

Abstract
The molecular basis of nonlinear optical (NLO) chiral effects in the amide I region of type I collagen was investigated using sum-frequency generation vibrational spectroscopy; chiral and achiral tensor elements were separated using different input/output beam polarization conditions. Spectra were obtained from native rat tail tendon (RTT) collagen and from cholesteric liquid crystal-like (LC) type I collagen films. Although RTT and LC collagen both possess long-range order, LC collagen lacks the complex hierarchical organization of RTT collagen. Their spectra were compared to assess the role of such organization in NLO chirality. No significant differences were observed between RTT and LC with respect to chiral or achiral spectra. These findings suggest that amide I NLO chiral effects in type I collagen assemblies arise predominantly from the chiral organization of amide chromophores within individual collagen molecules, rather than from supramolecular structures. The study suggests that sum-frequency generation vibrational spectroscopy may be uniquely valuable in exploring fundamental aspects of chiral nonlinearity in complex macromolecular structures.
Introduction
Collagen, the most abundant protein in virtually all higher life forms, was discovered to have nonlinear optical properties <10 years after the discovery of the laser (1). However, with the exception of early seminal studies by Roth and Freund (2–5), there were few detailed investigations of these properties until the development of ultrashort pulse lasers. Collagen, as well as other noncentrosymmetric, highly ordered biological macromolecules, is characterized by second-order nonlinear optical (NLO) susceptibility; that is, it is capable of undergoing nonlinear electric polarization under the influence of an intense, coherent optical field. The induced electric dipole moment leads to the emission of a secondary electromagnetic wave, a macroscopic event that represents the collective interaction of electric dipoles induced at the molecular level. In sum-frequency generation (SFG), a sample is irradiated with two incident beams, each at a different wavelength, of high intensity coherent light; the induced dipole results in the emission of a secondary beam with a frequency equal to the sum of the two incident beams. In second harmonic generation (SHG), a degenerate form of SFG, a hyperpolarized state is induced with an incident beam at a single wavelength; the secondary beam emitted by the sample has a frequency twice that of the incident beam. Both SFG and SHG have been particularly fruitful with respect to the development of new, to our knowledge, methods for analyzing structure, organization, and function in biological macromolecules with NLO properties.
SFG can also be used to perform polarization sensitive, resonance-enhanced spectroscopic analysis, a capability first demonstrated by Shen and co-workers (6–8). This technique, usually referred to as SFG vibrational (SFG-V) spectroscopy, is increasingly being used to analyze complex biological structures; studies have included analyses of avidin-biotin ligand binding (9), misfolding of key biological molecules, such as amyloid proteins (10), and conformation of peptide backbones at interfaces (11–15). A key feature of SFG-V is its ability to probe specific tensor elements through the use of different combinations of input and output polarizations, a capability that can be used to identify molecular resonances and molecular symmetries underlying the second-order NLO susceptibility of organic molecules. It should be noted, however, that higher energy dipolar contributions, which are not probed by SFG-V, do contribute to second-order NLO properties of nonresonant SHG. Although it is not known how these various effects collectively contribute to the SHG signal, SFG-V may provide useful information about specific aspects of SHG. For example, SFG-V studies performed by our group, in which we identified methylene and peptide groups as the dominant molecular resonances in collagen (14), provided experimental evidence consistent with a recent theoretical study showing that methylene groups dominate the polarization dependence of SHG measured in collagen (16). In this study, we use SFG-V to investigate the basis of the NLO chiral effects that appear to dominate the amide I region of native rat tail tendon (RTT), an observation we reported in our previous study (14).
The molecular basis of nonlinear chiral phenomena has been a topic of periodic discussions in the literature, with little consensus as to the fundamental mechanisms. In a very early study, Meijer et al. (17) reported on chiral nonlinearity arising from magnetic dipole effects in centrosymmetric crystals. Persoons and co-workers (18) have reported on detailed analyses of magnetic dipole contributions to chiral nonlinearity, although other investigators have suggested that interference between electric and magnetic dipole interactions may contribute to generation of chiral effects (19). In many studies, however, magnetic dipole effects have been shown to be orders of magnitude smaller than electric dipole effects on nonlinearity. Over the last several years, there has been growing interest by several different groups in the role of supramolecular orientational effects, stimulated in part by the study of Persoons et al. (20) showing that the presence of supramolecular chiral structures increased optical nonlinear effects of helicene enantiomers 50-fold. Burke et al. (21) presented experimental data showing that achiral charge-transfer chromophores assembled on chiral templates generated large chiral effects arising from the supramolecular chiral structures produced by the templating process. In ab initio studies, Simpson and co-workers (13) have reported on the contribution of orientational chirality to total SFG and SHG signal in different polypeptide secondary structures, including α-helices, 310-helices, and pleated sheets. Simpson has speculated that orientational chirality in helical polypeptides may arise from changes in rotation of the torsion angle ψ in successive amides, resulting in changes in their twist angles; the successively twisted planar moieties form a macroscopic chiral structure (13,22).
In this study we experimentally investigate the molecular resonant origins of NLO chirality in RTT collagen, using SFG-V spectroscopy to probe the fine structure of the amide I region. We use different combinations of beam input/output polarizations to acquire spectra with selected chiral or achiral tensor elements. We also investigate to what extent the chromophores organized at the supramolecular level may contribute to NLO chiral effects by comparing RTT and liquid crystal-like (LC) spectra. Despite inherent limitations in this approach due to the long-range order in LC collagen, it does allow us to determine the relative contributions to NLO chirality of the densely hierarchical supramolecular structures in RTT.
Theoretical background
This study was designed to use SFG-V spectroscopy to investigate the structural basis of the strong chiral signal in the amide I region of collagen that we had observed in our earlier study (14). We therefore used an infrared (IR) beam tuned from 1500 to 1750 cm−1, a range that encompasses the amide I region. When the frequency of the IR incident beam matches a molecular vibrational resonance, the SFG signal is enhanced, provided the transition is also Raman active. The SFG hyperpolarizability tensor can be expressed as a tensor product of the IR transition dipole moment and the Raman polarizability tensor:
| (1) |
where the subscripts i,j,k can each represent any of the axes defining the molecular frame (a, b, or c, see Fig. 1), is the IR dipole transition moment between the ground state and the nth energy level, polarized in the k direction, is the ij component of the anti-Stokes Raman polarizability tensor, is the frequency corresponding to the vibrational resonance, is the incident IR frequency, is the incident visible frequency, is the frequency of the generated signal, and is the relaxation time of the vibrationally excited state involved in the resonance.
Figure 1.
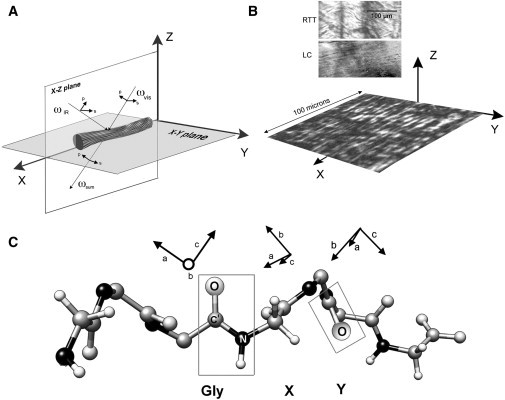
The geometry of experimental SFG-V spectroscopy. (A) The laboratory frame (X, Y, Z): the X-Z plane designates the plane of incidence within which the visible and IR incident beams are oriented orthogonally to each other and at a 45° angle to the collagen fibril, which also lies in the X-Z plane, parallel to the X axis. The polarization states of the input/output beams are defined relative to the X-Z plane, as indicated by the small black arrows; (B) Orientation of LC collagen film in the laboratory frame: a photomicrograph (20×) under polarized light of cholesteric LC collagen film, 100 × 100 microns, shows parallel bands with alternating light and dark stripes oriented longitudinally along the X axis and orthogonal to the Z axis; the inset above the laboratory frame shows that LC collagen lacks the crimp pattern seen in RTT collagen; (C) The molecular frame (a, b, c): the a and c axes designate the orientation of the two orthogonal dipoles of the amide bond, with the a axis parallel to the C-N bond on the polypeptide backbone. The molecular frames corresponding to the three peptide bonds in the collagen tripeptide Gly-X-Y are shown above a model of the polypeptide backbone. The chain is oriented such that the two-dimensional conformation of the chromophores is clearly seen in the Gly amide chromophore.
In the limit of negligible coupling between chromophores, the macroscopic second-order susceptibility tensor elements are expressed as the sum of the values for each element projected into the laboratory frame:
| (2) |
where N is the number density of the molecular group being accessed and I,J,K are placeholders, each of which can represent any of the axes defining the laboratory reference frame, X, Y, or Z, as shown in Fig. 1. The angular bracketed term, , is the orientational average of the Euler rotation matrix projecting each molecular hyperpolarizability term defined in the molecular frame (a, b, c) into the laboratory frame (X, Y, Z). The susceptibility tensor element subscripts correspond to the polarizations of the sum-frequency, visible, and IR beams, respectively. The beam geometry of the spectroscopic setup—which defines the laboratory frame—is shown in Fig. 1 A; a RTT fibril is shown oriented parallel to the X axis; the incidence plane is defined by the X-Z axes. Similarly, Fig. 1 B illustrates the orientation of a LC cholesteric film within the laboratory frame; the LC film lies normal to the Z axis with the long cholesteric bands oriented parallel to the X axis. Fig. 1 C illustrates the molecular frames of the three major chromophores responsible for generating the SFG-V signal. In the amide I region of proteins, it is the carbonyl moiety within the peptide bond whose vibrational transitions at specific IR frequencies are capable of enhancing hyperpolarizability of the SFG dipole, whose major axis is generally parallel to the C-N bond. IR data suggest that the amide I region in collagen is dominated by a recurring structural motif known as the Gly-X-Y tripeptide. Although the X and Y positions are most commonly occupied by proline and hydroxyproline residues, respectively, other residues may also be present. Studies with model peptides suggest that the position of the residue relative to the glycine may exert a greater influence on its physicochemical properties than its structure (23–25). The molecular frames for the three tripeptide residues are shown in Fig. 1 C above the corresponding region of the collagen polypeptide backbone. The essentially two-dimensional property of the amide chromophore is illustrated by the Gly chromophore in the middle of the chain: note that the a and c axes lie in the plane of the page, whereas the b axis is normal to it. Although the X and Y chromophores are also coplanar with the backbone, all three axes are visible as the chain twists in and out of the page.
Assuming that collagen fibrils are characterized by cylindrical rod symmetry, the number of nonzero second-order tensor elements reduces to seven independent elements: (26,27), when the symmetry axis of the collagen fibrils is parallel to the X axis of the laboratory frame. To ensure that the collagen fascicle is correctly positioned in the scanning apparatus, orientation of fibrillar collagen within each sample is analyzed using polarization-modulated SHG imaging, as described in detail previously (14).
Associations between molecular vibrational modes and specific nonzero tensor elements are determined by obtaining spectra of the amide I region using different polarization combinations for the input (visible and IR) and output (SFG) beams. Each spectrum is designated by three letters, which refer to the polarization state of the input and output beams. In this study, we indicate beam polarization in the following order: SFG (output), visible (input), IR (input). Thus, for example, the spectrum SPP is characterized by a s-polarized SFG beam, p-polarized visible beam, and p-polarized IR beam. Three spectra (SPP, PSP, PPS), are dependent only on chiral tensor elements ( with ). Four spectra (PPP, SPS, PSS, and SSP) result only from the achiral NLO properties of the amide chromophores. As expected with cylindrical symmetry, the SSS polarization state produced no significant SFG signal.
Materials and Methods
Preparation of native RTT
RTT fascicles were obtained from the tails of adult (3–6 month old) Sprague-Dawley rats, which had served as control subjects in other, unrelated studies and had been frozen before use in this study. For spectroscopy, the fascicles were cut into sections 5 cm long and ∼0.5 mm wide and placed onto microscope slides. SFG-V spectroscopy was conducted on three different RTT samples; five complete scans were performed on each sample to determine within-sample variance.
Preparation of cholesteric LC collagen fibrils
As noted earlier, one goal of this study was to determine if the supramolecular structures observed in native RTT collagen contribute to chiral NLO effects. We postulated that SFG-V spectra obtained from LC collagen subjected to velocity gradients during preparation would provide useful information concerning the contributions of RTT supramolecular organization to chiral NLO effects. This preparation method results in the formation of liquid crystalline assemblies characterized by a highly oriented cholesteric liquid crystal band aligned with the long axis of the deposited collagen; such bands are capable of generating detectable SHG and SFG signals; however, despite their long-range ordering, structural studies (28) have shown that these cholesteric LC collagen films lack the hierarchical supramolecular chiral structures that have been identified in native RTT collagen.
The preparation of soluble collagen has been described in detail previously (29). Minced, washed RTT samples are digested in pepsin (1:10) at 4°C in ice cold 0.5 M acetic acid for 2 days; the pepsin was then inactivated by raising the pH > 6 with NaOH. Type I collagen was selectively precipitated from solution by addition of solid NaCl, recovered by vacuum filtration, and resuspended in 0.01 M HCl. After reprecipitation and recovery, the purified, soluble collagen was solubilized in 0.01 M HCl at a concentration of 5 mg/ml. The collagen solutions were concentrated by reverse dialysis in 0.01 HCL/50% polyethylene glycol, Mr 20,000 (Sigma-Aldrich, St. Louis, MO) as described by Knight (30). Aliquots were periodically removed from the dialysis tubing to measure collagen concentration (31). Sample concentrations of 80 mg/ml were used to prepare the LC films; the concentrated solution was extruded from the dialysis tubing directly into 1 ml syringes equipped with 22 gauge needles. The collagen was deposited onto cleaned glass slides by placing the needle tip, angled parallel to the slide surface, at one end of the slide, and pushing on the syringe while drawing the needle down the length of the slide in the opposite direction. The velocity gradients resulting from the combination of extensional and pressure-driven flow have previously been determined to induce unidirectional orientation of the collagen molecules during deposition (28). After deposition, the film was rapidly desiccated under laminar flow to minimize orientational relaxation. Before SFG-V spectral analysis, LC preparations were analyzed microscopically using a Nikon Eclipse microscope (Nikon, Melville, NY) under white light and polarized light to confirm the presence of cholesteric bands; typical results are shown in the 100 × 100 μm film in Fig. 1 B, in which alternating light and dark bands are aligned parallel to each other, identical to the preparations described by Kirkwood and Fuller (28). The inset in Fig. 1 B shows that the LC collagen films completely lack the undulating crimp pattern present in native RTT collagen. In addition, LC samples were analyzed using polarization-modulated SHG imaging to confirm that orientation was parallel to the long axis of the slide. Three LC collagen films were prepared for spectral analysis. However, the LC collagen films did not generate nearly as strong a signal as native RTT collagen, and we were unable to obtain SFG-V spectral data from all three LC samples.
Raman spectroscopy
Fourier transform Raman spectroscopy was carried out in reflection mode using a Bruker RFS 100/S and Bruker RamanScope III (Bruker Optics, Ettlingen, Germany) equipped with a Coherent 1064-500N laser (Coherent, Santa Clara, CA). Data acquisition was performed by Bruker OPUS 5.5 data acquisition software (Bruker Optics). The spectrum of each sample was obtained from 6000 scans over the range 3500–500 cm−1 at a resolution of 4 cm−1 with the laser power set at 350 mW by the OPUS software. The integrity of collagen structure was maintained under these scanning conditions, confirmed by the reproducibility of repeated scan of each sample.
Advanced Chemistry Development (ACD) Labs UV-IR Processor software (ACD/Labs, Toronto, Ontario, Canada) was used to view and analyze the data. Baseline subtraction was carried out using the built-in automatic baseline subtraction settings (end to end, snap to spectrum, negative values set to zero). Spectra were smoothed using the program's trapezoidal FFT filter function with automatic settings.
Sum-frequency vibrational spectroscopy
The apparatus for performing SFG-V spectroscopy is similar to the one described in detail previously (14), but includes a modification in the detection system. Measurements of collagen samples were made in transmission mode using a 532 nm visible beam produced by a frequency-doubled Nd:YAG laser with 30 ps pulses at a 10 Hz repetition rates (model No. PL2143C, EKSPLA, Vilnius-53, Lithuania) and an IR beam produced by an OPG/OPA/DFG unit (EKSPLA PG501 VIR/DFG) tuned across the wavelengths associated with the amide I band of polypeptides (5.7–6.7 μm, in 5 nm steps). The two beams were overlapped spatially and temporally on the collagen sample. The IR and visible beams were weakly focused (∼0.5 and ∼0.32 mm beam waists, respectively) onto the sample at 45° relative to the axis normal to the sample and 90° relative to each other, as shown in Fig. 1 A. The energy fluence in the collagen sample was kept constant by setting the energy of the IR and visible beams between 15 and 22 μJ and 41 and 50 μJ, respectively. A polarizer and a λ/2 plate were used to attenuate and set the polarization (P/S) of the visible beam, after which the visible beam was focused onto the collagen sample with a 15 cm focal length lens. The IR P/S polarization was set using a ZnSe IR-coated Fresnel rhomb (II-VI Corporation, Saxonburg, PA) and the beam was focused on the sample using a ZnSe 25 cm focal length lens. A set of lenses were used to collect the scattered signal from the sample and relay the light to the detection system, allowing the SFG beam to be collected without the necessity of tracking its direction. An analyzer was placed after the sample to characterize the polarization of the SFG signal. An interference filter (Notch-Plus; Kaiser Optical Systems, Ann Arbor, MI) was used to reject the scattered 532 nm beam. The detection system was modified by using a HORIBA Jobin Yvon 750 SPEX monochromator (HORIBA Jobin Yvon, Edison, NJ) as a tunable passband filter to replace the sharp-cut-edge filters we had used previously to filter out spurious signals from the OPA and 532 nm beams. Output from the monochromator was passed directly into a photomultiplier tube (PMT, model No. R928; Hamamatsu, Bridgewater, NJ) biased at 800 V, which replaced the PMT used in previous experiments. A gated integrator (model No. SR250; Stanford Research Systems, Sunnyvale, CA) was used to convert the signal from the PMT into an exponential moving average with a weighting factor α = 1.98 × 10−3 and to perform baseline subtraction to reduce the background noise floor. As a result of the implementation of the monochromator and the exponential moving average, an average signal/noise ratio better than 22 dB was achieved.
SHG polarization modulation imaging (32) was performed on both RTT and LC collagen samples using the experimental apparatus described previously (14) to determine the orientation of collagen fibrils within the samples. Within RTT samples, fibrils were well aligned, with a standard distribution of ± 11° over dimensions comparable to the overlap of the visible and IR beams (∼0.45 mm, as determined by the projection of the visible beam onto the sample at 45°.) The overlap of the beams is more than twice the periodicity of the crimp structure in RTT (∼0.2 mm), but given the alignment of the fibrils within the overlap region, it is unlikely that variations in fibril orientation introduced by crimp structure will significantly affect our ability to assess spectral features.
Peak fitting
The SFG-V spectral peaks were fitted using an unconstrained nonlinear optimization algorithm based on a modification of the Nelder-Mead simplex technique; a program was written in MATLAB for this purpose (2010a; The MathWorks, Natick, MA). Peaks were assumed to be Lorentzian. Spectra were examined in the wavenumber range 1610–1690 cm−1 to detect the presence of broad peak shoulders that have been reported in collagen at both the high-frequency and low-frequency regions of the amide I region in collagen (33). At convergence, the peak-fitting algorithm generated 3–4 peaks in the amide I band region. The resolution of these peaks met both the Sparrow and Houston criteria for resolving spectral lines; these criteria are based on the spectral bandwidth of the SFG beam in our apparatus.
Statistical analysis
Within-sample variance (comparison of spectra obtained by repeatedly scanning a single sample) was determined for both RTT and LC spectra. Between-sample variance (comparison of spectra from separate samples) was determined for all RTT spectral data. However, because the signal from LC collagen was considerably weaker than that from RTT collagen, spectral data were only able to be obtained from a single sample, thus precluding determination of between-sample variance. Peak areas corresponding to the three residues comprising the Gly-X-Y tripeptide motif, which had been determined by curve-fitting, were analyzed by analysis of variance, with significance set at p < 0.05. For analysis of within-sample variance, five scans were analyzed for both RTT and LC samples; for between-sample variance, spectral data from three different samples of native RTT were analyzed.
Results and Discussion
Raman spectra
Raman spectra were obtained from both native RTT and LC collagen. The results, shown in Fig. 2, confirm that the preparation procedure for LC collagen did not interfere with the basic structural features of collagen; the amide I contour, shown in the inset, is consistent with published reports for vibrational spectra of collagen (33,34).
Figure 2.
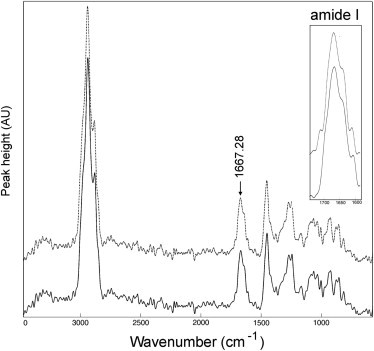
Comparison of Raman spectra from normal RTT and from LC collagen. Raman spectra from RTT (solid line) and LC collagen (dotted line) are shown, demonstrating that the LC collagen preparation process did not alter Raman spectral properties. The inset shows a detailed view of the amide I band.
SFG-V spectra
Comparison between chiral and achiral spectra
Different combinations of input/output beam polarizations were used to separate achiral tensor elements from chiral elements, resulting in a total of seven spectra with nonzero tensor elements. Within a given polarization group (chiral or achiral), there were few differences among the individual spectra, despite the differences in the I/O beam polarizations. There were, however, marked differences between chiral spectra and achiral spectra. Somewhat surprisingly, the intragroup similarities and intergroup differences observed in spectra from native RTT closely resembled those seen in spectra from LC collagen. These observations are shown in Fig. 3. The four superimposed spectra include PPS (chiral) and PSS (achiral) from both native RTT and LC collagen. All spectra are normalized such that the maximum peak height for each spectrum is set equal to 1. Absolute values of peak heights and areas cannot be used for analysis, because suitable internal standards have not been definitively identified that would allow comparison among spectra.
Figure 3.
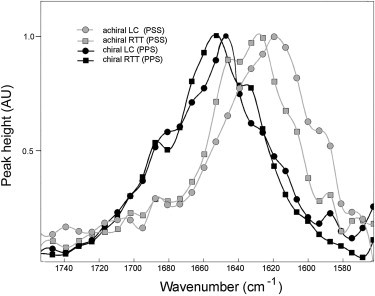
Comparison of chiral and achiral spectra from RTT and LC collagen. Four spectral plots are overlaid to illustrate the similarities between RTT and LC collagen and the differences between chiral and achiral spectra. Note that chiral spectra from RTT (black squares) and LC collagen (black circles) have nearly overlapping profiles, predominantly in the high-frequency region, whereas achiral spectra lie in the lower frequency region.
The data shown in Fig. 3 provide clues as to which regions of the amide I band are most likely to contribute to chiral NLO effects. First, the signal in the chiral spectra arises predominantly from higher frequency regions than does the achiral signal. Fourth quartile peak maxima values for the chiral peaks occur between 1640 and 1668 cm−1, whereas maxima for the achiral peaks range from 1603 to 1638 cm−1. Second, the chiral spectra show a large, distinct peak between 1680 and 1690 cm−1; in the achiral spectra, however, there are, at most, shoulder regions that appear too close to baseline noise to be unambiguously identified.
Comparison between chiral and achiral spectra after curve-fitting
Currently, there have been few studies addressing the selection of appropriate curve-fitting methods for SFG-V spectra, although investigators have noted pitfalls in treating SFG-V spectra as analogous to IR/Raman spectra (35–37). We chose to use a flexible constraint-free approach for nonlinear optimization, a modification of the Nelder-Mead simplex technique.
Curve-fit data were consistent with the raw spectral data shown in Fig. 3; in addition, we detected differences between native RTT collagen and LC collagen that had not previously been apparent. Fig. 4 shows curve-fit peaks for chiral and achiral spectra from both native RTT and LC collagen. In each panel, the curve-fit peaks (white) are overlaid on the amide I contour generated by the spectral data (black). The wavenumber for each peak maximum is indicated above the peak. The data shown in Fig. 4 again confirm that significant differences exist between chiral and achiral spectra for both native RTT and LC collagen. Both chiral and achiral spectra have peaks at wavenumbers ∼1628–1635 cm−1 and at ∼1639–1645 cm−1. Occasionally, very small high-frequency peaks were observed in both chiral and achiral spectra (∼1680–1695 cm−1). Chiral spectra are characterized by a third peak, absent in achiral spectra, which we observed over a wider range (∼1655–1665 cm−1) than the lower frequency peaks. We also detected one difference between RTT and LC collagen spectra: a low-frequency peak at ∼1615–1620 cm−1 was detectable in achiral spectra from RTT collagen, but not in spectra from LC collagen.
Figure 4.
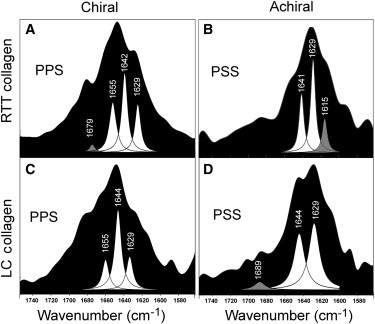
Curve-fitting of SFG-V spectra. Curve-fitting for all spectra was performed using a modification of the Nelder-Mead simplex technique. Representative results for chiral and achiral spectra for both native RTT and LC collagen are presented in panels A–D. Chiral spectra (A and C) from both RTT and LC contain three major peaks, at ∼1629 cm−1, ∼1643 cm−1, and ∼1655–1665 cm−1. Achiral spectra (B and D) from both RTT and LC collagen have two major peaks, but both lack the high-frequency peak. Achiral RTT (B) contains a peak at ∼1615 cm−1 that is absent in the LC achiral spectra (D).
We also compared RTT and LC spectra with respect to peak ratios. We selected the 1643 cm−1 subband as the reference peak because it is present in both achiral and chiral spectra. In this analysis we include only the three subbands definitively identified in model peptide studies as being present in the collagen amide I contour: those with wavenumbers at ∼1629 cm−1, ∼1643 cm−1, and ∼1655–1665 cm−1. Peak ratio comparisons between native RTT collagen and LC collagen are shown in Fig. 5, revealing striking similarities between the RTT and LC spectra.
Figure 5.
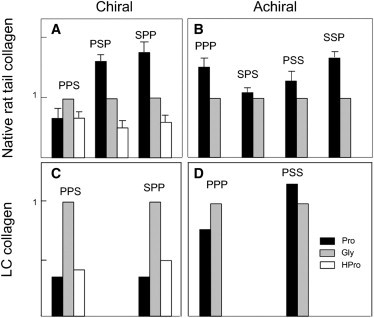
Relative amounts of amide I subbands for achiral and chiral spectra in RTT and LC collagen. Peak areas corresponding to the three major amide I subbands present in collagen were computed based on curve-fitting data (Fig. 4). Based on analogy to IR spectra, the band assignments correspond to the collagen tripeptide residues: Gly (mid-frequency, gray bar), Pro (low-frequency, black bar), Hpro (high-frequency, white bar). Error bars in 5 A and 5 B represent the standard deviation (determined from three RTT samples). The peak area of Gly was arbitrarily set to 1, and the areas for the other subbands were adjusted accordingly. Data for chiral and achiral spectra for both native RTT and LC collagen are shown in the four panels: (A) chiral spectra, native RTT; (B) achiral spectra, native RTT; (C) chiral spectra, LC collagen; (D) achiral spectra, LC collagen. Note the absence of Hpro in all achiral spectra.
Interpretation of data: assignment of subband identities
The amide I band of collagen-like polypeptides differs from that of alpha helical and pleated sheet polypeptides in its dependence on primary structure (38). This property may contribute to the somewhat surprising finding that collagen's amide I contour may comprise as few as three major subbands (15,33,37,39,40). The peak maxima occur at ∼1629–1635 cm−1, ∼1639–1645 cm−1, and ∼1657–1675 cm−1. There is considerable evidence, based on studies of collagen model peptides, that the three amide I subbands represent the carbonyl groups in the Gly-X-Y tripeptide (33,39,41). In these studies, the low-frequency peak (1628–1635 cm−1) is identified as the X carbonyl; the mid-frequency peak (1639–1648 cm−1) as the Gly carbonyl, and the high-frequency peak (1657–1675 cm−1) as the Y carbonyl. By convention, the name of the carbonyl group is derived from the residue contributing the amine group to the peptide bond.
Based on the similarities between IR spectra of collagen model peptides and our SFG-V spectra we provisionally assign the low-, mid-, and high-frequency peaks to the X, Gly, and Y carbonyl groups, respectively. The variability in peak wavenumber, particularly the chiral high-frequency peak, is most likely attributable to the large number of environmental variables that can affect spectral profiles. Veis (40) suggested that such variability may result from interactions among factors known to affect vibrational modes, including carbonyl basicity, number and strength of inter- and intramolecular hydrogen bonding, subtle local conformational fluctuations, and coupling between similar carbonyl stretching modes. Lazarev and co-workers (42) determined that changes in relative hydration can produce shifts in wavenumber of up to 20 cm−1 for the high-frequency amide I subband present in collagen. Other factors affecting amide I subbands include bound water, temperature, pH, amino acid composition, nearest neighbor effects, chain conformation, and degree of order in molecular packing (33,40,42).
We cannot definitively assign the identity of the low-frequency peak at 1615 cm−1 in achiral RTT collagen. In IR studies of collagen-like model peptides this peak was detected in (Gly-Pro-Hyp)n but not in (Gly-Pro-Pro)n, consistent with the presence of hydrogen bonding between carbonyl residues in this peak and hydroxyproline residues (38). Differences in hydrogen bonding networks may account for the absence of this peak in LC collagen spectra. Other studies suggest this peak may represent a region of partial denaturation and aggregation (43), or, alternatively, the presence of glucopyranose derivatives associated with nonenzymatic glycation (44). Either of these possibilities is consistent with our findings.
A major goal of this study was to determine if specific structural features of collagen could be correlated with NLO chiral effects in the amide I region. Although it is not feasible to determine absolute peak values for carbonyl residues, as noted previously, computation of peak ratios provides an alternative approach to analysis (35). We chose the Gly carbonyl as the reference peak by setting its value to 1 and expressing the X and Y peak areas relative to Gly. Normalized X and Y carbonyl values for RTT chiral and achiral spectra are shown in Fig. 5, A and B, respectively. The height of each bar represents the mean value of the peak area obtained from scans of three separate samples; the error bar indicates the standard deviation. Differences among the seven spectra with respect to the mean values for the X peaks and the mean values of the Y peaks were assessed by analysis of variance, with significance set at p < 0.05. With respect to the X carbonyl peak area, there were few differences between achiral and chiral spectra. As shown in Fig. 5, A and B, the X carbonyl has the highest value in six of the seven spectra, and a value close to 80% of the maximum in the seventh spectrum (PPS). With respect to the values for the Y carbonyl, however, a striking and unexpected difference between chiral and achiral RTT spectra was observed. No Y carbonyl peak was detected in any of the four achiral spectra, although all three chiral spectra had readily detectable Y carbonyl peaks. Furthermore, there were no significant differences among the three chiral spectra with respect to the value for the Y/Gly ratio.
X and Y carbonyl peak ratios were also obtained from chiral and achiral spectra LC collagen, shown in Fig. 5, C and D. Signal strength from the LC collagen was not as strong as from RTT collagen, and we were unable to obtain spectra from more than one sample using our present instrument configuration, nor were we able to obtain a detectable signal for all seven spectra. Multiple scans from the single sample showed very low within-sample variance (p < 0.001). The LC data are of interest, however, in that they also show an absence of the Y carbonyl peak in all of the detectable achiral spectra. With respect to the X carbonyl ratios similarities (PSS, PPS) and differences (SPP) were observed. Whether or not these are statistically significant differences will require acquisition of complete spectral profiles from multiple LC samples.
Structural basis of NLO chiral effects
The data presented previously allow us to draw some preliminary conclusions concerning the structural basis of NLO chiral effects. First, the presence of all three carbonyl groups in the chiral spectra strongly suggests that we cannot associate NLO chirality with the properties of a specific carbonyl group in the tripeptide. Rather, all three carbonyl groups appear to contribute to chiral effects through one of several possible mechanisms. 1), Each carbonyl group contributes to NLO chirality through its intrinsic chiral properties; 2), the carbonyl groups on a given collagen molecule function as if they comprised a single structure with intrinsic chirality; 3), interactions between carbonyl groups on different molecules create supramolecular structures with intrinsic chirality. In the section below we evaluate these mechanisms in the context of recent crystallographic and ab initio studies of the collagen tripeptide carbonyl groups.
Intrinsic chromophore chirality
Crystallographic data obtained from collagen model peptides have been reported by several groups. The three nonequivalent carbonyl groups differ with respect to several key structural parameters, including torsion angles, particularly helical twist, hydrogen bonding, hydrogen bond length, proline ring puckering, and hydration (45–47). Both the Gly carbonyl and the X carbonyl have very low twist angles, resulting in close alignment with the plane of the polypeptide backbone, a conformation inconsistent with intrinsic chirality. In contrast, the Y carbonyl has a high twist angle. In addition, Tuer et al. (48,49) have conducted quantum mechanical studies of collagen hyperpolarizability in which they observed that of the three carbonyl groups, only the Y carbonyl dipole is out-of-plane with respect to the C-N backbone, a conformation consistent with intrinsic chirality. Although it is possible that the Y carbonyl represents the primary source of the observed chiral NLO effects, this interpretation seems unlikely in view of the presence of the two other carbonyl groups in all chiral spectra, as well as the low signal strength of the Y carbonyl group relative to that of the other two carbonyls (Fig. 5).
The apparent absence of the high-frequency Y carbonyl band in achiral spectra, shown in Fig. 5 in the left-hand panels, was unexpected. The ab initio studies of Tuer et al. (48,49) have shown that, of the three tripeptide residues, hydroxyproline is associated with the largest SHG dipole. Thus, it seems unlikely that lack of signal strength is responsible for our observations, although it is recognized that these dipole effects are not the major contribution to signal strength in SFG-V spectra. Perhaps more pertinent, Tuer et al. have noted that symmetry restrictions allow only the N-C dipole of hydroxyproline in the Y position to contribute to NLO effects, whereas proline, in the X position, is unrestricted; possibly the symmetry constraints of hydroxyproline in the Y position in conjunction with its out-of-plane conformation are related to the absence of detectable signal. In most spectra we observed that the X carbonyl was predominant. This observation is consistent with Tuer's observation that proline is the only residue in which both dipoles contribute to NLO effects. This distinctive attribute may account for the dominance of the X carbonyl peak in these spectra, regardless of beam polarization. In short, crystallographic and computation data suggest that intrinsic chirality of individual chromophores is unlikely.
Molecular chirality
In contrast to intrinsic chirality, the presence of molecular chirality is supported by our data. The consistent ratios among the three carbonyl groups in chiral spectra are compatible with Simpson's concept of orientational chirality, in which the carbonyl chromophores within a given polypeptide are considered to function as a single structure whose properties derive from the conformation of the polypeptide. In helical proteins such as collagen, Simpson compares the amide chromophores to a series of propellers attached to a shaft with progressive changes in their pitch and tilt angles (50). Although an individual propeller is an achiral object, a group of progressively offset propellers possesses intrinsic chirality.
Supramolecular chirality
We now consider the third proposed mechanism, which is essentially an extension of the second mechanism to the supramolecular domain. Because crystallographic studies have shown that Y carbonyl groups are capable of forming intermolecular hydrogen bonds (51), it is possible that carbonyl-based chiral structures might exist on a supramolecular scale. However, the spectral data we obtained from the LC collagen films suggest that the multicarbonyl structures contributing to chiral NLO effects are more likely to be molecular in scale, rather than supramolecular. Although the LC collagen films are characterized by long-range ordering of individual collagen molecules into chiral cholesteric bands, there is no evidence, based on analyses of cholesteric liquid crystal morphologies (52), that these structures are morphologically comparable to the multimolecular unit cell structures of fibrillar collagen proposed by Orgel based on detailed x-ray fiber diffraction studies of native RTT (53). If such supramolecular organization allowed the carbonyl groups from multiple collagen molecules to function as a single chiral entity, we would expect significant differences between the carbonyl ratios from the RTT collagen and the LC collagen. Given the similarities in carbonyl ratios between RTT and LC collagen, shown in Fig. 5, it seems most likely that the chiral NLO effects arise from the structural component that is common to both RTT and LC collagen: the chiral arrangement of carbonyl groups on individual collagen molecules.
Conclusions
In summary, our study represents, to our knowledge, the first report in which the fine structure of the amide I band in native type I collagen was analyzed using SFG-V spectroscopy under different conditions of input/output beam polarization. Our data showed an unexpected absence of the Y carbonyl group in all four achiral spectra. Based on a comparison of the data from native RTT with those from LC collagen, we conclude that NLO chiral effects most likely are attributable to orientational chirality at the molecular scale. Given the challenging nature of interpreting SFG-V spectra in native collagen, it seems likely that significant advances will depend on complementary studies of SFG-V in collagen model peptides and ab initio calculations of the IR and Raman polarizability tensors in collagen model peptide sequences.
References
- 1.Fine S., Hansen W.P. Optical second harmonic generation in biological systems. Appl. Opt. 1971;10:2350–2353. doi: 10.1364/AO.10.002350. [DOI] [PubMed] [Google Scholar]
- 2.Freund I., Deutsch M. Macroscopic polarity of connective tissue is due to discrete polar structures. Biopolymers. 1986;25:601–606. doi: 10.1002/bip.360250406. [DOI] [PubMed] [Google Scholar]
- 3.Freund I., Deutsch M., Sprecher A. Connective tissue polarity. Optical second-harmonic microscopy, crossed-beam summation, and small-angle scattering in rat-tail tendon. Biophys. J. 1986;50:693–712. doi: 10.1016/S0006-3495(86)83510-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roth S., Freund I. Second harmonic generation in collagen. J. Chem. Phys. 1979;70:1637–1643. [Google Scholar]
- 5.Roth S., Freund I. Optical second-harmonic scattering in rat-tail tendon. Biopolymers. 1981;20:1271–1290. doi: 10.1002/bip.1981.360200613. [DOI] [PubMed] [Google Scholar]
- 6.Belkin M.A., Shen Y.R. Non-linear optical spectroscopy as a novel probe for molecular chirality. Int. Rev. Phys. Chem. 2005;24:257–299. [Google Scholar]
- 7.Shen Y.R. A few selected applications of surface nonlinear optical spectroscopy. Proc. Natl. Acad. Sci. USA. 1996;93:12104–12111. doi: 10.1073/pnas.93.22.12104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu X.D., Suhr H., Shen Y.R. Surface vibrational spectroscopy by infrared-visible sum frequency generation. Phys. Rev. B Condens. Matter. 1987;35:3047–3050. doi: 10.1103/physrevb.35.3047. [DOI] [PubMed] [Google Scholar]
- 9.Dreesen L., Sartenaer Y., Peremans A. Probing ligand-protein recognition with sum-frequency generation spectroscopy: the avidin-biocytin case. ChemPhysChem. 2004;5:1719–1725. doi: 10.1002/cphc.200400213. [DOI] [PubMed] [Google Scholar]
- 10.Fu L., Liu J., Yan E.C.Y. Chiral sum frequency generation spectroscopy for characterizing protein secondary structures at interfaces. J. Am. Chem. Soc. 2011;133:8094–8097. doi: 10.1021/ja201575e. [DOI] [PubMed] [Google Scholar]
- 11.Knoesen A., Pakalnis S., Frank C.W. Sum-frequency spectroscopy and imaging of aligned helical polypeptides. IEEE. J. Sel. Top. Quant. 2004;10:1154–1163. [Google Scholar]
- 12.Nguyen K.T., King J.T., Chen Z. Orientation determination of interfacial beta-sheet structures in situ. J. Phys. Chem. B. 2010;114:8291–8300. doi: 10.1021/jp102343h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perry J.M., Moad A.J., Simpson G.J. Electronic and vibrational second-order nonlinear optical properties of protein secondary structural motifs. J. Phys. Chem. B. 2005;109:20009–20026. doi: 10.1021/jp0506888. [DOI] [PubMed] [Google Scholar]
- 14.Rocha-Mendoza I., Yankelevich D.R., Knoesen A. Sum frequency vibrational spectroscopy: the molecular origins of the optical second-order nonlinearity of collagen. Biophys. J. 2007;93:4433–4444. doi: 10.1529/biophysj.107.111047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J., Even M.A., Chen Z. Detection of amide I signals of interfacial proteins in situ using SFG. J. Am. Chem. Soc. 2003;125:9914–9915. doi: 10.1021/ja036373s. [DOI] [PubMed] [Google Scholar]
- 16.Su P.-J., Chen W.-L., Dong C.Y. Determination of collagen nanostructure from second-order susceptibility tensor analysis. Biophys. J. 2011;100:2053–2062. doi: 10.1016/j.bpj.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meijer E.W., Havinga E.E., Rikken G.L. Second-harmonic generation in centrosymmetric crystals of chiral molecules. Phys. Rev. Lett. 1990;65:37–39. doi: 10.1103/PhysRevLett.65.37. [DOI] [PubMed] [Google Scholar]
- 18.Kauranen M., Maki J.J., Persoons A. Quantitative determination of electric and magnetic second-order susceptibility tensors of chiral surfaces. Phys. Rev. B. 1997;55:R1985–R1988. [Google Scholar]
- 19.Schanne-Klein M.C., Boulesteix T., Andraud C. Strong chiroptical effects in surface second harmonic generation obtained for molecules exhibiting excitonic coupling chirality. Chem. Phys. Lett. 2002;362:103–108. [Google Scholar]
- 20.Verbiest T., Elshocht S.V., Persoons A. Strong enhancement of nonlinear optical properties through supramolecular chirality. Science. 1998;282:913–915. doi: 10.1126/science.282.5390.913. [DOI] [PubMed] [Google Scholar]
- 21.Burke B.J., Moad A.J., Simpson G.J. Experimental confirmation of the importance of orientation in the anomalous chiral sensitivity of second harmonic generation. J. Am. Chem. Soc. 2003;125:9111–9115. doi: 10.1021/ja0298500. [DOI] [PubMed] [Google Scholar]
- 22.Haupert L.M., Simpson G.J. Chirality in nonlinear optics. Ann. Rev. Phys. Chem. 2009;115:345–365. doi: 10.1146/annurev.physchem.59.032607.093712. [DOI] [PubMed] [Google Scholar]
- 23.Bretscher L.E., Jenkins C.L., Raines R.T. Conformational stability of collagen relies on a stereoelectronic effect. J. Am. Chem. Soc. 2001;123:777–778. doi: 10.1021/ja005542v. [DOI] [PubMed] [Google Scholar]
- 24.Kramer R.Z., Bella J., Berman H.M. Sequence dependent conformational variations of collagen triple-helical structure. Nat. Struct. Biol. 1999;6:454–457. doi: 10.1038/8259. [DOI] [PubMed] [Google Scholar]
- 25.Persikov A.V., Ramshaw J.A.M., Brodsky B. Amino acid propensities for the collagen triple-helix. Biochemistry. 2000;39:14960–14967. doi: 10.1021/bi001560d. [DOI] [PubMed] [Google Scholar]
- 26.Dick B. Irreducible tensor analysis of sum-frequency and difference-frequency generation in partially oriented samples. Chem. Phys. 1985;96:199–215. [Google Scholar]
- 27.Fischer P., Hache F. Nonlinear optical spectroscopy of chiral molecules. Chirality. 2005;17:421–437. doi: 10.1002/chir.20179. [DOI] [PubMed] [Google Scholar]
- 28.Kirkwood J.E., Fuller G.G. Liquid crystalline collagen: a self-assembled morphology for the orientation of mammalian cells. Langmuir. 2009;25:3200–3206. doi: 10.1021/la803736x. [DOI] [PubMed] [Google Scholar]
- 29.Buckingham B., Reiser K.M. Relationship between the content of lysyl oxidase-dependent cross-links in skin collagen, nonenzymatic glycosylation, and long-term complications in type I diabetes mellitus. J. Clin. Invest. 1990;86:1046–1054. doi: 10.1172/JCI114807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Knight D.P., Nash L., Ho M.W. In vitro formation by reverse dialysis of collagen gels containing highly oriented arrays of fibrils. J. Biomed. Mater. Res. 1998;41:185–191. doi: 10.1002/(sici)1097-4636(199808)41:2<185::aid-jbm2>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 31.Woessner J.F. The determination of hydroxyproline in tissue and protein samples containing small proportions of this imino acid. Arch. Biochem. Biophys. 1961;93:440–447. doi: 10.1016/0003-9861(61)90291-0. [DOI] [PubMed] [Google Scholar]
- 32.Stoller P., Reiser K.M., Rubenchik A.M. Polarization-modulated second harmonic generation in collagen. Biophys. J. 2002;82:3330–3342. doi: 10.1016/S0006-3495(02)75673-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lazarev Y.A., Grishkovsky B.A., Khromova T.B. Amide I band of IR spectrum and structure of collagen and related polypeptides. Biopolymers. 1985;24:1449–1478. doi: 10.1002/bip.360240804. [DOI] [PubMed] [Google Scholar]
- 34.Frushour B.G., Koenig J.L. Raman scattering of collagen, gelatin, and elastin. Biopolymers. 1975;14:379–391. doi: 10.1002/bip.1975.360140211. [DOI] [PubMed] [Google Scholar]
- 35.Bain C.D., Davies P.B., Brown M.A. Quantitative-analysis of monolayer composition by sum-frequency vibrational spectroscopy. Langmuir. 1991;7:1563–1566. [Google Scholar]
- 36.McGall S.J., Davies P.B., Neivandt D.J. Interference effects in sum frequency vibrational spectra of thin polymer films: an experimental and modeling investigation. J. Phys. Chem. B. 2004;108:16030–16039. [Google Scholar]
- 37.Miragliotta J., Polizzotti R.S., Hall R.B. IR-visible sum-frequency generation study of methanol adsorption and reaction on NI(100) Chem. Phys. 1990;143:123–130. [Google Scholar]
- 38.Doyle B.B., Bendit E.G., Blout E.R. Infrared spectroscopy of collagen and collagen-like polypeptides. Biopolymers. 1975;14:937–957. doi: 10.1002/bip.1975.360140505. [DOI] [PubMed] [Google Scholar]
- 39.Bryan M.A., Brauner J.W., Mendelsohn R. FTIR studies of collagen model peptides: complementary experimental and simulation approaches to conformation and unfolding. J. Am. Chem. Soc. 2007;129:7877–7884. doi: 10.1021/ja071154i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Payne K.J., Veis A. Fourier transform IR spectroscopy of collagen and gelatin solutions: deconvolution of the amide I band for conformational studies. Biopolymers. 1988;27:1749–1760. doi: 10.1002/bip.360271105. [DOI] [PubMed] [Google Scholar]
- 41.Juszczak L.J. Comparative vibrational spectroscopy of intracellular tau and extracellular collagen I reveals parallels of gelation and fibrillar structure. J. Biol. Chem. 2004;279:7395–7404. doi: 10.1074/jbc.M309971200. [DOI] [PubMed] [Google Scholar]
- 42.Lazarev Y.A., Grishkovsky B.A., Grechishko V.S. Bound water in the collagen-like triple-helical structure. Biopolymers. 1992;32:189–195. doi: 10.1002/bip.360320209. [DOI] [PubMed] [Google Scholar]
- 43.Lin S.-Y., Lee S.-M., Liang R.C. Fourier transform infrared spectral evidences for protein conformational changes in immature cataractous human lens capsules accelerated by myopia and/or systemic hypertension. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1997;53A:1507–1513. doi: 10.1016/s1386-1425(97)00077-2. [DOI] [PubMed] [Google Scholar]
- 44.Tipson, R. S. 1968. Infrared spectroscopy of carbohydrates: review of the literature. National Bureau of Standards Monograph 110. N. B. O. Standards. U.S. Government Printing Office, Washington, DC.
- 45.Bella J. A new method for describing the helical conformation of collagen: dependence of the triple helical twist on amino acid sequence. J. Struct. Biol. 2010;170:377–391. doi: 10.1016/j.jsb.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 46.Erdmann R.S., Wennemers H. Importance of ring puckering versus interstrand hydrogen bonds for the conformational stability of collagen. Angew. Chem. Int. Ed. Engl. 2011;50:6835–6838. doi: 10.1002/anie.201008118. [DOI] [PubMed] [Google Scholar]
- 47.Okuyama K. Revisiting the molecular structure of collagen. Connect. Tissue Res. 2008;49:299–310. doi: 10.1080/03008200802325110. [DOI] [PubMed] [Google Scholar]
- 48.Tuer A., Krouglov S., Barzda V. Three-dimensional visualization of the first hyperpolarizability tensor. J. Comput. Chem. 2011;32:1128–1134. doi: 10.1002/jcc.21694. [DOI] [PubMed] [Google Scholar]
- 49.Tuer A.E., Krouglov S., Barzda V. Nonlinear optical properties of type I collagen fibers studied by polarization dependent second harmonic generation microscopy. J. Phys. Chem. B. 2011;115:12759–12769. doi: 10.1021/jp206308k. [DOI] [PubMed] [Google Scholar]
- 50.Simpson G.J., Perry J.M., Wampler R.D. Uncoupled oscillator model for interpreting second harmonic generation measurements of oriented chiral systems. Chem. Phys. Lett. 2004;399:26–32. [Google Scholar]
- 51.Bella J., Eaton M., Berman H.M. Crystal and molecular structure of a collagen-like peptide at 1.9 A resolution. Science. 1994;266:75–81. doi: 10.1126/science.7695699. [DOI] [PubMed] [Google Scholar]
- 52.Dierking I. John Wiley & Sons; Hoboken, NJ: 2006. Textures of Liquid Crystals. [Google Scholar]
- 53.Orgel J.P.R.O., Irving T.C., Wess T.J. Microfibrillar structure of type I collagen in situ. Proc. Natl. Acad. Sci. USA. 2006;103:9001–9005. doi: 10.1073/pnas.0502718103. [DOI] [PMC free article] [PubMed] [Google Scholar]