

Abstract
Many intrinsically disordered proteins (IDPs) are significantly unstructured under physiological conditions. A number of these IDPs have been shown to undergo coupled folding and binding reactions whereby they can gain structure upon association with an appropriate partner protein. In general, these systems display weaker binding affinities than do systems with association between completely structured domains, with micromolar Kd values appearing typical. One such system is the association between α- and β-spectrin, where two partially structured, incomplete domains associate to form a fully structured, three-helix bundle, the spectrin tetramerization domain. Here, we use this model system to demonstrate a method for fitting association and dissociation kinetic traces where, using typical biophysical concentrations, the association reactions are expected to be highly reversible. We elucidate the unusually slow, two-state kinetics of spectrin assembly in solution. The advantages of studying kinetics in this regime include the potential for gaining equilibrium constants as well as rate constants, and for performing experiments with low protein concentrations. We suggest that this approach would be particularly appropriate for high-throughput mutational analysis of two-state reversible binding processes.
Introduction
The association and dissociation of protein complexes is of enormous biological significance, especially because most proteins exist in such forms in vivo, with monomers accounting for only 19% of proteins in Escherichia coli (1). The study of protein-protein association kinetics initially focused on interactions between folded, fully structured proteins. However, more recently, there has been increasing interest in association of intrinsically disordered proteins (IDPs), proteins that are largely unstructured in the unbound state and that form structure only upon binding another protein (2–4). These coupled folding and binding reactions are predicted to be widespread in biology (5), with important examples found in signaling (3), transcription (6), apoptosis regulation (7), and disease-related proteins (8).
It has been suggested that some protein-protein interactions have evolved to include this coupled folding and binding to separate specificity from the strength of binding, i.e., to allow for many residue-specific interactions at an interface without necessarily having tight binding (3). This could be of particular utility in signaling pathways that may require more transient interactions. Thus, combining many of the published equilibrium constants (Kds) for hetero-/homodimeric systems, several of which have been tabulated previously by Huang et al. (9) and Qin et al. (10), show that, on average, systems where one or both proteins undergo substantial folding upon association have significantly weaker binding than those with two structured proteins (p ≤ 0.001) (Fig. 1 A). It was recently suggested that such a difference in binding strength would most likely be due to a change in the dissociation rate constants (11), and the data presented here show this to be the case. The dissociation rate constants are statistically significantly different between the two groups (p ≤ 0.0001) (Fig. 1 C), but the association rate constants are not (p ≈ 0.5, see Methods) (Fig. 1 B).
Figure 1.
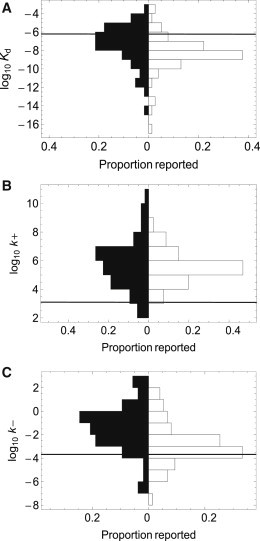
Literature values of the equilibrium dissociation constant (A), association rate constant (B), and dissociation rate constant (C) for various ordered (open bars) and disordered (closed bars) dimeric complexes are displayed in paired histograms. Complexes between two ordered proteins are, in general more stable, with lower dissociation rate constants. The values used to generate these plots are shown in Table S1 and Table S2. A detailed description of the statistical methods used for comparing the data sets is found in the Methods. For comparison, the horizontal lines indicate the values determined for the spectrin tetramerization complex in this study, using the reversible model.
It is imperative to consider the equilibrium constant (relative to the protein concentration) before choosing experimental and fitting strategies. The importance of Kd in experimental design is clear in equilibrium denaturation, the standard method for obtaining equilibrium constants for protein folding and homodimer association (12). In Fig. 2, we illustrate the effect of denaturant (urea) on the formation of a hypothetical two-state dimeric complex with a Kd of 0.39 μM and meq = 1.20 kcal mol−1 M−1 at equimolar concentrations of the two components, between 1 and 40 μM, typical for a biophysical investigation. These equilibrium curves cannot be fit to achieve accurate estimates of the equilibrium constant, because under these micromolar protein concentrations, a significant proportion of the protein is uncomplexed, even at 0 M denaturant (Fig. 2). Indeed, for a protein with this Kd we would need 3.9 mM of each subunit to complex 99% of the protein.
Figure 2.
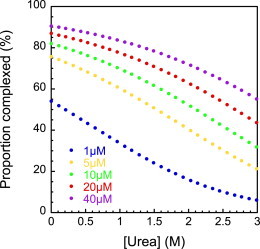
Equilibrium denaturation curve modeled for the spectrin tetramerization domain using values of Kd (0.39 μM) and meq (1.21 kcal−1 mol−1 M−1) obtained from the kinetic data. The range of urea concentrations shown is that accessible for the study of the protein-protein association reaction, since above 3 M urea the neighboring α1 domain begins to unfold. Note that under these conditions we never achieved 100% complex or 100% disassociated.
There is now a wealth of data on the thermodynamics of IDP binding, but few kinetic studies. In particular, there are few studies that assess the effects of mutagenesis on the kinetics of binding—analogous to protein folding Φ-value analysis. Recent studies on the artificial model system, S-peptide and S-protein (13), and various peptides binding PDZ domains (14,15) are likely to pave the way for such studies to begin. In these previous studies, measurements were made in a single set of solvent conditions using pseudo-first-order kinetics. To do a similar study under a number of solvent conditions, in which, for example, denaturant concentration, viscosity, and ionic strength are varied, would be prohibitively time- and protein-consuming. Thus, we sought to find an alternative, more efficient method of data collection and analysis.
Given the typical micromolar equilibrium constants for IDPs and the concentration ranges suitable for biophysical studies, it is likely that under standard experimental conditions, the exchange between dissociated and complexed protein will be observably reversible for equimolar mixing. This means that individual kinetic traces should not be fitted to the standard single-exponential function used in unimolecular protein-folding studies, or to hyperbolic functions used for effectively irreversible association in heterodimeric systems. Such approaches would give incorrect estimates of the association rate constants, which do not properly account for the apparent concentration dependence of the reaction. In the Supporting Material, we review approaches that have been used to investigate reversible heterodimer association, for extracting both kinetic and equilibrium constants. Most groups have utilized pseudo-first-order conditions, and extracted rate constants by conducting experiments at a range of subunit concentrations. In this work, we used an alternative approach. We show, for one IDP system, that estimates for the association and dissociation rate constants, and for the equilibrium dissociation constant for the complex, can be extracted from individual kinetic traces fitted to an equation appropriate for a two-state reversible process. This approach is fully suitable where high-throughput analysis is required, such as in large-scale mutational analyses and condition screening, because it is cost-effective in terms of both time and protein. We note that a significant advantage to using this approach to study two-state association kinetics is the quick access to an estimate of Kd for the complex, without the need for high concentrations and/or isothermal titration calorimetry (ITC) equipment. This also allows for Kd to be obtained in solutions that are unsuitable for ITC, such as those with high concentrations of denaturant or where higher-order aggregation is an issue. Furthermore, we show that, although unfolding kinetic traces may fit well to single-exponential functions (as might be expected to demonstrate operation of an irreversible, first-order process), ignoring the association reaction in this manner can lead to misleadingly high values for the dissociation rate constant (and therefore also to artificially low and high values for the unfolding m-value and Kd, respectively).
One system with approximately average binding strength (for a disordered protein complex) is the head-to-head dimerization reaction that forms the erythroid spectrin tetramerization domain. Various estimates have been made of the equilibrium constant for the heterodimeric spectrin complex formed by the association of α- and β-spectrin (16–26). These suggest that under the typical concentrations used in biophysical studies the association is relatively reversible, containing a significant back reaction. Full-length eyrthyroid α- and β-spectrin are large, membrane-bound, multidomain proteins containing 21 and 16 linked spectrin repeats, respectively. Each spectrin repeat is a folded, 106-amino-acid (aa), three-helix bundle, and the equilibrium stability and kinetics of folding of such domains have been studied extensively (27–32). The long α- and β-spectrin associate to form α2β2 tetramers on the intracellular side of the cell membranes, where they perform a structural role, providing a resilient cytoskeleton that can withstand deformations during circulation (33,34). Many mutations have been reported that affect this critical interaction and lead to disease (16,35). To form the functional tetramer, the α- and β-spectrin associate side to side to form antiparallel αβ heterodimers, which then associate head to head to form the α2β2 tetramer. A crystal structure recently obtained (36) demonstrates that the head-to-head interaction involves a dimerization reaction between the ∼30-aa N-terminus of α-spectrin and the ∼70-aa of the C-terminus of β-spectrin to form a three-helix bundle that resembles a fully folded spectrin domain. Constructs α0α1 and β16β17 are used in this study, both of which contain the sequence required to form the tetramerization domain (α0 and β17) plus one neighboring, folded spectrin domain (α1 and β16) (Fig. 3). These constructs undergo the head-to-head dimerization.
Figure 3.
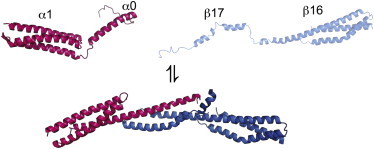
Cartoon depicting association of α0α1 (pink) and β16β17 (blue). Cartoon of unbound α0α1 is based on an NMR structure (pdb 1OWA). No structural information is available for unbound β16β17; a representation was made using Swiss-pdb. The cartoon of the final bound complex is based on a crystal structure (pdb 3LBX). The figure was prepared using PyMol.
Here, we report a complete analysis of the kinetics (and associated thermodynamics) of association of recombinantly expressed human eyrthrocyte α- and β-spectrin constructs that model the formation of the spectrin tetramerization domain using stopped-flow experiments. We show that use of a reversible model to analyze the data gives reliable, reproducible estimates of kinetic and thermodynamic parameters and their denaturant dependence.
Materials and Methods
Histograms and statistical analyses of assembled literature equilibrium and rate constants
The histograms were generated from values we found reported in the literature and are not exhaustive (see Table S1 and Table S2). Nontwo-state results were excluded from the tables. Where multiple estimates had been made for the same protein system, an average was taken. Where multiple mutational analyses had been performed, results were only included for the wild-type proteins. This approach was taken to avoid introducing significant bias into the result. Although bias as a result of the choice of systems reported in the literature cannot be ruled out, this factor might be considered to affect both groups in roughly the same way.
We compared the two groups (ordered and disordered subunits) to find out if the apparent differences in the rate and equilibrium constants were statistically significant. We investigated which statistical tests would be appropriate to compare the groups, as follows: The standardly applied t-test approach was not appropriate in this case, because the data sets are not well described by a normal distribution (according to the Kolmogorov-Smirnov test). The variances of the groups are not statistically significant at the 5% level for either Kd (p = 0.095) or k− (p = 0.19) according to a Conover test, demonstrating that the Kruskal-Wallis test was appropriate in these cases. Hence, statistical significances between the (log values of the) two groups were tested using the Kruskal-Wallis test, and the resulting p-values, both of which demonstrated statistical significance, are reported in the Results section. The variances of the disordered and ordered groups were statistically significant for k+ (p = 0.0014). However, we obtained very similar p-values using both the Kruskal-Wallis test (p = 0.55), which assumes similar variances between the two groups, and a Welch’s t-test (p = 0.54), which assumes normality but is appropriate for unequal populations with unequal variances. We therefore conclude that the difference in association rate constant between the two groups is not statistically significant. All statistical calculations were performed using Mathematica 8.0 (Wolfram Research, Champaign, IL).
Protein expression and purification
Synthetic genes for α0α1 (αI2–163) and β16β17 (βI1898–2083) were obtained from Genscript USA and inserted into a modified version of the pRSETA vector that encodes an N-terminal histidine tag with a thrombin cleavage site between the tag and the protein. Protein expression was carried out in E. coli C41 (DE3) (37) grown in 2× TY media at 37°C. Expression was induced once the cells reached an optical density at 600 nm of 0.4–0.6 AU by adding IPTG to a final concentration of 100 μg ml−1. The cells were grown overnight at 25°C, harvested by centrifugation, and resuspended in phosphate-buffered saline (PBS) (50 mM sodium phosphate and 150 mM NaCl, pH 7.0). The harvested cells were sonicated and centrifuged, and the protein was purified from the soluble fraction by affinity chromatography on Ni2+-agarose resin. Bound protein was removed by thrombin cleavage and further purified by gel filtration using a 26/60 G75 Superdex column (GE Healthcare, Milwaukee, WI). All proteins were stored at 4°C in PBS.
Kinetics of complex association and dissociation
Complex association and dissociation were monitored by following the change in intrinsic fluorescence on an SX18 or an SX20 stopped-flow spectrometer (Applied Photophysics, Leatherhead, United Kingdom) upon 1:1 volume mixing. An excitation wavelength of 280 nm was used with a 320 nm cut-off filter, and the temperature was maintained at 25 ± 0.1°C. All experiments were carried out in PBS + 5 mM dithiothreitol, and at least three traces were averaged for a typical measurement. For each association experiment, solutions of α0α1 and β16β17 at equal concentration were mixed. For each dissociation experiment, a solution of α0α1 and β16β17 at 1:1 molar ratio was pre-equilibrated for 2 h before mixing with an appropriate denaturant solution. Kinetic traces from association experiments were analyzed using Kaleidagraph (Synergy Software, Ann Arbor, MI), and unfolding was analyzed with ProFit (Quantumsoft, Tomsk, Russia) with the equations described in the text.
Circular dichroism
Circular dichroism (CD) spectra of each protein mixture in PBS + 5 mM dithiothreitol were obtained with an AP Photophysics Chirascan CD spectrophotometer at 25°C in a 2-mm-pathlength cuvette.
Fitting kinetics traces
Irreversible association model
| (Scheme 1) |
For irreversible association described by Scheme 1, and when the total concentration of protein A is equal to that of protein B, the concentration of the complex is as previously described (38), where k+ is the association rate constant, [A]T is the total concentration of A, and t is time. The fluorescence throughout the time course can simply be described by Eq. 1:
| (1) |
where F0 is the fluorescence of 1 M A + B, and ΔF is the change in fluorescence when 1 M of A + B is converted to 1 M AB. Combining these results, it is clear that the fluorescence throughout the reaction may be described by Eq. 2:
| (2) |
Reversible association and dissociation
| (Scheme 2) |
According to a reversible model, as described by Scheme 2, where k+ and k− are the association and dissociation rate constants, respectively, the rate of complex formation is given by Eq. 3:
| (3) |
When the experimental constraints are applied, i.e., [A]T = [B]T, identifying the equilibrium constant as Kd = k−/k+, and requiring mass balance, i.e., [A] = [B] = [A]T − [AB] leads to Eq. 4:
| (4) |
Integration of Eq. 4, evaluation of the integration constant at time zero, and combining Eq. 4 with Eq. 1 leads to Eq. 5, a general equation for describing the system fluorescence in a reversible system, which applies equally for association and dissociation kinetic traces,
| (5) |
where , , and [AB]0 is the concentration of complex at the start of the reaction. For kinetic traces where the complex was pre-equilibrated before dissociation, the concentration of complex in the syringe ([AB]syr) was calculated using Eq. 6, where [A]T,syr is the total concentration of protein A in the syringe:
| (6) |
Irreversible dissociation
If complex dissociation is effectively irreversible, then the process can be fit to Eq. 7:
| (7) |
Isothermal titration calorimetry
ITC experiments to determine tetramer interaction stability were performed on a VP-ITC from MicroCal (GE). β16β17, 16-50 μM, was titrated 14–20 injections, at 1000-s intervals, of α0α1 (150–600 μM). Protein concentrations were confirmed by absorbance spectroscopy, using a Cary 400 spectrophotometer (Varian, Palo Alto, CA). The injection profile was integrated and the resulting data fitted with a one-site binding model in Origin 7 for MicroCal (Northampton, MA).
Results
Kinetics of association in absence of denaturant
When studying homodimers, it is necessary to displace the equilibrium with temperature or denaturing cosolvents. One advantage of heterodimeric systems is that the kinetics of association can be followed by mixing of the two subunits without having to address these requirements. Consequently, any signal change observed when subunits are mixed is entirely a result of their interaction, without the complication of possible conformational changes in the individual subunits. Association of the heterodimeric spectrin complex was followed by measuring the (tryptophan) fluorescence quenching when α0α1 and β16β17 domains were mixed together in a 1:1 molar ratio in a stopped-flow spectrophotometer. Under the conditions used (5–40 μM range of protein concentration), the spectrin subunits were seen to associate slowly (Fig. 4 A), and the data fit poorly to the simple irreversible model (Scheme 1, Eq. 2 (38)), as judged by systematic deviations apparent in the residuals, and by the overall χ2 value for the fit. As described earlier, this is unsurprising; a significant back reaction is expected, given the Kd values for almost equivalent pairs of domains of 0.4 ± 0.1 μM (16), 1.1 ± 0.1 μM (18), and 0.84 μM (25) that have been reported previously. Instead, we considered a reversible kinetic model (Scheme 2, Materials and Methods), where a significant dissociation reaction is also in operation. Here, the time-dependent fluorescent signal is described by Eq. 5, which reflects the reversible nature of the process itself by being equally applicable for describing the dissociation of preformed complex induced, for example, by rapid mixing with a solution of denaturant. In the case of 1:1 molar mixing, this equation may be simplified by identifying [AB]0 = 0, providing an analogous equation to that used by Wendt et al. for describing the association of a heterodimeric leucine zipper (39). Eq. 5 reduces to Eq. 2 in the limit that k− approaches zero. We found that the kinetic traces were well fit by Eq. 5, which contains only four free variables; ΔF, F0, k+, and Kd (the natural variables for association), without the requirement for further fixing. This is in contrast to previous attempts to fit such equations by Wendt et al. (39) and Milla et al. (40) for a homodimeric system, which have required at least one further variable to be fixed (typically ΔF or F0).
Figure 4.
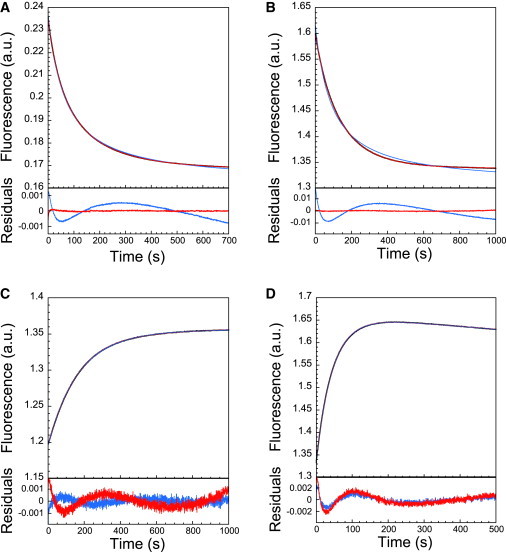
(Intrinsic tryptophan) fluorescence stopped-flow traces of the association and dissociation of the spectrin tetramerization domain and accompanying fit residuals for irreversible (blue; Eq. 2 and single-exponential equation) and reversible (red; Eq. 5) schemes. (A and B) Association of domains after 1:1 mixing of α0α1 and β16β17 domains at 1:1 molar ratios at 0 M urea (A) and 1.4 M urea (B). (C and D) Dissociation of preformed complex (preequilibrated for 2 h without urea) after rapid dilution into urea to final urea concentrations of 1.5 M (C) and 3.0 M (D). In fitting C and D, a linear drift term was included. Note that although the reversible fit is significantly better for association, the irreversible (single-exponential) fit is apparently as good for dissociation. However, the fitted parameters from the irreversible fits are concentration-dependent (see Results and Fig. S1), indicating that the method is nonetheless inappropriate.
The rate constant for association, k+, may then be estimated from single kinetic traces. The values of k+ obtained from this fitting method (Fig. S1) were similar for a range of different protein concentrations <k+> = 650 ± 70 M−1 s−1.
Promisingly, the Kd values obtained from each individual trace (0.1–0.6 μM) are in good agreement with the equilibrium values obtained in previous studies (16–26) and appeared independent of protein concentration, again in accordance with a two-state mechanism. The dissociation rate constant can be extracted from the association kinetic traces (k− = Kdk+), providing estimates of k− (<k−> = (2.6 ± 1.2) × 10−4 s−1), which are also independent of concentration. Interestingly, the kinetics of association for a similar pair of constructs was investigated using surface-attached surface plasmon resonance spectroscopy (25). Although the Kd reported, 0.84 μM, is close to what we observed, both k+ and k− were lower by an order of magnitude. Possible reasons for this discrepancy include different buffer conditions and effects due to the attachment of one partner to a surface.
Association and folding in the presence of denaturant
Denaturants such as urea have been shown to slow the folding and binding of dimeric proteins through the destabilization of partially structured transition states. The two spectrin subunits were pre-equilibrated in the same concentration of urea for 30 min and then mixed in equal amounts. As in the absence of denaturant, the kinetic traces obtained were well fit by Eq. 5 (see, e.g., Fig. 4 B). The log of the association rate constant was observed to decrease linearly with urea concentration (Fig. 5 A). It is important to note that the apparent noisiness of the data comes from the fact that each data point represents fitting of an individual kinetic trace. We have included even very low concentration data points in the figure legend to demonstrate that all the resultant parameters are essentially independent of protein concentration. m-values for association (m+) can be determined from the gradient in these k+ plots (Fig. 5 A) multiplied by RT. This is similar to a kinetic m-value in protein folding and m+ values were consistent for all concentrations investigated, around <m+>= −0.40 ± 0.03 kcal mol−1 M−1. By providing estimates for k+ at multiple denaturant concentrations, k+(H2O) and Kd(H2O) values are better approximated. Again, there was no significant concentration dependence displayed, the weighted averages for the equilibrium constant and association rate constant being <Kd(H2O)> = 0.39 ± 0.02 μM and <k+> = 630 ± 20 M−1 s−1, respectively. Using the well established technique of ITC, we also estimated the equilibrium constant (Fig. S2) as 0.68 ± 0.46 μM, in excellent agreement with the value we obtained by fitting the association kinetics traces.
Figure 5.
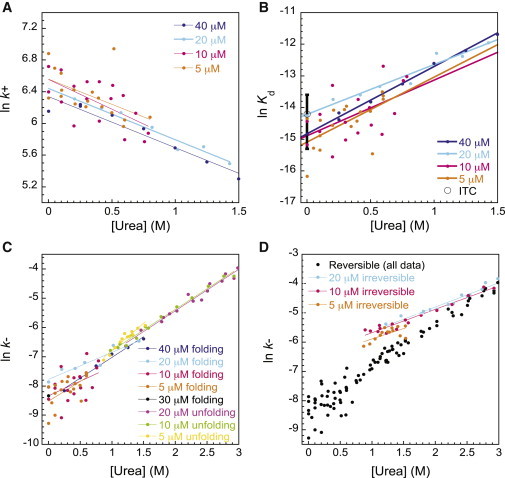
(A–C) Dependence of the association rate constant (A), equilibrium dissociation constant (B), and dissociation rate constant (C) on urea concentration. Values are extracted from fitting the reversible model. (D) Dissociation rate constants obtained from the reversible model are compared to apparent rate constants extracted from fits to an irreversible model. Note that the apparent noisiness of the data comes from the fact that each data point represents fitting of an individual kinetic trace. We have included even very low concentration data points in the figure legend to demonstrate that all the resultant parameters are essentially protein concentration independent. In (B), the Kd from ITC experiments is included for comparison, the error bar represents the standard deviation for three independent experiments.
As expected, urea destabilized the final structured complex relative to the dissociated subunits, with ln(Kd) increasing linearly with denaturant concentration (Fig. 5 B), the gradients suggesting that meq = 1.20 ± 0.03 kcal mol−1 M−1. As previously mentioned, the fit k+ and Kd values can be used to calculate the dissociation rate constant, k− (Fig. 5 C) for each reaction: <k−> = (2.64 ± 0.15) × 10−4 s−1, and ln(k−) also increased linearly with denaturant, to give (<m−> = 0.74 ± 0.04 kcal mol−1 M−1), in good support of the two-state model.
Unfolding and dissociation
Dissociation of the spectrin complex was induced by pre-equilibrating a 1:1 molar mixture of α0α1 and β16β17 in the absence of denaturant for 2 h followed by rapid mixing to varying concentrations of urea. There was an upper limit to the final concentration of urea investigated as, above 3 M urea, the neighboring folded spectrin (α1) began to unfold significantly (data not shown). All unfolding kinetic traces experienced some drift in fluorescence signal and all fits included a linear drift term.
Since Eq. 5 can take into account a nonzero initial concentration of complex, it theoretically can be used directly to fit kinetic traces for dissociation as well as association. However, to aid the fitting process, we rewrote the exponential rate constant in terms of k− rather than k+, identifying . Again, in the limit as k+ approaches zero, Eq. 5 simply reduces to the equation for an irreversible process (Eq. 7). Unfortunately, if left to fit Kd and [AB]0, the errors on these variables were prohibitively large. The fitting was greatly improved by fixing [AB]0 and the Kd using expected values from the association kinetic traces. Although all data presented were fit using weighted values of Kd(H2O) and meq from all data sets, the quality of fit and the values obtained were relatively insensitive to the folding data set chosen (data not shown). In additional support of the two-state reversible model, the k− values and m− values from the unfolding data (Fig. 5 C), which have weighted averages of <k−> = (2.61 ± 0.16) × 10−4 s−1 and <m−> = 0.85 ± 0.02 kcal mol−1 M−1, agree very well with those calculated from the folding kinetic traces (Fig. S1, E and F).
We found that the data for the urea-induced dissociation of the spectrin tetramerization domain also fit surprisingly well to a single-exponential function (with linear drift term), even when a significant reverse (association) rate was expected (Fig. 4, C and D). However, the rate constants obtained in this manner show poorer linearity with urea and display greater sensitivity to the total spectrin concentration, particularly under conditions where tighter binding is expected, as in the case of lower concentrations of urea (Fig. 5 D).
Discussion
More complex analysis is required for studying the binding kinetics of two associating proteins at concentrations around their equilibrium constant than for studying effectively irreversible tight binding, but the former has the potential to yield more information. Considering the typical concentration range for biophysical protein-folding studies, together with the average equilibrium constant for disordered protein complexes (Fig. 1 A), it is clear that this is an area that merits investigation. We have successfully fit the kinetics of such a system, the dimerization of α- and β-spectrin termini to form a folded three-helix bundle known as the spectrin tetramerization domain. Using the unique properties of a reversible second-order system, we fit each individual association kinetic trace to provide concentration-independent estimates of k+ and Kd (and k−).
All of the data presented here for the heterodimeric spectrin complex are consistent with a two-state model for folding, without a populated intermediate. Aside from the good fits of the data to Eq. 5, the estimated values of k+(H2O), Kd(H2O) (k−(H2O)), m+, and m− are all independent of protein concentration within error. In general, this would not be expected to be the case if the model were incorrect. An example of this is found in the irreversible fits to the folding and unfolding data presented here, where the values do appear to depend upon the concentration employed in the experiment. In addition, the Kd we obtained from our fitting matched very well with that from the well established equilibrium technique of ITC. Both estimates are also close to that obtained for a very similar pair of spectrin domains by Gaetani et al. (16) (0.4 ± 0.1 μM at 23°C).
The linear dependence of ln(k+) on denaturant concentration, as is typically found in monomeric protein folding, has also been seen previously for the association of both homodimers (12) and heterodimers (41). It has been shown empirically that the kinetic and equilibrium m-values are linearly related to the change in solvent-accessible surface area (ΔSASA) between unfolded and folded states (42,43). In the dimerization reaction, it is difficult to distinguish whether the urea dependence of the association rate is due to the denaturant-destabilizing structure formation in the transition state (as for monomeric protein folding), or to the denaturant-favoring conformations in the individual subunits that are less competent to associate. The magnitude of the experimental m-value can be used to assess the degree of structure formation upon association. Using the GETAREA (44) algorithm, and the crystal structure for the bound complex (pdb 3LBX), it is possible to calculate an estimate for ΔSASA upon dissociation, keeping all helices intact. The calculated value of 4212 Å2 was used to estimate the m-value for this dissociation (42), 0.82 ± 0.04 kcal mol−1 M−1. This is significantly lower than the experimental weighted average of <meq> = 1.21 ± 0.03 kcal mol−1 M−1 and suggests that the final dissociated subunits have some loss of helical character to account for this exposure of surface area. The experimental value of meq is similar to the values obtained from the unfolding of isolated spectrin domains R15 (1.8 ± 0.1 kcal mol−1 M−1), R16 (1.9 ± 0.1 kcal mol−1 M−1), and R17 (2.0 ± 0.1 kcal mol−1 M−1) determined by equilibrium denaturation (29). Also, we may be confident that there is some significant folding taking place during the association reaction, since the α-helical content is increased in the complex compared with the individual subunits according to circular dichroism measurements (Fig. S3). The dimerization of the spectrin tetramerization domain can therefore be justifiably classified as a coupled folding and binding process.
The association rate constant of the spectrin domains (630 M−1 s−1, an order of magnitude faster than that determined using surface plasmon resonance spectroscopy, where the interactions take place on a surface) is notable in itself. The association is markedly slower than a purely diffusion-limited reaction, which we would expect to be in the range of 105–109 M−1 s−1 (45–47), even when electrostatics and orientation are taken into account (estimated at 4 × 105 M−1 s−1 using the TransComp web server (10) based on the crystal structure of the tetramerization domain (36) (pdb 3LBX). It is also lower than most previously recorded values for other homo- and heterodimers, which can range from 102–1010 M−1 s−1 (Fig. 1 B) (9,12). Using our data, it is not possible to distinguish between the proposed mechanisms of association for IDPs, that is, induced fit or conformational selection (48). This slow association may reflect the presence of a significant folding event (and, therefore, energy barrier) between the relatively unstructured subunits and the final associated domain. Alternatively, a specific orientation of the two subunits or particularly low-population conformation in one or both of the subunits may be required for a productive collision. It is interesting that, given the importance of maintaining the integrity of red blood cells, Nature would evolve a pair of proteins for which the association rate constant is abnormally slow and, consequently, binding is relatively weak. The importance of this interaction in vivo is manifested by the fact that most mutations of spectrin domains that are related to hereditary hemolytic anaemias are associated with this tetramerization site (16). In vivo, these proteins are bound to the membrane, which perhaps accelerates their association by positioning the proteins in an orientation favorable for binding. Favorable side-to-side interaction between the full-length spectrin domains was not considered here (22). Furthermore, it is not clear what exactly the effective concentration of these domains is within the context of the membrane, so it is difficult to interpret the equilibrium constant in this case. Perhaps this result highlights the limitations of studying some interactions outside of their cellular context. Alternatively, it has been suggested that this weak binding could have a functional role in the unusual deformability of the erythrocyte cell membrane (34). We note that the association of nonerythroid spectrin results in a significantly stronger complex and faster association kinetics (25).
Reflecting the reversible nature of the process, the same equation as that used for association was used here to describe the dissociation of preformed spectrin complex, induced by dilution into denaturant. This provided estimates of the dissociation rate constant and its linear response to denaturant that were consistent with those from the association reaction, in good support of a two-state reversible model for the interaction. Unfolding of similar homo- and heterodimeric complexes is often fit to single-exponential decay functions appropriate for effectively irreversible systems (Eq. 7). We found that this method could obtain comparably good fits to the individual kinetic traces, although the estimated dissociation rates were not concentration-independent and did not match well with those estimated from the association experiments. In fact, detailed inspection of Eq. 5 shows that fitting with the irreversible model will always overestimate the dissociation rate constant, to an extent that depends on the relative sizes of Kd and protein concentration. This highlights the need to exert caution when using a simple single-exponential fit for unfolding data of dimeric systems, since k− values may deviate from actual values while retaining a reasonable fit.
Conclusions
Here, we show that by deliberately working at protein concentrations near the Kd, where association and dissociation reactions are highly reversible, kinetic and thermodynamic parameters can be determined from individual kinetic traces with confidence. This may prove to be a useful method for systems with low binding affinities, such as the binding of IDPs, and this has been demonstrated for the coupled folding and binding of the spectrin tetramerization domain. Provided that the system is demonstrably two-state, the approach described here for obtaining equilibrium constants can provide a convenient alternative to ITC and equilibrium denaturation (49). It requires only stopped-flow apparatus and avoids the requirement for high protein concentrations that may be experimentally inaccessible where higher-order aggregation is an issue.
Acknowledgments
This work was supported by the Wellcome Trust (grant number WT095195) and the Division of Intramural Research at the National Heart, Lung, and Blood Institute, National Institutes of Health. J.C. is a Wellcome Trust Senior Research Fellow. J.M.R. is supported by a Biotechnology and Biological Sciences Research Council studentship. S.A.H. is supported by a National Institutes of Health Cambridge Partnership fellowship and the Cambridge Overseas Trust.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References
- 1.Goodsell D.S., Olson A.J. Structural symmetry and protein function. Annu. Rev. Biophys. Biomol. Struct. 2000;29:105–153. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
- 2.Dyson H.J., Wright P.E. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 3.Wright P.E., Dyson H.J. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 4.Wright P.E., Dyson H.J. Linking folding and binding. Curr. Opin. Struct. Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oldfield C.J., Cheng Y., Dunker A.K. Coupled folding and binding with α-helix-forming molecular recognition elements. Biochemistry. 2005;44:12454–12470. doi: 10.1021/bi050736e. [DOI] [PubMed] [Google Scholar]
- 6.Radivojac P., Iakoucheva L.M., Dunker A.K. Intrinsic disorder and functional proteomics. Biophys. J. 2007;92:1439–1456. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hinds M.G., Smits C., Day C.L. Bim, Bad and Bmf: intrinsically unstructured BH3-only proteins that undergo a localized conformational change upon binding to prosurvival Bcl-2 targets. Cell Death Differ. 2007;14:128–136. doi: 10.1038/sj.cdd.4401934. [DOI] [PubMed] [Google Scholar]
- 8.Uversky V.N., Oldfield C.J., Dunker A.K. Unfoldomics of human diseases: linking protein intrinsic disorder with diseases. BMC Genomics. 2009;10(Suppl 1):S7. doi: 10.1186/1471-2164-10-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang Y.Q., Liu Z.R. Kinetic advantage of intrinsically disordered proteins in coupled folding-binding process: a critical assessment of the “fly-casting” mechanism. J. Mol. Biol. 2009;393:1143–1159. doi: 10.1016/j.jmb.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 10.Qin S., Pang X., Zhou H.X. Automated prediction of protein association rate constants. Structure. 2011;19:1744–1751. doi: 10.1016/j.str.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou H.X. Intrinsic disorder: signaling via highly specific but short-lived association. Trends Biochem. Sci. 2012;37:43–48. doi: 10.1016/j.tibs.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rumfeldt J.A.O., Galvagnion C., Meiering E.M. Conformational stability and folding mechanisms of dimeric proteins. Prog. Biophys. Mol. Biol. 2008;98:61–84. doi: 10.1016/j.pbiomolbio.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Bachmann A., Wildemann D., Kiefhaber T. Mapping backbone and side-chain interactions in the transition state of a coupled protein folding and binding reaction. Proc. Natl. Acad. Sci. USA. 2011;108:3952–3957. doi: 10.1073/pnas.1012668108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karlsson O.A., Chi C.N., Jemth P. The transition state of coupled folding and binding for a flexible β-finger. J. Mol. Biol. 2012;417:253–261. doi: 10.1016/j.jmb.2012.01.042. [DOI] [PubMed] [Google Scholar]
- 15.Haq S.R., Chi C.N., Jemth P. Side-chain interactions form late and cooperatively in the binding reaction between disordered peptides and PDZ domains. J. Am. Chem. Soc. 2012;134:599–605. doi: 10.1021/ja209341w. [DOI] [PubMed] [Google Scholar]
- 16.Gaetani M., Mootien S., Speicher D.W. Structural and functional effects of hereditary hemolytic anemia-associated point mutations in the α spectrin tetramer site. Blood. 2008;111:5712–5720. doi: 10.1182/blood-2007-11-122457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DeSilva T.M., Peng K.C., Speicher D.W. Analysis of human red cell spectrin tetramer (head-to-head) assembly using complementary univalent peptides. Biochemistry. 1992;31:10872–10878. doi: 10.1021/bi00159a030. [DOI] [PubMed] [Google Scholar]
- 18.Mehboob S., Jacob J., Fung L.W. Structural analysis of the α N-terminal region of erythroid and nonerythroid spectrins by small-angle x-ray scattering. Biochemistry. 2003;42:14702–14710. doi: 10.1021/bi0353833. [DOI] [PubMed] [Google Scholar]
- 19.Lam V.Q., Antoniou C., Fung L.W. Association studies of erythroid α-spectrin at the tetramerization site. Br. J. Haematol. 2009;147:392–395. doi: 10.1111/j.1365-2141.2009.07876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Long F., McElheny D., Fung L.W. Conformational change of erythroid α-spectrin at the tetramerization site upon binding β-spectrin. Protein Sci. 2007;16:2519–2530. doi: 10.1110/ps.073115307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mehboob S., Luo B.H., Fung L.W. αβ Spectrin coiled coil association at the tetramerization site. Biochemistry. 2001;40:12457–12464. doi: 10.1021/bi010984k. [DOI] [PubMed] [Google Scholar]
- 22.Cherry L., Menhart N., Fung L.W.M. Interactions of the α-spectrin N-terminal region with β-spectrin. Implications for the spectrin tetramerization reaction. J. Biol. Chem. 1999;274:2077–2084. doi: 10.1074/jbc.274.4.2077. [DOI] [PubMed] [Google Scholar]
- 23.Mehboob S., Song Y.L., Fung L.W. Crystal structure of the nonerythroid α-spectrin tetramerization site reveals differences between erythroid and nonerythroid spectrin tetramer formation. J. Biol. Chem. 2010;285:14572–14584. doi: 10.1074/jbc.M109.080028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park S., Johnson M.E., Fung L.W.M. Nuclear magnetic resonance studies of mutations at the tetramerization region of human α spectrin. Blood. 2002;100:283–288. doi: 10.1182/blood.v100.1.283. [DOI] [PubMed] [Google Scholar]
- 25.Bignone P.A., Baines A.J. Spectrin α II and β II isoforms interact with high affinity at the tetramerization site. Biochem. J. 2003;374:613–624. doi: 10.1042/BJ20030507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henniker A., Ralston G.B. Reinvestigation of the thermodynamics of spectrin self-association. Biophys. Chem. 1994;52:251–258. doi: 10.1016/0301-4622(94)00094-z. [DOI] [PubMed] [Google Scholar]
- 27.Wensley B.G., Gärtner M., Clarke J. Different members of a simple three-helix bundle protein family have very different folding rate constants and fold by different mechanisms. J. Mol. Biol. 2009;390:1074–1085. doi: 10.1016/j.jmb.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wensley B.G., Batey S., Clarke J. Experimental evidence for a frustrated energy landscape in a three-helix-bundle protein family. Nature. 2010;463:685–688. doi: 10.1038/nature08743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scott K.A., Batey S., Clarke J. The folding of spectrin domains I: wild-type domains have the same stability but very different kinetic properties. J. Mol. Biol. 2004;344:195–205. doi: 10.1016/j.jmb.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 30.Scott K.A., Clarke J. Spectrin R16: broad energy barrier or sequential transition states? Protein Sci. 2005;14:1617–1629. doi: 10.1110/ps.051377105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scott K.A., Randles L.G., Clarke J. The folding of spectrin domains II: Φ-value analysis of R16. J. Mol. Biol. 2004;344:207–221. doi: 10.1016/j.jmb.2004.09.023. [DOI] [PubMed] [Google Scholar]
- 32.Scott K.A., Randles L.G., Clarke J. The folding pathway of spectrin R17 from experiment and simulation: using experimentally validated MD simulations to characterize states hinted at by experiment. J. Mol. Biol. 2006;359:159–173. doi: 10.1016/j.jmb.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 33.Bennett V., Healy J. Organizing the fluid membrane bilayer: diseases linked to spectrin and ankyrin. Trends Mol. Med. 2008;14:28–36. doi: 10.1016/j.molmed.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 34.Salomao M., An X., Baines A.J. Mammalian α I-spectrin is a neofunctionalized polypeptide adapted to small highly deformable erythrocytes. Proc. Natl. Acad. Sci. USA. 2006;103:643–648. doi: 10.1073/pnas.0507661103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nicolas G., Pedroni S., Lecomte M.C. Spectrin self-association site: characterization and study of β-spectrin mutations associated with hereditary elliptocytosis. Biochem. J. 1998;332:81–89. doi: 10.1042/bj3320081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ipsaro J.J., Harper S.L., Speicher D.W. Crystal structure and functional interpretation of the erythrocyte spectrin tetramerization domain complex. Blood. 2010;115:4843–4852. doi: 10.1182/blood-2010-01-261396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miroux B., Walker J.E. Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J. Mol. Biol. 1996;260:289–298. doi: 10.1006/jmbi.1996.0399. [DOI] [PubMed] [Google Scholar]
- 38.Schreiber G., Fersht A.R. Interaction of barnase with its polypeptide inhibitor barstar studied by protein engineering. Biochemistry. 1993;32:5145–5150. doi: 10.1021/bi00070a025. [DOI] [PubMed] [Google Scholar]
- 39.Wendt H., Leder L., Bosshard H.R. Very rapid, ionic strength-dependent association and folding of a heterodimeric leucine zipper. Biochemistry. 1997;36:204–213. doi: 10.1021/bi961672y. [DOI] [PubMed] [Google Scholar]
- 40.Milla M.E., Sauer R.T. P22 Arc repressor: folding kinetics of a single-domain, dimeric protein. Biochemistry. 1994;33:1125–1133. doi: 10.1021/bi00171a011. [DOI] [PubMed] [Google Scholar]
- 41.Goldberg J.M., Baldwin R.L. Kinetic mechanism of a partial folding reaction. 1. Properties of the reaction and effects of denaturants. Biochemistry. 1998;37:2546–2555. doi: 10.1021/bi972402y. [DOI] [PubMed] [Google Scholar]
- 42.Geierhaas C.D., Nickson A.A., Vendruscolo M. BPPred: a Web-based computational tool for predicting biophysical parameters of proteins. Protein Sci. 2007;16:125–134. doi: 10.1110/ps.062383807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Myers J.K., Pace C.N., Scholtz J.M. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rychkov G., Petukhov M. Joint neighbors approximation of macromolecular solvent accessible surface area. J. Comput. Chem. 2007;28:1974–1989. doi: 10.1002/jcc.20550. [DOI] [PubMed] [Google Scholar]
- 45.Northrup S.H., Erickson H.P. Kinetics of protein-protein association explained by Brownian dynamics computer simulation. Proc. Natl. Acad. Sci. USA. 1992;89:3338–3342. doi: 10.1073/pnas.89.8.3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berg O.G., von Hippel P.H. Diffusion-controlled macromolecular interactions. Annu. Rev. Biophys. Biophys. Chem. 1985;14:131–160. doi: 10.1146/annurev.bb.14.060185.001023. [DOI] [PubMed] [Google Scholar]
- 47.Janin J. The kinetics of protein-protein recognition. Proteins. 1997;28:153–161. doi: 10.1002/(sici)1097-0134(199706)28:2<153::aid-prot4>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 48.Zhou H.X., Pang X., Lu C. Rate constants and mechanisms of intrinsically disordered proteins binding to structured targets. Phys. Chem. Chem. Phys. 2012;14:10466–10476. doi: 10.1039/c2cp41196b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pace C.N. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 50.Panayotou G., Gish G., Waterfield M.D. Interactions between SH2 domains and tyrosine-phosphorylated platelet-derived growth factor β-receptor sequences: analysis of kinetic parameters by a novel biosensor-based approach. Mol. Cell. Biol. 1993;13:3567–3576. doi: 10.1128/mcb.13.6.3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ladbury J.E., Hensmann M., Campbell I.D. Alternative modes of tyrosyl phosphopeptide binding to a Src family SH2 domain: implications for regulation of tyrosine kinase activity. Biochemistry. 1996;35:11062–11069. doi: 10.1021/bi960543e. [DOI] [PubMed] [Google Scholar]
- 52.Chen T., Repetto B., Gilfillan A.M. Interaction of phosphorylated FcεRIγ immunoglobulin receptor tyrosine activation motif-based peptides with dual and single SH2 domains of p72syk. Assessment of binding parameters and real time binding kinetics. J. Biol. Chem. 1996;271:25308–25315. doi: 10.1074/jbc.271.41.25308. [DOI] [PubMed] [Google Scholar]
- 53.Chook Y.M., Gish G.D., Pawson T. The Grb2-mSos1 complex binds phosphopeptides with higher affinity than Grb2. J. Biol. Chem. 1996;271:30472–30478. doi: 10.1074/jbc.271.48.30472. [DOI] [PubMed] [Google Scholar]
- 54.Nominé Y., Botuyan M.V., Mer G. Kinetic analysis of interaction of BRCA1 tandem breast cancer C-terminal domains with phosphorylated peptides reveals two binding conformations. Biochemistry. 2008;47:9866–9879. doi: 10.1021/bi702247d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rudolph M.G., Bayer P., Wittinghofer A. The Cdc42/Rac interactive binding region motif of the Wiskott Aldrich syndrome protein (WASP) is necessary but not sufficient for tight binding to Cdc42 and structure formation. J. Biol. Chem. 1998;273:18067–18076. doi: 10.1074/jbc.273.29.18067. [DOI] [PubMed] [Google Scholar]
- 56.Yu X., Wang Q.Y., Rong L. Kinetic analysis of binding interaction between the subgroup A Rous sarcoma virus glycoprotein SU and its cognate receptor Tva: calcium is not required for ligand binding. J. Virol. 2003;77:7517–7526. doi: 10.1128/JVI.77.13.7517-7526.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Waxham M.N., Tsai A.L., Putkey J.A. A mechanism for calmodulin (CaM) trapping by CaM-kinase II defined by a family of CaM-binding peptides. J. Biol. Chem. 1998;273:17579–17584. doi: 10.1074/jbc.273.28.17579. [DOI] [PubMed] [Google Scholar]
- 58.Anderluh G., Gökçe I., Lakey J.H. A natively unfolded toxin domain uses its receptor as a folding template. J. Biol. Chem. 2004;279:22002–22009. doi: 10.1074/jbc.M313603200. [DOI] [PubMed] [Google Scholar]
- 59.Anderluh G., Hong Q., Lakey J.H. Concerted folding and binding of a flexible colicin domain to its periplasmic receptor TolA. J. Biol. Chem. 2003;278:21860–21868. doi: 10.1074/jbc.M300411200. [DOI] [PubMed] [Google Scholar]
- 60.Garcia K.C., Radu C.G., Ward E.S. Kinetics and thermodynamics of T cell receptor- autoantigen interactions in murine experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA. 2001;98:6818–6823. doi: 10.1073/pnas.111161198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woodside D.G., Obergfell A., Ginsberg M.H. The N-terminal SH2 domains of Syk and ZAP-70 mediate phosphotyrosine-independent binding to integrin β cytoplasmic domains. J. Biol. Chem. 2002;277:39401–39408. doi: 10.1074/jbc.M207657200. [DOI] [PubMed] [Google Scholar]
- 62.Sugase K., Lansing J.C., Wright P.E. Tailoring relaxation dispersion experiments for fast-associating protein complexes. J. Am. Chem. Soc. 2007;129:13406–13407. doi: 10.1021/ja0762238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krantz B.A., Moran L.B., Sosnick T.R. D/H amide kinetic isotope effects reveal when hydrogen bonds form during protein folding. Nat. Struct. Biol. 2000;7:62–71. doi: 10.1038/71265. [DOI] [PubMed] [Google Scholar]
- 64.Dürr E., Jelesarov I., Bosshard H.R. Extremely fast folding of a very stable leucine zipper with a strengthened hydrophobic core and lacking electrostatic interactions between helices. Biochemistry. 1999;38:870–880. doi: 10.1021/bi981891e. [DOI] [PubMed] [Google Scholar]
- 65.Ibarra-Molero B., Makhatadze G.I., Matthews C.R. Mapping the energy surface for the folding reaction of the coiled-coil peptide GCN4-p1. Biochemistry. 2001;40:719–731. doi: 10.1021/bi001438e. [DOI] [PubMed] [Google Scholar]
- 66.Steinmetz M.O., Jelesarov I., Kammerer R.A. Molecular basis of coiled-coil formation. Proc. Natl. Acad. Sci. USA. 2007;104:7062–7067. doi: 10.1073/pnas.0700321104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wendt H., Berger C., Bosshard H.R. Kinetics of folding of leucine zipper domains. Biochemistry. 1995;34:4097–4107. doi: 10.1021/bi00012a028. [DOI] [PubMed] [Google Scholar]
- 68.Matsuno H., Furusawa H., Okahata Y. Kinetic study of phosphorylation-dependent complex formation between the kinase-inducible domain (KID) of CREB and the KIX domain of CBP on a quartz crystal microbalance. Chemistry. 2004;10:6172–6178. doi: 10.1002/chem.200400030. [DOI] [PubMed] [Google Scholar]
- 69.Lengyel C.S., Willis L.J., McFarland B.J. Mutations designed to destabilize the receptor-bound conformation increase MICA-NKG2D association rate and affinity. J. Biol. Chem. 2007;282:30658–30666. doi: 10.1074/jbc.M704513200. [DOI] [PubMed] [Google Scholar]
- 70.Lavery D.N., McEwan I.J. Functional characterization of the native NH2-terminal transactivation domain of the human androgen receptor: binding kinetics for interactions with TFIIF and SRC-1a. Biochemistry. 2008;47:3352–3359. doi: 10.1021/bi702220p. [DOI] [PubMed] [Google Scholar]
- 71.Zanier K., Charbonnier S., Travé G. Kinetic analysis of the interactions of human papillomavirus E6 oncoproteins with the ubiquitin ligase E6AP using surface plasmon resonance. J. Mol. Biol. 2005;349:401–412. doi: 10.1016/j.jmb.2005.03.071. [DOI] [PubMed] [Google Scholar]
- 72.Schon O., Friedler A., Fersht A.R. Molecular mechanism of the interaction between MDM2 and p53. J. Mol. Biol. 2002;323:491–501. doi: 10.1016/s0022-2836(02)00852-5. [DOI] [PubMed] [Google Scholar]
- 73.Catimel B., Teh T., Kobe B. Biophysical characterization of interactions involving importin-α during nuclear import. J. Biol. Chem. 2001;276:34189–34198. doi: 10.1074/jbc.M103531200. [DOI] [PubMed] [Google Scholar]
- 74.Gianni S., Engström A., Jemth P. The kinetics of PDZ domain-ligand interactions and implications for the binding mechanism. J. Biol. Chem. 2005;280:34805–34812. doi: 10.1074/jbc.M506017200. [DOI] [PubMed] [Google Scholar]
- 75.Papadakos G., Housden N.G., Kleanthous C. Kinetic basis for the competitive recruitment of TolB by the intrinsically disordered translocation domain of colicin E9. J. Mol. Biol. 2012;418:269–280. doi: 10.1016/j.jmb.2012.01.039. [DOI] [PubMed] [Google Scholar]
- 76.Kleerekoper Q.K., Putkey J.A. PEP-19, an intrinsically disordered regulator of calmodulin signaling. J. Biol. Chem. 2009;284:7455–7464. doi: 10.1074/jbc.M808067200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Choi H.J., Huber A.H., Weis W.I. Thermodynamics of β-catenin-ligand interactions: the roles of the N- and C-terminal tails in modulating binding affinity. J. Biol. Chem. 2006;281:1027–1038. doi: 10.1074/jbc.M511338200. [DOI] [PubMed] [Google Scholar]
- 78.Chemes L.B., Sánchez I.E., de Prat-Gay G. Kinetic recognition of the retinoblastoma tumor suppressor by a specific protein target. J. Mol. Biol. 2011;412:267–284. doi: 10.1016/j.jmb.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 79.Reference deleted in proof.
- 80.Arai M., Ferreon J.C., Wright P.E. Quantitative analysis of multisite protein-ligand interactions by NMR: binding of intrinsically disordered p53 transactivation subdomains with the TAZ2 domain of CBP. J. Am. Chem. Soc. 2012;134:3792–3803. doi: 10.1021/ja209936u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zitzewitz J.A., Bilsel O., Matthews C.R. Probing the folding mechanism of a leucine zipper peptide by stopped-flow circular dichroism spectroscopy. Biochemistry. 1995;34:12812–12819. doi: 10.1021/bi00039a042. [DOI] [PubMed] [Google Scholar]
- 82.Rosengarth A., Rösgen J., Hinz H.J. Slow unfolding and refolding kinetics of the mesophilic Rop wild-type protein in the transition range. Eur. J. Biochem. 1999;264:989–995. doi: 10.1046/j.1432-1327.1999.00712.x. [DOI] [PubMed] [Google Scholar]
- 83.Topping T.B., Gloss L.M. Stability and folding mechanism of mesophilic, thermophilic and hyperthermophilic archael histones: the importance of folding intermediates. J. Mol. Biol. 2004;342:247–260. doi: 10.1016/j.jmb.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 84.Bowie J.U., Sauer R.T. Equilibrium dissociation and unfolding of the Arc repressor dimer. Biochemistry. 1989;28:7139–7143. doi: 10.1021/bi00444a001. [DOI] [PubMed] [Google Scholar]
- 85.Wales T.E., Richardson J.S., Fitzgerald M.C. Facile chemical synthesis and equilibrium unfolding properties of CopG. Protein Sci. 2004;13:1918–1926. doi: 10.1110/ps.04671804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zeeb M., Lipps G., Balbach J. Folding and association of an extremely stable dimeric protein from Sulfolobus islandicus. J. Mol. Biol. 2004;336:227–240. doi: 10.1016/j.jmb.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 87.Liang H., Terwilliger T.C. Reversible denaturation of the gene V protein of bacteriophage f1. Biochemistry. 1991;30:2772–2782. doi: 10.1021/bi00225a006. [DOI] [PubMed] [Google Scholar]
- 88.Grant S.K., Deckman I.C., Meek T.D. Use of protein unfolding studies to determine the conformational and dimeric stabilities of HIV-1 and SIV proteases. Biochemistry. 1992;31:9491–9501. doi: 10.1021/bi00154a023. [DOI] [PubMed] [Google Scholar]
- 89.Varghese L.T., Sinha R.K., Irudayaraj J. Study of binding and denaturation dynamics of IgG and anti-IgG using dual color fluorescence correlation spectroscopy. Anal. Chim. Acta. 2008;625:103–109. doi: 10.1016/j.aca.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 90.Castro M.J., Anderson S. Alanine point-mutations in the reactive region of bovine pancreatic trypsin inhibitor: effects on the kinetics and thermodynamics of binding to β-trypsin and α-chymotrypsin. Biochemistry. 1996;35:11435–11446. doi: 10.1021/bi960515w. [DOI] [PubMed] [Google Scholar]
- 91.Tello D., Eisenstein E., Poljak R.J. Structural and physicochemical analysis of the reaction between the anti-lysozyme antibody D1.3 and the anti-idiotopic antibodies E225 and E5.2. J. Mol. Recognit. 1994;7:57–62. doi: 10.1002/jmr.300070108. [DOI] [PubMed] [Google Scholar]
- 92.Kelley R.F., Costas K.E., Lazarus R.A. Analysis of the factor VIIa binding site on human tissue factor: effects of tissue factor mutations on the kinetics and thermodynamics of binding. Biochemistry. 1995;34:10383–10392. doi: 10.1021/bi00033a009. [DOI] [PubMed] [Google Scholar]
- 93.Johanson K., Appelbaum E., Doyle M., Hensley P., Zhao B., Abdel-Meguid S.S., Young P., Cook R., Carr S., Matico R. Binding interactions of human interleukin 5 with its receptor α subunit. Large scale production, structural, and functional studies of Drosophila-expressed recombinant proteins. J. Biol. Chem. 1995;270:9459–9471. doi: 10.1074/jbc.270.16.9459. [DOI] [PubMed] [Google Scholar]
- 94.Cunningham B.C., Wells J.A. Comparison of a structural and a functional epitope. J. Mol. Biol. 1993;234:554–563. doi: 10.1006/jmbi.1993.1611. [DOI] [PubMed] [Google Scholar]
- 95.Wu H., Myszka D.G., Hendrickson W.A. Kinetic and structural analysis of mutant CD4 receptors that are defective in HIV gp120 binding. Proc. Natl. Acad. Sci. USA. 1996;93:15030–15035. doi: 10.1073/pnas.93.26.15030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Myszka D.G., Sweet R.W., Doyle M.L. Energetics of the HIV gp120-CD4 binding reaction. Proc. Natl. Acad. Sci. USA. 2000;97:9026–9031. doi: 10.1073/pnas.97.16.9026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Singha N.C., Vlamis-Gardikas A., Holmgren A. Real-time kinetics of the interaction between the two subunits, Escherichia coli thioredoxin and gene 5 protein of phage T7 DNA polymerase. J. Biol. Chem. 2003;278:21421–21428. doi: 10.1074/jbc.M302310200. [DOI] [PubMed] [Google Scholar]
- 98.Raman C.S., Jemmerson R., Allen M.J. Diffusion-limited rates for monoclonal antibody binding to cytochrome c. Biochemistry. 1992;31:10370–10379. doi: 10.1021/bi00157a027. [DOI] [PubMed] [Google Scholar]
- 99.Potempa J., Kwon K., Travis J. Inter-α-trypsin inhibitor. Inhibition spectrum of native and derived forms. J. Biol. Chem. 1989;264:15109–15114. [PubMed] [Google Scholar]
- 100.Nielsen P.K., Bønsager B.C., Svensson B. Kinetics and energetics of the binding between barley α-amylase/subtilisin inhibitor and barley α-amylase 2 analyzed by surface plasmon resonance and isothermal titration calorimetry. Biochemistry. 2003;42:1478–1487. doi: 10.1021/bi020508+. [DOI] [PubMed] [Google Scholar]
- 101.Bowman B.F., Peterson J.A., Stull J.T. Pre-steady-state kinetics of the activation of rabbit skeletal muscle myosin light chain kinase by Ca2+/calmodulin. J. Biol. Chem. 1992;267:5346–5354. [PubMed] [Google Scholar]
- 102.England P., Brégégère F., Bedouelle H. Energetic and kinetic contributions of contact residues of antibody D1.3 in the interaction with lysozyme. Biochemistry. 1997;36:164–172. doi: 10.1021/bi961419y. [DOI] [PubMed] [Google Scholar]
- 103.Silversmith R.E., Levin M.D., Bourret R.B. Kinetic characterization of catalysis by the chemotaxis phosphatase CheZ. Modulation of activity by the phosphorylated CheY substrate. J. Biol. Chem. 2008;283:756–765. doi: 10.1074/jbc.M704400200. [DOI] [PubMed] [Google Scholar]
- 104.Xavier K.A., McDonald S.M., Willson R.C. Association and dissociation kinetics of bobwhite quail lysozyme with monoclonal antibody HyHEL-5. Protein Eng. 1999;12:79–83. doi: 10.1093/protein/12.1.79. [DOI] [PubMed] [Google Scholar]
- 105.Li X.L., Fong C.C., Yang M.S. Measurement of binding kinetics between PI3-K and phosphorylated IGF-1R using a surface plasmon resonance biosensor. Microchim. Acta. 2008;162:253–260. [Google Scholar]
- 106.Fridmann Y., Kafri G., Horovitz A. Dissociation of the GroEL-GroES asymmetric complex is accelerated by increased cooperativity in ATP binding to the GroEL ring distal to GroES. Biochemistry. 2002;41:5938–5944. doi: 10.1021/bi020117v. [DOI] [PubMed] [Google Scholar]
- 107.Murphy A.J., Kemp F., Love J. Surface plasmon resonance characterization of calspermin-calmodulin binding kinetics. Anal. Biochem. 2008;376:61–72. doi: 10.1016/j.ab.2008.01.023. [DOI] [PubMed] [Google Scholar]
- 108.Razai A., Garcia-Rodriguez C., Marks J.D. Molecular evolution of antibody affinity for sensitive detection of botulinum neurotoxin type A. J. Mol. Biol. 2005;351:158–169. doi: 10.1016/j.jmb.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 109.Wu H., Pfarr D.S., Young J.F. Ultra-potent antibodies against respiratory syncytial virus: effects of binding kinetics and binding valence on viral neutralization. J. Mol. Biol. 2005;350:126–144. doi: 10.1016/j.jmb.2005.04.049. [DOI] [PubMed] [Google Scholar]
- 110.Solheim S.A., Petsalaki E., Berge T. Interactions between the Fyn SH3-domain and adaptor protein Cbp/PAG derived ligands, effects on kinase activity and affinity. FEBS J. 2008;275:4863–4874. doi: 10.1111/j.1742-4658.2008.06626.x. [DOI] [PubMed] [Google Scholar]
- 111.Swope S.L., Huganir R.L. Binding of the nicotinic acetylcholine receptor to SH2 domains of Fyn and Fyk protein tyrosine kinases. J. Biol. Chem. 1994;269:29817–29824. [PubMed] [Google Scholar]
- 112.Rich R.L., Cannon M.J., Myszka D.G. Extracting kinetic rate constants from surface plasmon resonance array systems. Anal. Biochem. 2008;373:112–120. doi: 10.1016/j.ab.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 113.Lambert B., Buckle M. Characterisation of the interface between nucleophosmin (NPM) and p53: potential role in p53 stabilisation. FEBS Lett. 2006;580:345–350. doi: 10.1016/j.febslet.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 114.Spoerner M., Herrmann C., Wittinghofer A. Dynamic properties of the Ras switch I region and its importance for binding to effectors. Proc. Natl. Acad. Sci. USA. 2001;98:4944–4949. doi: 10.1073/pnas.081441398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Curmi P.A., Andersen S.S., Sobel A. The stathmin/tubulin interaction in vitro. J. Biol. Chem. 1997;272:25029–25036. doi: 10.1074/jbc.272.40.25029. [DOI] [PubMed] [Google Scholar]
- 116.Ishino T., Pasut G., Chaiken I. Kinetic interaction analysis of human interleukin 5 receptor α mutants reveals a unique binding topology and charge distribution for cytokine recognition. J. Biol. Chem. 2004;279:9547–9556. doi: 10.1074/jbc.M309327200. [DOI] [PubMed] [Google Scholar]
- 117.Abdiche Y., Malashock D., Pons J. Determining kinetics and affinities of protein interactions using a parallel real-time label-free biosensor, the Octet. Anal. Biochem. 2008;377:209–217. doi: 10.1016/j.ab.2008.03.035. [DOI] [PubMed] [Google Scholar]
- 118.Keeler C., Jablonski E.M., Hodsdon M.E. The kinetics of binding human prolactin, but not growth hormone, to the prolactin receptor vary over a physiologic pH range. Biochemistry. 2007;46:2398–2410. doi: 10.1021/bi061958v. [DOI] [PubMed] [Google Scholar]
- 119.Zaccheo O.J., Prince S.N., Hassan A.B. Kinetics of insulin-like growth factor II (IGF-II) interaction with domain 11 of the human IGF-II/mannose 6-phosphate receptor: function of CD and AB loop solvent-exposed residues. J. Mol. Biol. 2006;359:403–421. doi: 10.1016/j.jmb.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 120.Albeck S., Schreiber G. Biophysical characterization of the interaction of the β-lactamase TEM-1 with its protein inhibitor BLIP. Biochemistry. 1999;38:11–21. doi: 10.1021/bi981772z. [DOI] [PubMed] [Google Scholar]
- 121.Wohlgemuth S., Kiel C., Herrmann C. Recognizing and defining true Ras binding domains I: biochemical analysis. J. Mol. Biol. 2005;348:741–758. doi: 10.1016/j.jmb.2005.02.048. [DOI] [PubMed] [Google Scholar]
- 122.Nakajima H., Cocquerel L., Levy S. Kinetics of HCV envelope proteins’ interaction with CD81 large extracellular loop. Biochem. Biophys. Res. Commun. 2005;328:1091–1100. doi: 10.1016/j.bbrc.2005.01.056. [DOI] [PubMed] [Google Scholar]
- 123.Portmann R., Magnani D., Solioz M. Interaction kinetics of the copper-responsive CopY repressor with the cop promoter of Enterococcus hirae. J. Biol. Inorg. Chem. 2004;9:396–402. doi: 10.1007/s00775-004-0536-1. [DOI] [PubMed] [Google Scholar]
- 124.Selve N., Wegner A. pH-dependent rate of formation of the gelsolin-actin complex from gelsolin and monomeric actin. Eur. J. Biochem. 1987;168:111–115. doi: 10.1111/j.1432-1033.1987.tb13394.x. [DOI] [PubMed] [Google Scholar]
- 125.Troeberg L., Tanaka M., Nagase H. E. coli expression of TIMP-4 and comparative kinetic studies with TIMP-1 and TIMP-2: insights into the interactions of TIMPs and matrix metalloproteinase 2 (gelatinase A) Biochemistry. 2002;41:15025–15035. doi: 10.1021/bi026454l. [DOI] [PubMed] [Google Scholar]
- 126.Martel V., Filhol O., Cochet C. Dynamic localization/association of protein kinase CK2 subunits in living cells: a role in its cellular regulation? Ann. N. Y. Acad. Sci. 2002;973:272–277. doi: 10.1111/j.1749-6632.2002.tb04648.x. [DOI] [PubMed] [Google Scholar]
- 127.Li W., Zhang C., Farzan M. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24:1634–1643. doi: 10.1038/sj.emboj.7600640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kirby I., Lord R., Santis G. Adenovirus type 9 fiber knob binds to the coxsackie B virus-adenovirus receptor (CAR) with lower affinity than fiber knobs of other CAR-binding adenovirus serotypes. J. Virol. 2001;75:7210–7214. doi: 10.1128/JVI.75.15.7210-7214.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dooley H., Stanfield R.L., Flajnik M.F. First molecular and biochemical analysis of in vivo affinity maturation in an ectothermic vertebrate. Proc. Natl. Acad. Sci. USA. 2006;103:1846–1851. doi: 10.1073/pnas.0508341103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Malchiodi E.L., Eisenstein E., Mariuzza R.A. Superantigen binding to a T cell receptor β chain of known three-dimensional structure. J. Exp. Med. 1995;182:1833–1845. doi: 10.1084/jem.182.6.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wigelsworth D.J., Krantz B.A., Collier R.J. Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, CMG2, with protective antigen. J. Biol. Chem. 2004;279:23349–23356. doi: 10.1074/jbc.M401292200. [DOI] [PubMed] [Google Scholar]
- 132.Fleury D., Wharton S.A., Bizebard T. Antigen distortion allows influenza virus to escape neutralization. Nat. Struct. Biol. 1998;5:119–123. doi: 10.1038/nsb0298-119. [DOI] [PubMed] [Google Scholar]
- 133.Lavergne J.P., Jault J.M., Galinier A. Insights into the functioning of Bacillus subtilis HPr kinase/phosphatase: affinity for its protein substrates and role of cations and phosphate. Biochemistry. 2002;41:6218–6225. doi: 10.1021/bi025613y. [DOI] [PubMed] [Google Scholar]
- 134.Shimaoka M., Xiao T., Springer T.A. Structures of the α L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell. 2003;112:99–111. doi: 10.1016/s0092-8674(02)01257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wyer J.R., Willcox B.E., Jakobsen B.K. T cell receptor and coreceptor CD8 αα bind peptide-MHC independently and with distinct kinetics. Immunity. 1999;10:219–225. doi: 10.1016/s1074-7613(00)80022-9. [DOI] [PubMed] [Google Scholar]
- 136.Olson M.W., Gervasi D.C., Fridman R. Kinetic analysis of the binding of human matrix metalloproteinase-2 and -9 to tissue inhibitor of metalloproteinase (TIMP)-1 and TIMP-2. J. Biol. Chem. 1997;272:29975–29983. doi: 10.1074/jbc.272.47.29975. [DOI] [PubMed] [Google Scholar]
- 137.Lauwereys M., Arbabi Ghahroudi M., Muyldermans S. Potent enzyme inhibitors derived from dromedary heavy-chain antibodies. EMBO J. 1998;17:3512–3520. doi: 10.1093/emboj/17.13.3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Monaco-Malbet S., Berthet-Colominas C., Cusack S. Mutual conformational adaptations in antigen and antibody upon complex formation between an Fab and HIV-1 capsid protein p24. Structure. 2000;8:1069–1077. doi: 10.1016/s0969-2126(00)00507-4. [DOI] [PubMed] [Google Scholar]
- 139.Fierens K., Gils A., Delcour J.A. His374 of wheat endoxylanase inhibitor TAXI-I stabilizes complex formation with glycoside hydrolase family 11 endoxylanases. FEBS J. 2005;272:5872–5882. doi: 10.1111/j.1742-4658.2005.04987.x. [DOI] [PubMed] [Google Scholar]
- 140.van der Merwe P.A., Barclay A.N., Davis S.J. Human cell-adhesion molecule CD2 binds CD58 (LFA-3) with a very low affinity and an extremely fast dissociation rate but does not bind CD48 or CD59. Biochemistry. 1994;33:10149–10160. doi: 10.1021/bi00199a043. [DOI] [PubMed] [Google Scholar]
- 141.Maenaka K., van der Merwe P.A., Sondermann P. The human low affinity Fcγ receptors IIa, IIb, and III bind IgG with fast kinetics and distinct thermodynamic properties. J. Biol. Chem. 2001;276:44898–44904. doi: 10.1074/jbc.M106819200. [DOI] [PubMed] [Google Scholar]
- 142.Tsiang M., Jones G.S., Geleziunas R. Affinities between the binding partners of the HIV-1 integrase dimer-lens epithelium-derived growth factor (IN dimer-LEDGF) complex. J. Biol. Chem. 2009;284:33580–33599. doi: 10.1074/jbc.M109.040121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Jonsson A., Dogan J., Nygren P.A. Engineering of a femtomolar affinity binding protein to human serum albumin. Protein Eng. Des. Sel. 2008;21:515–527. doi: 10.1093/protein/gzn028. [DOI] [PubMed] [Google Scholar]
- 144.Baardsnes J., Hinck C.S., O’Connor-McCourt M.D. TβR-II discriminates the high- and low-affinity TGF-β isoforms via two hydrogen-bonded ion pairs. Biochemistry. 2009;48:2146–2155. doi: 10.1021/bi8019004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kellenberger C., Boudier C., Hietter H. Serine protease inhibition by insect peptides containing a cysteine knot and a triple-stranded β-sheet. J. Biol. Chem. 1995;270:25514–25519. doi: 10.1074/jbc.270.43.25514. [DOI] [PubMed] [Google Scholar]
- 146.Eggers C.T., Wang S.X., Craik C.S. The role of ecotin dimerization in protease inhibition. J. Mol. Biol. 2001;308:975–991. doi: 10.1006/jmbi.2001.4754. [DOI] [PubMed] [Google Scholar]
- 147.Presta L., Sims P., Kirchhofer D. Generation of a humanized, high affinity anti-tissue factor antibody for use as a novel antithrombotic therapeutic. Thromb. Haemost. 2001;85:379–389. [PubMed] [Google Scholar]
- 148.Nam T.W., Jung H.I., Cha S.S. Analyses of Mlc-IIBGlc interaction and a plausible molecular mechanism of Mlc inactivation by membrane sequestration. Proc. Natl. Acad. Sci. USA. 2008;105:3751–3756. doi: 10.1073/pnas.0709295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Ardelt W., Laskowski M., Jr. Turkey ovomucoid third domain inhibits eight different serine proteinases of varied specificity on the same …Leu18-Glu19 … reactive site. Biochemistry. 1985;24:5313–5320. doi: 10.1021/bi00341a007. [DOI] [PubMed] [Google Scholar]
- 150.Foote J., Winter G. Antibody framework residues affecting the conformation of the hypervariable loops. J. Mol. Biol. 1992;224:487–499. doi: 10.1016/0022-2836(92)91010-m. [DOI] [PubMed] [Google Scholar]
- 151.Lan K.L., Zhong H., Neubig R.R. Rapid kinetics of regulator of G-protein signaling (RGS)-mediated Gαi and Gαo deactivation. Gα specificity of RGS4 AND RGS7. J. Biol. Chem. 2000;275:33497–33503. doi: 10.1074/jbc.M005785200. [DOI] [PubMed] [Google Scholar]
- 152.Ying Q.L., Simon S.R. Kinetics of the inhibition of human leukocyte elastase by elafin, a 6-kilodalton elastase-specific inhibitor from human skin. Biochemistry. 1993;32:1866–1874. doi: 10.1021/bi00058a021. [DOI] [PubMed] [Google Scholar]
- 153.Wen K.K., McKane M., Rubenstein P.A. Control of the ability of profilin to bind and facilitate nucleotide exchange from G-actin. J. Biol. Chem. 2008;283:9444–9453. doi: 10.1074/jbc.M709806200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Radić Z., Kirchhoff P.D., Taylor P. Electrostatic influence on the kinetics of ligand binding to acetylcholinesterase. Distinctions between active center ligands and fasciculin. J. Biol. Chem. 1997;272:23265–23277. doi: 10.1074/jbc.272.37.23265. [DOI] [PubMed] [Google Scholar]
- 155.Cederholm-Williams S.A., De Cock F., Collen D. Kinetics of the reactions between streptokinase, plasmin and α 2-antiplasmin. Eur. J. Biochem. 1979;100:125–132. doi: 10.1111/j.1432-1033.1979.tb02040.x. [DOI] [PubMed] [Google Scholar]
- 156.Stewart R.C., Van Bruggen R. Association and dissociation kinetics for CheY interacting with the P2 domain of CheA. J. Mol. Biol. 2004;336:287–301. doi: 10.1016/j.jmb.2003.11.059. [DOI] [PubMed] [Google Scholar]
- 157.Kannt A.Y., Bendall S., D.S. The role of acidic residues of plastocyanin in its interaction with cytochrome ƒ. Biochim. Biophys. Acta. 1996;1277:115–126. [PubMed] [Google Scholar]
- 158.Jana R., Hazbun T.R., Mossing M.C. A folded monomeric intermediate in the formation of λ Cro dimer-DNA complexes. J. Mol. Biol. 1997;273:402–416. doi: 10.1006/jmbi.1997.1256. [DOI] [PubMed] [Google Scholar]
- 159.Bajaj K., Chakshusmathi G., Varadarajan R. Thermodynamic characterization of monomeric and dimeric forms of CcdB (controller of cell division or death B protein) Biochem. J. 2004;380:409–417. doi: 10.1042/BJ20031528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Talbott M., Hare M., Barbar E. Folding is coupled to dimerization of Tctex-1 dynein light chain. Biochemistry. 2006;45:6793–6800. doi: 10.1021/bi0600345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Schreiber G., Fersht A.R. Rapid, electrostatically assisted association of proteins. Nat. Struct. Biol. 1996;3:427–431. doi: 10.1038/nsb0596-427. [DOI] [PubMed] [Google Scholar]
- 162.Venezia C.F., Meany B.J., Barkley M.D. Kinetics of association and dissociation of HIV-1 reverse transcriptase subunits. Biochemistry. 2009;48:9084–9093. doi: 10.1021/bi9010495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Meisner W.K., Sosnick T.R. Fast folding of a helical protein initiated by the collision of unstructured chains. Proc. Natl. Acad. Sci. USA. 2004;101:13478–13482. doi: 10.1073/pnas.0404057101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kippen A.D., Fersht A.R. Analysis of the mechanism of assembly of cleaved barnase from two peptide fragments and its relevance to the folding pathway of uncleaved barnase. Biochemistry. 1995;34:1464–1468. doi: 10.1021/bi00004a042. [DOI] [PubMed] [Google Scholar]
- 165.Reference deleted in proof.
- 166.Kiel C., Selzer T., Herrmann C. Electrostatically optimized Ras-binding Ral guanine dissociation stimulator mutants increase the rate of association by stabilizing the encounter complex. Proc. Natl. Acad. Sci. USA. 2004;101:9223–9228. doi: 10.1073/pnas.0401160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kiefhaber T., Bachmann A., Jensen K.S. Dynamics and mechanisms of coupled protein folding and binding reactions. Curr. Opin. Struct. Biol. 2012;22:21–29. doi: 10.1016/j.sbi.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 168.Malatesta F. The study of bimolecular reactions under non-pseudo-first order conditions. Biophys. Chem. 2005;116:251–256. doi: 10.1016/j.bpc.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 169.Chi C.N., Engström A., Jemth P. Two conserved residues govern the salt and pH dependencies of the binding reaction of a PDZ domain. J. Biol. Chem. 2006;281:36811–36818. doi: 10.1074/jbc.M607883200. [DOI] [PubMed] [Google Scholar]
- 170.Onitsuka M., Kamikubo H., Kataoka M. Mechanism of induced folding: Both folding before binding and binding before folding can be realized in staphylococcal nuclease mutants. Proteins. 2008;72:837–847. doi: 10.1002/prot.21978. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.