. However, no information on changes in specific interactions involving residues in this functionally important region is available from the crystal structures (Table 2), as the electron density for these residues is not visible. The MD simulation throws light on the dynamics of this region. In camphor-bound CYP101, Tyr96, located at the C-terminus of the B′ helix, is oriented toward the active site, making a strong hydrogen bond with the oxygen atom of the camphor. However, removal of the hydrogen bond in the substrate-free form leads to fluctuation of Tyr96 side chain between the active site and the channel opening, occasionally forming a hydrogen bond with the carbonyl oxygen atom of Met184 (see Fig. S7). This significantly increased conformational flexibility of Tyr96 in substrate-free CYP101 is consistent with severe line-broadening observed for the resonance of this residue in the NMR spectrum. Similar NMR line-broadening in the substrate-free form was also observed for several other residues in the B′ helix that are involved in interactions with the G-helix as described above, including Ala95 and Asp97, supporting the existence of the increased associated fluctuations observed in the simulation.
In substrate-free CYP101, increased distances and fluctuations were also observed in the simulation for several hydrogen bonds that help stabilize the B/C loop in the camphor-bound form. These include the hydrogen bonds between the side chains of Ser83 and Asp104 and between the backbone oxygen of Phe87 and the side chain of Gln317 (see Fig. 4 e and Fig. S9). Furthermore, the C-terminus of the B/C loop (just before the beginning of the C-helix) was observed to be highly fluctuating in the substrate-free form (Fig. 4 a), owing to the breaking of the hydrogen bond between Ser83 and Asp104 (Table 2). Finally, in substrate-free CYP101, Arg90 present at the beginning of the B′ helix was found to extend its side chain, exhibiting dynamic interactions with surrounding residues Gly315 and Gln317, and the hydrogen-bonding interactions involved are weaker than those in the camphor-bound form (Table 2), which greatly contributes to the higher flexibility of the β4 sheet in the substrate-free form.
Discussion
Although structural studies of various P450s in presence of various substrates have been performed previously and provide a qualitative indication of which regions in a protein are disordered through crystallographic temperature factors (7–10,45), the relative contributions of specific regions to dynamic changes of a P450 induced by substrate binding have not been characterized in detail. NMR spectroscopy can potentially provide such dynamic information on an atomic level, although it can be challenging to collect extensive data (especially involving side chains) on proteins as large as P450s and only sparse data are usually available due to limited assignments and resonance overlap (39).
This situation is likely to be exacerbated in human P450s where the complexities are magnified due to increasing size and limited solubility of the proteins. Such systems would largely benefit from the synergistic use of spectroscopic and simulation techniques in characterization of dynamical amplitudes, timescales, and coupling, especially in the context of drug design. The incorporation of MD simulation augments the information that can be extracted from NMR and neutron scattering spectra by identifying which parts of the protein are involved in short-timescale (ps-ns) diffusive motions in a quantitative manner. In turn, the use of complementary experimental techniques alleviates force-field limitations and model approximations inherent to MD, allowing a more complete and reliable physical picture to be obtained of both the global structural flexibility and local fluctuations in the protein.
Conformations accessible to CYP101 for substrate binding
Recently solved x-ray crystal structures of CYP101 with and without camphor show that significant conformational differences are present between the two forms of the protein (10). The crystal structures, along with additional structures of CYP101 with camphor ligand analogs, indicate that the putative substrate access channel of CYP101 can adopt open, partially open, and closed conformations which allow closing of the channel via a multistep closure pathway involving movements of the F- and G-helices (7–10). Studies performed here also support the presence of distinct conformations in solution for the substrate-free and camphor-bound forms, based on the large chemical shift changes and exchange-broadening observed for several residues in the two-dimensional 15N-1H HSQC NMR spectra of the two forms.
These changes are found to be specific to substrate access regions involving the F/G loop, the B/C loop containing the B′ helix, and the β4 and β5 sheets, similar to those regions identified in the crystal structures. In the present MD simulations, the camphor-bound CYP101 clearly stays in the closed conformation with only small deviations around the average channel opening, whereas in the substrate-free form the channel opening exhibits large fluctuations and decreases by ∼3 Å on average during the course of the simulation (see Fig. S6 a), consistent with a transition toward the partially open conformation from the open conformation in substrate-free CYP101. These results are in at least qualitative accord with recent adaptive accelerated MD simulations (14), which suggested that, when initiated from the open conformation, substrate-free CYP101 rapidly fluctuates back and forth between open and partially open conformations, but never to the closed conformation. However, longer direct MD simulations would be required to observe full conversion between the two conformations, as the timescale of this process is likely to be considerably larger than that simulated here (approximately microseconds). These results suggest that binding of diverse substrates to CYP101 may proceed partially via conformational selection of appropriate conformational substates sampled by the substrate-free form, but may, depending partially on the substrate shape and size, also involve induced fit.
Contribution of CYP101 structural flexibility to substrate adaptation
Both NMR and neutron scattering demonstrate that substrate-free CYP101 exhibits globally increased dynamics relative to the camphor-bound form. Decomposition of the neutron spectra using MD revealed that the underlying basis for this higher flexibility is the increased jump motions and localized diffusions of the nonexchangeable hydrogen atoms (e.g., Tyr96 shown in Fig. S7) within the five protein regions important for substrate binding (i.e., the B/C and F/G loops, the I-helix kink near the active site, and the β4 and β5 sheets). These sample a larger conformational space in substrate-free CYP101 (see Fig. 4 d) due to weaker salt-bridge, hydrogen-bonding, and hydrophobic interactions. This higher structural flexibility of substrate-free CYP101 leads to an increase in the number of accessible conformations for binding of diverse substrates, as has previously been observed from x-ray crystallography (10), fluorescence (46), Fourier transform infrared spectroscopy (47), and MD simulations (13,48), as well as studies on other P450s (45,49).
The overall architecture of the above five regions identified here is highly conserved among P450s (45,50), suggesting a common mode for P450 substrate binding that involves dynamic modulation of coupled flexible regions via a network of key residue interactions (see Fig. 4 e and Table 2. Based on our observations that the inherent flexibility of the substrate-free form is mediated by these interactions, we hypothesize that coupled conformational transitions related to diffusive motions might be functionally important in substrate adaptation. These transitions may be modified in strength depending on the type of substrate, thus enabling the recognition and adaptation of substrates of varying shape and size. In the case of CYP101, the effects of camphor binding are likely transmitted throughout the flexible protein regions upon formation of a hydrogen bond between the –OH group of Tyr96 and the oxygen atom of camphor, with concomitant reduction in fluctuations and structural ordering of the B′ helix.
Indeed, the largest dynamic changes as discerned from the decomposition of the neutron scattering spectra are observed for the B/C loop containing the B′ helix (Table 1) and are likely to be a primary dynamic determinant in inducing the closure of the substrate access channel. This is done by strengthening the residue interactions between the B′ and G helices (e.g., Asp97-Lys197, Asp97-Tyr201, and Ala95-Phe193 in Table 2), which in turn leads to further closure of the F/G loop and the F-helix. Strengthening of the interactions between the β5 sheet and the F-helix in the camphor-bound form (the Val396-Arg186 hydrogen bond in Table 2) results in the β5 sheet moving in concert with the F-helix, accompanied by changes in the I-helix kink region. The B′-helix perturbation can also be transmitted to the β4 region via Phe87 and Arg90, which primarily interact with Gly315 and Gln317 from the β3 sheet (near C-terminus of the β4 sheet) through weak hydrogen-bonding interactions (Fig. 4 e). Such weak interactions between the B/C loop and the β3, β4 sheets are also found in several other P450s, and are often replaced by hydrophobic interactions (50–52). We therefore anticipate similar dynamic coupling of the flexible regions in other P450s.
Conclusion
This article demonstrates how the combination of complementary global (neutron) and local (NMR) experimental spectroscopies with MD simulation can provide a detailed description of changes in protein dynamics accompanying changes in functional states. This type of information is particularly valuable in cases where it may not be feasible to crystallize and solve large numbers of structures, both in substrate-free form and with various substrates or substrate analogs. Although it has been possible to solve multiple structures of CYP101 (7–10), the problem is often encountered in the case of human P450s. Hence, the application of the methodology presented in this article may be of particular help in drug design against these targets.
Acknowledgments
We thank Madhu Tyagi for assistance with HFBS experiments, Eugene Mamontov with BASIS experiments, and Georg Ehlers with CNCS experiments. We also thank Liang Hong and Nikolai Smolin for providing code for analysis, and for valuable discussions.
This project was supported by the National Science Foundation (award No. MCB-0842871). Computing time was provided in part by a National Science Foundation TeraGrid award (grant No. TG-MCA08X032) on the Kraken supercomputer and a National Energy Research Scientific Computing Center award (project No. M906) on the Franklin and Hopper supercomputers.
Contributor Information
Nitin Jain, Email: njain@utk.edu.
Jeremy C. Smith, Email: smithjc@ornl.gov.
Supporting Material
References
- 1.Sligar S.G. Chemistry. Glimpsing the critical intermediate in cytochrome P450 oxidations. Science. 2010;330:924–925. doi: 10.1126/science.1197881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaik S., Cohen S., Thiel W. P450 enzymes: their structure, reactivity, and selectivity modeled by QM/MM calculations. Chem. Rev. 2010;110:949–1017. doi: 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]
- 3.Schlichting I., Berendzen J., Sligar S.G. The catalytic pathway of cytochrome p450cam at atomic resolution. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- 4.Poulos T.L., Finzel B.C., Howard A.J. Crystal structure of substrate-free Pseudomonas putida cytochrome P-450. Biochemistry. 1986;25:5314–5322. doi: 10.1021/bi00366a049. [DOI] [PubMed] [Google Scholar]
- 5.Poulos T.L., Finzel B.C., Howard A.J. High-resolution crystal structure of cytochrome P450cam. J. Mol. Biol. 1987;195:687–700. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- 6.Cojocaru V., Winn P.J., Wade R.C. The ins and outs of cytochrome P450s. Biochim. Biophys. Acta. 2007;1770:390–401. doi: 10.1016/j.bbagen.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 7.Hays A.M.A., Dunn A.R., Goodin D.B. Conformational states of cytochrome P450cam revealed by trapping of synthetic molecular wires. J. Mol. Biol. 2004;344:455–469. doi: 10.1016/j.jmb.2004.09.046. [DOI] [PubMed] [Google Scholar]
- 8.Dunn A.R., Dmochowski I.J., Crane B.R. Probing the open state of cytochrome P450cam with ruthenium-linker substrates. Proc. Natl. Acad. Sci. USA. 2001;98:12420–12425. doi: 10.1073/pnas.221297998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee Y.-T., Glazer E.C., Goodin D.B. Three clusters of conformational states in p450cam reveal a multistep pathway for closing of the substrate access channel. Biochemistry. 2011;50:693–703. doi: 10.1021/bi101726d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee Y.T., Wilson R.F., Goodin D.B. P450cam visits an open conformation in the absence of substrate. Biochemistry. 2010;49:3412–3419. doi: 10.1021/bi100183g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lüdemann S.K., Lounnas V., Wade R.C. How do substrates enter and products exit the buried active site of cytochrome P450cam? 1. Random expulsion molecular dynamics investigation of ligand access channels and mechanisms. J. Mol. Biol. 2000;303:797–811. doi: 10.1006/jmbi.2000.4154. [DOI] [PubMed] [Google Scholar]
- 12.Lüdemann S.K., Lounnas V., Wade R.C. How do substrates enter and products exit the buried active site of cytochrome P450cam? 2. Steered molecular dynamics and adiabatic mapping of substrate pathways. J. Mol. Biol. 2000;303:813–830. doi: 10.1006/jmbi.2000.4155. [DOI] [PubMed] [Google Scholar]
- 13.Ludemann, S. K., O. Carugo, and R. C. Wade. 1997. Substrate access to Cytochrome P450cam: a comparison of a thermal motion pathway analysis with molecular dynamics simulation data. 11th Molecular Modeling Workshop, Darmstadt, Germany. 369–374.
- 14.Markwick P.R.L., Pierce L.C.T., McCammon J.A. Adaptive accelerated molecular dynamics (Ad-AMD) revealing the molecular plasticity of P450cam. J. Phys. Chem. Lett. 2011;2:158–164. doi: 10.1021/jz101462n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miao Y., Baudry J. Active-site hydration and water diffusion in cytochrome P450cam: a highly dynamic process. Biophys. J. 2011;101:1493–1503. doi: 10.1016/j.bpj.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doster W., Cusack S., Petry W. Dynamical transition of myoglobin revealed by inelastic neutron scattering. Nature. 1989;337:754–756. doi: 10.1038/337754a0. [DOI] [PubMed] [Google Scholar]
- 17.Mittermaier A., Kay L.E. New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312:224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 18.Henzler-Wildman K., Kern D. Dynamic personalities of proteins. Nature. 2007;450:964–972. doi: 10.1038/nature06522. [DOI] [PubMed] [Google Scholar]
- 19.Roh J.H., Curtis J.E., Sokolov A.P. Influence of hydration on the dynamics of lysozyme. Biophys. J. 2006;91:2573–2588. doi: 10.1529/biophysj.106.082214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wood K., Frölich A., Weik M. Coincidence of dynamical transitions in a soluble protein and its hydration water: direct measurements by neutron scattering and MD simulations. J. Am. Chem. Soc. 2008;130:4586–4587. doi: 10.1021/ja710526r. [DOI] [PubMed] [Google Scholar]
- 21.Chen S.H., Lagi M., Baglioni P. Dynamics of a globular protein and its hydration water studied by neutron scattering and MD simulations. Spectrosc. Int. J. 2010;24:1–24. [Google Scholar]
- 22.Jasnin M., Moulin M., Tehei M. In vivo measurement of internal and global macromolecular motions in Escherichia coli. Biophys. J. 2008;95:857–864. doi: 10.1529/biophysj.107.124420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith J.C. Protein dynamics: comparison of simulations with inelastic neutron scattering experiments. Q. Rev. Biophys. 1991;24:227–291. doi: 10.1017/s0033583500003723. [DOI] [PubMed] [Google Scholar]
- 24.Lipari G., Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982;104:4546–4559. [Google Scholar]
- 25.Zanotti J.M., Bellissent-Funel M.C., Parello J. Hydration-coupled dynamics in proteins studied by neutron scattering and NMR: the case of the typical EF-hand calcium-binding parvalbumin. Biophys. J. 1999;76:2390–2411. doi: 10.1016/S0006-3495(99)77395-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markwick P.R., Bouvignies G., Blackledge M. Exploring multiple timescale motions in protein GB3 using accelerated molecular dynamics and NMR spectroscopy. J. Am. Chem. Soc. 2007;129:4724–4730. doi: 10.1021/ja0687668. [DOI] [PubMed] [Google Scholar]
- 27.Zhang W., Pochapsky S.S., Jain N.U. Solution NMR structure of putidaredoxin-cytochrome P450cam complex via a combined residual dipolar coupling-spin labeling approach suggests a role for Trp106 of putidaredoxin in complex formation. J. Mol. Biol. 2008;384:349–363. doi: 10.1016/j.jmb.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mamontov E., Herwig K.W. A time-of-flight backscattering spectrometer at the Spallation Neutron Source, BASIS. Rev. Sci. Instrum. 2011;82:085109. doi: 10.1063/1.3626214. [DOI] [PubMed] [Google Scholar]
- 29.Ehlers G., Podlesnyak A.A., Sokol P.E. The new cold neutron chopper spectrometer at the Spallation Neutron Source: design and performance. Rev. Sci. Instrum. 2011;82:085108. doi: 10.1063/1.3626935. [DOI] [PubMed] [Google Scholar]
- 30.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yi Z., Miao Y., Smith J.C. Derivation of mean-square displacements for protein dynamics from elastic incoherent neutron scattering. J. Phys. Chem. B. 2012 doi: 10.1021/jp2102868. [DOI] [PubMed] [Google Scholar]
- 32.MacKerell A.D., Bashford D., Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 33.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 34.Zheng J.J., Altun A., Thiel W. Common system setup for the entire catalytic cycle of cytochrome P450(cam) in quantum mechanical/molecular mechanical studies. J. Comput. Chem. 2007;28:2147–2158. doi: 10.1002/jcc.20701. [DOI] [PubMed] [Google Scholar]
- 35.Schöneboom J.C., Lin H., Shaik S. The elusive oxidant species of cytochrome P450 enzymes: characterization by combined quantum mechanical/molecular mechanical (QM/MM) calculations. J. Am. Chem. Soc. 2002;124:8142–8151. doi: 10.1021/ja026279w. [DOI] [PubMed] [Google Scholar]
- 36.Lindner B., Smith J.C. SASSENA—x-ray and neutron scattering calculated from molecular dynamics trajectories using massively parallel computers. Comput. Phys. Commun. 2012;183:1491–1501. [Google Scholar]
- 37.Lipari G., Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 1982;104:4559–4570. [Google Scholar]
- 38.Williams G., Watts D.C. Non-symmetrical dielectric relaxation behavior arising from a simple empirical decay function. Trans. Faraday Soc. 1970;66:80–85. [Google Scholar]
- 39.Pochapsky S.S., Pochapsky T.C., Wei J.W. A model for effector activity in a highly specific biological electron transfer complex: the cytochrome P450(cam)-putidaredoxin couple. Biochemistry. 2003;42:5649–5656. doi: 10.1021/bi034263s. [DOI] [PubMed] [Google Scholar]
- 40.Hong L., Smolin N., Smith J.C. Three classes of motion in the dynamic neutron-scattering susceptibility of a globular protein. Phys. Rev. Lett. 2011;107:148102. doi: 10.1103/PhysRevLett.107.148102. [DOI] [PubMed] [Google Scholar]
- 41.Xue Y., Pavlova M.S., Skrynnikov N.R. Methyl rotation barriers in proteins from 2H relaxation data. Implications for protein structure. J. Am. Chem. Soc. 2007;129:6827–6838. doi: 10.1021/ja0702061. [DOI] [PubMed] [Google Scholar]
- 42.Baudry J. Van der Waals interactions and decrease of the rotational barrier of methyl-sized rotators: a theoretical study. J. Am. Chem. Soc. 2006;128:11088–11093. doi: 10.1021/ja060731k. [DOI] [PubMed] [Google Scholar]
- 43.Lounnas V., Wade R.C. Exceptionally stable salt bridges in cytochrome P450cam have functional roles. Biochemistry. 1997;36:5402–5417. doi: 10.1021/bi9622940. [DOI] [PubMed] [Google Scholar]
- 44.Wade R.C., Gabdoulline R.R., Lounnas V. Electrostatic steering and ionic tethering in enzyme-ligand binding: insights from simulations. Proc. Natl. Acad. Sci. USA. 1998;95:5942–5949. doi: 10.1073/pnas.95.11.5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poulos T.L. Cytochrome P450 flexibility. Proc. Natl. Acad. Sci. USA. 2003;100:13121–13122. doi: 10.1073/pnas.2336095100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prasad S., Mazumdar S., Mitra S. Binding of camphor to Pseudomonas putida cytochrome p450(cam): steady-state and picosecond time-resolved fluorescence studies. FEBS Lett. 2000;477:157–160. doi: 10.1016/s0014-5793(00)01745-2. [DOI] [PubMed] [Google Scholar]
- 47.Jung C. Insight into protein structure and protein-ligand recognition by Fourier transform infrared spectroscopy. J. Mol. Recognit. 2000;13:325–351. doi: 10.1002/1099-1352(200011/12)13:6<325::AID-JMR507>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 48.Asciutto E.K., Dang M., Pochapsky T.C. Experimentally restrained molecular dynamics simulations for characterizing the open states of cytochrome P450cam. Biochemistry. 2011;50:1664–1671. doi: 10.1021/bi101820d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li X.C., Baudry J., Schuler M.A. Structural and functional divergence of insect CYP6B proteins: from specialist to generalist cytochrome P450. Proc. Natl. Acad. Sci. USA. 2004;101:2939–2944. doi: 10.1073/pnas.0308691101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williams P.A., Cosme J., Jhoti H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature. 2003;424:464–468. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 51.Haines D.C., Chen B., Peterson J.A. Crystal structure of inhibitor-bound P450BM-3 reveals open conformation of substrate access channel. Biochemistry. 2008;47:3662–3670. doi: 10.1021/bi7023964. [DOI] [PubMed] [Google Scholar]
- 52.Williams P.A., Cosme J., Jhoti H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.