

Abstract
Humoral molecules can trigger injury on mechanically stressed and damaged tissue. We have studied the role of complement 3 (C3) in a mouse model of ventilator-induced lung injury (VILI). Compared with sham-treated wild type (WT) mice, ventilated WT mice have reduced total bronchoalveolar lavage (BAL) cells; and elevated activities of thrombin and matrix metalloproteinases (MMPs), such as gelatinase/collagenase in the BAL fluid. In contrast, these parameters in ventilated C3 null mice are not significantly different from sham-treated WT and C3 null mice. In mechanically ventilated mice, thrombin activity and MMPs are lower in C3 null mice than in WT mice and are inversely correlated with total single BAL cells. C3 activation is associated with MMP activation in vitro. Pretreatment of WT mice with humanized cobra venom factor, which inactivates C3, reduces C3 deposition in the lung and increases total BAL cells in VILI. We propose that C3 is involved with VILI and inhibition of complement activation may be a potential therapeutic strategy.
Keywords: Complement, Coagulation, Matrix metalloproteinase, Complement, Acute lung injury, Humanized cobra venom factor
1. Introduction
Ventilator-induced lung injury (VILI) is a major cause of morbidity and mortality in critically ill patients [1]. There are no biomarkers in identifying patients who would develop VILI during and following mechanical ventilation. Furthermore, not all patients with similar ventilator settings develop VILI, suggesting that genetic factors are involved. Such factors could be linked to molecules of the innate immune system. This system consists of cellular and humoral components, the former including epithelial cells and phagocytes and the latter comprising pattern recognition molecules and peptidases [2, 3]. These peptidases include serine proteases found in complement pathways and coagulation system and also in metal-requiring proteinases, like matrix metalloproteinases (MMPs). Activation of MMPs leads to digestion of pulmonary interstitial proteins, resulting in destruction of tissue integrity [1, 4–6]. Alveolar coagulopathy has also been associated with acute lung inflammation/acute respiratory distress syndrome and VILI [7–11].
The interaction among complement pathways, coagulation cascade and MMPs interact have been increasingly clear [12–14], although their detailed interplay is not fully understood. Complement activation enhances thrombin production [15–17]. Thrombin regulates MMPs and vice versa [18–20]. A recent study has demonstrated that C3 is involved with mechanisms causing a secondary lung injury from intestinal ischemia–reperfusion injury [21]. Animal model studies demonstrated alterations of complement levels and expression in acute lung injury [22]. In addition, inactivation of C3 by pretreatment of mice with a humanized cobra venom factor (HCVF) attenuated myocardial reperfusion injury [23]. These observations support the hypothesis that complement activation could mediate VILI and that the mechanism would be involved with coagulation enzymes and MMPs.
As for cellular component, it has been shown that a decrease in total bronchoalveolar lavage (BAL) cells with sequestration of polymorphonuclear neutrophil (PMN) is a marker of early VILI [24]. Supporting this observation is the observation that potent PMN chemokines are upregulated early during VILI [25].
In this study, we hypothesize that the complement cascade is involved in the initial mechanism of VILI, and linked to the associated triggering of the coagulation cascade. To test this hypothesis, we subjected WT and C3 null mice to injurious mechanical ventilation and compared BAL cells and BAL enzyme activities, such as thrombin and gelatinase/collagenase, in those animals following a period of injurious ventilation. We also investigated the mechanisms in vitro by assessing the activity of gelatinase/collagenase in mouse sera.
2. Methods
2.1. Mice
Wild type (WT) and C3 null mice were on C57B/6J genetic background. Male mice with body weight between 24 and 30 g were used. C3 null mice breeders were previously provided by Dr. Michael C. Carroll, Immune Disease Institute, Harvard Medical School [26]. All mice were bred at the animal facility in Massachusetts General Hospital under specific pathogen free environment. All experiments were performed under an approved protocol by the Subcommittee on Research Animal Care at Massachusetts General Hospital, Boston, MA.
2.2. Ventilator-induced lung injury (VILI)
Anesthesia of mice was maintained with intraperitoneal injection of avertin (250 mg/kg) [27]. Tracheotomy was performed, followed by intubation with a 22 G IV catheter (Terumo, Japan), which was connected to a small animal mechanical ventilator (Harvard Apparatus, MA). Mice were ventilated for 2 h on a warm plate (37 °C). Mice were injected with additional avertin (125 mg/kg) and 1 ml of saline at 1 h after initiation of ventilation. Ventilation settings were 35 ml/kg of tidal volume, respiratory rate of 50 per min, fraction of inspired oxygen (FIO)2 =20% (room air), and 2 cm H2O positive end-expiratory pressure [28]. Mice in the sham group were intubated in the manner described above but did not undergo mechanical ventilation.
For inactivation of C3, mice were subcutaneously injected with 250 μg/kg of HCVF 1 h prior to VILI experiment [23].
2.3. Preparation of bronchial alveolar lavage fluid (BALF) and cells
After 2 h, the mice were sacrificed and BAL was performed with 1 ml of phosphate buffered saline (PBS) with 3 lavages to collect BALF and BAL cells. BALF was centrifuged at 1300 g for 10 min and supernatant was collected and stored in −80 °C freezer. Volume of the supernatant was recorded to calculate the total single BAL cells per lung. BAL cell pellet was resuspended with PBS and cells were manually counted under microscope using a hemocytometer. Remaining cells were individually spun onto a glass-slide using Cytopro (Wescor Inc., UT) to prepare single cell layers for microscopic analysis. These cells were air-dried, fixed and then stained using Diff-Quick staining kit (Dade Behring, DE) and were mounted in permount mounting media (Sigma, MO). The stained cells were digitally micrographed under the upright Nikon microscope at ×10 magnification (Nikon Eclips E800, Japan). Macrophages and polymorphonuclear neutrophil (PMN) were counted at ×40 magnification under the Nikon microscope. Three to five fields per slide were counted and combined. No lymphocyte was observed. The counts were used to calculate percentage of PMN in total single BAL cells.
Harvested lungs were slowly injected with 50% of TBS tissue freezing media (Electron Microscopy Sciences, PA) in milliQ water on ice and embedded into TBS freezing media by a snap frozen method in a liquid nitrogen. Lungs were sectioned at 10 μm thickness and stained with FITC-conjugated goat anti-mouse C3 antibody (Sigma-Aldrich, MO) and mounted in aqueous mounting media. Images of these lung sections were digitally captured under the Nikon microscope at ×20 magnification. C3 positive cells were counted per 1 mm2 in 3 different fields per lung and averages were used for analysis.
2.4. Assays of BALF
Thrombin and gelatinase/collagenase were measured using the rhodamine 110-based proteinase substrate R22124 (Invitrogen, CA) and EnzCheck gelatinase/collagenase assay kit (E-12055, Molecular Probes, OR), respectively, according to the manufacturer’s instructions. This E-12055 assay kit is designed to measure all gelatinases and collagenases as a fast screening for these activities. BALF was diluted to 50% with a buffer provided by the kit, mixed with 20 μl of either substrate in 384 well assay plates and then incubated at room temperature. Reaction was read at 500 nm excitation/520 nm emission using the SpectraMax M5 (Molecular Devices, CA) and was expressed as arbitrary activity units (AUs). The assay was performed in triplicates and repeated twice. Representative data was shown.
C5a was assayed as previously described [29]. Briefly, 96 well plates were coated with rat anti-mouse C5a antibody (BD Biosciences, CA) and diluted BALF (50%) in PBS was incubated. Captured C5a was detected by incubating with biotinylated rat anti-mouse C5a antibody (BD Biosciences, CA) followed by horseradish peroxidase-conjugated streptavidin and then OPD substrate (Sigma-Aldrich, MO). Reaction was read at 450 nm using the SpectraMax M5 (Molecular Devices, CA) and expressed as OD reading value.
2.5. Gelatinase/collagenase activation assay on zymosan
Gelatinase/collagenase activity was assayed on zymosan, which is widely used to assay C3 activation [30]. 384 well assay plates were coated with 107 particles of zymosan (Sigma, MO) in 40 μl of 20 mM carb-bicarbonate buffer, pH 9.5 and incubated at 4 °C overnight. After washing in 10 mM Tris, pH 7.4, 120 mM saline, 10 mM CaCl2 (TBS), pH 7.4, the plates were incubated with 20 μl of diluted mouse serum in TBS for 1 h at 37 °C. Then, the plates were added with 20 μl of diluted gelatinase/collagenase substrate as above and incubated for 5 h at room temperature. Pooled sera of three naïve mice were used for this assay. Reaction was read and expressed as AUs as above. The assay was performed in triplicates and repeated twice. Representative data was shown.
2.6. Statistical analysis
Statistical analysis was performed using JMP software (SAS Institute Inc., NC). Box plots (JMP software) were used to present data for total WBC cells and enzyme activities in the BALF. Means were compared by student-t test. P values less than 0.05 were considered significant.
3. Results
3.1. BAL cell aggregates in the BALF of ventilated WT mice
BAL cells of naïve mice normally display scattered single cells [31]. Cytological examination of BAL cells revealed such scattered single BAL cells in both WT and C3 null naïve mice as expected (Fig. 1A and B) [31]. In contrast, remarkably few scattered single BAL cells and many large cell aggregates were observed in the BALF of mechanically ventilated WT mice (Fig. 1C). Although there were large cell aggregates in the BALF of ventilated C3 null clearly more scattered single BAL cells were observed compared with mechanically ventilated WT mice (Fig. 1C vs. 1D). We postulated that biological responses to ventilation induced cell aggregation, which in turn might have reduced scattered single BAL cells. Therefore, we determined total numbers of single BAL cells in these mice.
Fig. 1.

Representative micrography of BAL cells. A) Sham wild type (WT) mouse, a representative of 7 mice. B) Sham C3 null mouse, a representative of 4 mice. C) Ventilated WT mouse, a representative of 7 mice. D) Ventilated C3 null mouse, a representative of 5 mice. White arrows indicate cell aggregates in 1C and 1D. Objective magnification was ×10 and ×40 for insets in C) and D).
3.2. Total single BAL cells in BALF significantly decrease in WT mice compared with C3 null mice following mechanical ventilation
Total single BAL cell counts were observed to be significantly lower in mechanically ventilated WT mice compared with all other groups, including C3 null mice (Fig. 2A). There was no statistical difference in total single BAL cell counts between sham and ventilated C3 null mice (Fig. 2A). Although the overall number of total single BAL cells decreased in ventilated WT mice, the percentage of PMN in BAL cells was slightly higher compared with all other groups (Table 1).
Fig. 2.
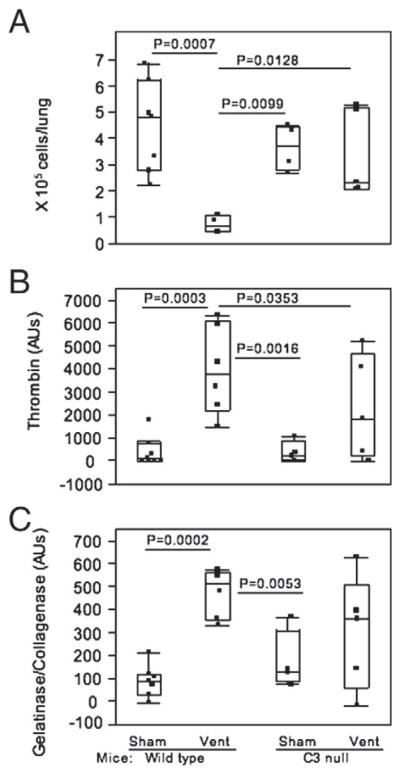
Examination of BAL cells and BALF. Dots in each box plot represent individual mouse. Vent=ventilated for 2 h as described in the Methods. A) Total single BAL cells in the BALF. Numbers of mice in each group were 7 wild type (WT) sham, 4 WT vent, 4 C3 null sham, and 5 C3 null vent. Statistical significance was against wild type ventilated. B) Thrombin activity. Numbers of mice in each group were 7 wild type (WT) sham, 6 WT vent, 4 C3 null sham, and 6 C3 null vent. C) Gelatinase/collagenase activity. Numbers of mice in each group were 7 wild type (WT) sham, 6 WT vent, 4 C3 null sham, and 5 C3 null vent.
Table 1.
Percentage of PMN in the total single BAL cells.
| Sham | Vent | |
|---|---|---|
| WT | 0.6±0.4 (7) | 2.8±1.4 (6) |
| C3 null | 1.3±1.1 (4) | 0.6±0.9 (5) |
Note: Data are expressed as mean±SEM. Numbers in parenthesis are animals used.
3.3. Upregulated activities of thrombin and gelatinase/collagenase in ventilated WT mice without C5a generation
Thrombin activity was significantly elevated in the BALF of mechanically ventilated WT mice compared to all other groups (Fig. 2B, P<0.05). In mechanically ventilated group, median thrombin activity was two-fold larger in WT than in C3 null mice (Fig. 2B). There was no significant difference between sham and mechanically ventilated groups in C3 null mice.
Similar to thrombin activity, gelatinase/collagenase activity was elevated in the BALF of ventilated WT mice compared to all other groups although it was not statistically significant (Fig. 2B and C). In mechanically ventilated mice, gelatinase/collagenase activity tended to be larger in WT than in C3 null mice (P=0.062). There was no statistical difference between sham C3 null and mechanically ventilated C3 null mice.
Likewise, C5a levels in BALF were comparable among these four groups: sham WT, mechanically ventilated WT, sham C3 null, and mechanically ventilated C3 null mice. C5a levels (mean±SEM) in these groups were 0.240±0.015, 0.239±0.033, 0.215±0.051, and 0.225±0.039, respectively.
3.4. Total single BAL cells in the BALF are inversely correlated with activities of gelatinase/collagenase
Correlation analysis between total single BAL cell counts and gelatinase/collagenase activity revealed that all sham treated animals and 2 out of 5 ventilated C3 null mice were sorted to the left upper corner where gelatinase/collagenase activities were low and total single BAL cells were high (Fig. 3A). The association was statistically significant (P=0.0049). A similar association was observed between total single BAL cell counts and thrombin activities (P=0.0416) (Fig. 3B).
Fig. 3.

Association analysis between total single BAL cells and gelatinase/collagenase or thrombin activity. Total single BAL cells were plotted against activities of gelatinase/collagenase (A) or thrombin (B) using the JMP 8 software. Each symbol represents individual mouse.
3.5. Lack of C3 fails to activate gelatinase/collagenase
Sera of naïve WT and C3 null mice were examined for activation of thrombin and gelatinase/collagenase upon incubation of sera on zymosan, which is a well-known and widely used activator of C3 [30, 32]. Thrombin activity was undetectable in both sera. In contrast, gelatinase/collagenase activity increased in WT sera in a dose-dependent manner whereas it was undetectable in C3 null sera (Fig. 4).
Fig. 4.

Gelatinase/collagenase activity in native mouse sera on zymosan-coated surface. Pooled sera of three naïve WT and C3 null mice were assayed at indicated dilutions for gelatinase/collagenase activities. Data were expressed as mean±SEM, many of which were smaller than symbols. *, P<0.0001.
3.6. HCVF-pretreatment inactivates C3 and increases BAL cells
Following injurious mechanical ventilation, punctate C3 staining was observed in the lungs of control (untreated) WT mice while it was essentially undetectable in FCVF-pretreated WT mice (Fig. 5A). Quantitative analysis of C3 positive cells demonstrated that C3 positive cells were significantly low in the lungs of HCVF-treated WT mice compared with untreated control WT mice (Fig. 5B). As expected, total BAL cells in HCVF-treated lungs were significantly higher than the controls (Fig. 5C).
Fig. 5.

HCVF-pretreatment. A) Immunohistochemical staining of C3. Lung sections were stained with FITC-conjugated goat anti-mouse C3 antibody. White arrows indicate C3 positive cells. Objective magnification ×20. 4 control and 3 HCVF-treated WT mice were used. B) Quantitative analysis of C3 positive cells in the lung. C3 positive cells were counted per 1 mm2 and expressed as mean±SEM. C) Total single BAL cells. Cells were counted as in Fig. 2A and expressed as mean±SEM.
4. Discussion
In this study, we focused on soluble factors and cells, in order to obtain objectively quantitative data, which could be easily statistically analyzed. Acute lung injury has been associated with reduced total single BAL cells and PMN infiltration as early markers of lung injury [24, 33, 34]. Our results demonstrate that in mechanically ventilated C3 null mice, total single BAL cells are comparable to those in sham treated C3 and WT mice. MMP activities are inversely correlated with total single BAL cells that are mostly alveolar macrophages (data not shown) [31]. In addition, there is a slight increase in PMN population in the total single BAL cells in WT mice but not in C3 null mice following mechanical ventilation. C3 deficiency did not alter lung pathology, as lungs of C3 null mice are reportedly normal in appearance [35]. Thus, our investigations, for the first time, provide in vivo evidence that C3 is involved with pathogenesis of VILI. The mechanisms of VILI are, in part, mediated by activation of C3, which further activates MMPs.
We presume that the loss of single BAL cells is due to increased cell aggregates. Indeed, large cell aggregates are clearly noticed from microscopic analysis of BAL cells obtained from mechanically ventilated WT mice, whereas fewer such aggregates were observed in BAL cell preparations of mechanically ventilated C3 null mice. Cell aggregation can be caused by up-regulation of cell adhesion molecules, which has been reported following VILI [24, 36, 37] and coagulation, which involves fibrin formation, thereby embedding cells into fibrin clots. Fibrin is formed from fibrinogen by thrombin. Consistent with this reasoning, thrombin activity was significantly higher in WT mice than in C3 null mice following mechanical ventilation.
Fibrin deposition has been suggested to be protective by localizing and minimizing injury at an early phase [38]. We speculated that elevated thrombin activity is in response to developing lung injury, so that fibrin clots can localize or contain injury. Lung injury could be caused by mechanical forces, but also by enzymatic digestion of tissue matrices. Previous studies have associated pathogenesis of acute lung injury with matrix metalloproteinase (MMP), including MMP-1 (collagenase), MMP-2 (gelatinase A) and MMP-9 (gelatinase B) [4–6, 8, 39]. In line with our hypothesis, MMP activities, measured as gelatinase/collagenase activity, are significantly elevated in BALF of WT mice compared with C3 null mice following mechanical ventilation. Taken together, these results suggest that C3 activation mediates injury by activating MMPs, which digest tissue matrix proteins, resulting in lung injury.
The logical extension is to determine whether gelatinase/collagenase is upregulated by activating C3. Sera of WT but not C3 null mice demonstrated gelatinase/collagenase activities in a dose dependent manner upon activation by zymosan, a well-known and widely used C3 activator [30]. These results suggest that C3 can be activated on damaged tissue by mechanical forces, which can be pressure and/or stretching by repeated expansions as lowering tidal volume reduced VILI and reperfusion injury [40]. As for endogenous C3 targets, there are many potential candidates that have been found in ischemic tissue injuries, such as cytokeratin and nonmuscle myosin [41, 42]. Further investigations are required to determine endogenous C3 targets, including cell types that would release MMPs.
HCVF has been developed for an intentional therapeutic use and it has been shown to attenuate reperfusion myocardial injury in mouse model study [23, 43]. Current study also demonstrates that HCVF treatment is effective in inactivating C3 in the lung during injurious mechanical ventilation as the HCVF treatment significantly reduces C3 deposition onto the lung of WT mice. Our results also suggest that the HCVF treatment attenuates acute lung injury as total BAL cells are increased significantly. C3 could be locally synthesized by tissue type cells and/or circulating C3 could be recruited to injured cells via activation of the lectin pathway [44–46]. Further studies are required to determine cell types that were deposited with C3. In addition, dosing and timing of HCVF treatment need to be optimized to maximize its therapeutic effect in VILI.
Lastly, lung injury also can be initiated by other causes, including inhalation of irritant/chemical and deposition of an immune complex [35]. The latter causes autoimmune disease [35]. Very recently, lung injury induced by paraquat (herbicide), was attenuated in C3 deficient mice, suggesting that C3 is also involved with chemically induced lung injury [47]. Interestingly, C3 deficiency was not protective in an IgG immune complex induced lung injury, in which thrombin was attributed to activate C5a, causing injury [35]. In our study, C5a levels in BALF were not affected by mechanical ventilation as expected. It is quite possible that tissue injuries would be mediated by different mechanisms depending on insults [48]. Examples are that C3 is activated by the alternative pathway in joints of an experimental autoimmune arthritis while it is mediated by the lectin pathway in a myocardial ischemic–reperfusion injury [46, 49]. The former is induced by the immune complex whereas the latter is triggered by a combination of hypoxia and reperfusion, which involves physical distress, similar to mechanical ventilation injury. Thus, understanding the upstream mechanisms of C3 activation requires further investigation in our mechanical lung injury model.
In conclusion, we propose that activation of C3 further induces activities of MMPs, which would destroy tissue by digesting tissue matrix. In response, coagulation cascade would be initiated in order to contain injury locally but aggregates BAL cells, in resulting decreased single BAL cells during the early phase of mechanical ventilation. Taken together, our findings and observations by others suggest that inhibitors of complement and MMPs may be potential adjunctive treatments for mechanical ventilator-induced acute lung injury.
Acknowledgments
The authors thank Dr. Michael Carroll for providing C3 null mice breeding pairs. This work was in part supported by NIH grants UO1 AI074503-01, R21 AI077081-01A1, and 5RO1HL086827.
References
- 1.Frank JA, Matthay MA. Science review: mechanisms of ventilator-induced injury. Crit Care. 2003;7:233–41. doi: 10.1186/cc1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Takahashi K, Ip WE, Michelow IC, Ezekowitz RA. The mannose-binding lectin: a prototypic pattern recognition molecule. Curr Opin Immunol. 2006;18:16–23. doi: 10.1016/j.coi.2005.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Markiewski MM, DeAngelis RA, Lambris JD. Complexity of complement activation in sepsis. J Cell Mol Med. 2008;12:2245–54. doi: 10.1111/j.1582-4934.2008.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foda HD, Rollo EE, Drews M, Conner C, Appelt K, Shalinsky DR, et al. Ventilator-induced lung injury upregulates and activates gelatinases and EMMPRIN: attenuation by the synthetic matrix metalloproteinase inhibitor, Prinomastat (AG3340) Am J Respir Cell Mol Biol. 2001;25:717–24. doi: 10.1165/ajrcmb.25.6.4558f. [DOI] [PubMed] [Google Scholar]
- 5.Delclaux C, d’Ortho MP, Delacourt C, Lebargy F, Brun-Buisson C, Brochard L, et al. Gelatinases in epithelial lining fluid of patients with adult respiratory distress syndrome. Am J Physiol. 1997;272:L442–51. doi: 10.1152/ajplung.1997.272.3.L442. [DOI] [PubMed] [Google Scholar]
- 6.Warner RL, Beltran L, Younkin EM, Lewis CS, Weiss SJ, Varani J, et al. Role of stromelysin 1 and gelatinase B in experimental acute lung injury. Am J Respir Cell Mol Biol. 2001;24:537–44. doi: 10.1165/ajrcmb.24.5.4160. [DOI] [PubMed] [Google Scholar]
- 7.Hofstra JJ, Juffermans NP, Schultz MJ, Zweers MM. Pulmonary coagulopathy as a new target in lung injury—a review of available pre-clinical models. Curr Med Chem. 2008;15:588–95. doi: 10.2174/092986708783769696. [DOI] [PubMed] [Google Scholar]
- 8.Schultz MJ, Haitsma JJ, Zhang H, Slutsky AS. Pulmonary coagulopathy as a new target in therapeutic studies of acute lung injury or pneumonia—a review. Crit Care Med. 2006;34:871–7. [PubMed] [Google Scholar]
- 9.Welty-Wolf KE, Carraway MS, Ortel TL, Ghio AJ, Idell S, Egan J, et al. Blockade of tissue factor-factor X binding attenuates sepsis-induced respiratory and renal failure. Am J Physiol Lung Cell Mol Physiol. 2006;290:L21–31. doi: 10.1152/ajplung.00155.2005. [DOI] [PubMed] [Google Scholar]
- 10.Chapman HA, Stahl M, Allen CL, Yee R, Fair DS. Regulation of the procoagulant activity within the bronchoalveolar compartment of normal human lung. Am Rev Respir Dis. 1988;137:1417–25. doi: 10.1164/ajrccm/137.6.1417. [DOI] [PubMed] [Google Scholar]
- 11.Gropper MA, Wiener-Kronish J. The epithelium in acute lung injury/acute respiratory distress syndrome. Curr Opin Crit Care. 2008;14:11–5. doi: 10.1097/MCC.0b013e3282f417a0. [DOI] [PubMed] [Google Scholar]
- 12.Haiko J, Suomalainen M, Ojala T, Lahteenmaki K, Korhonen TK. Invited review: breaking barriers—attack on innate immune defences by omptin surface proteases of enterobacterial pathogens. Innate Immun. 2009;15:67–80. doi: 10.1177/1753425909102559. [DOI] [PubMed] [Google Scholar]
- 13.Van de Wouwer M, Plaisance S, De Vriese A, Waelkens E, Collen D, Persson J, et al. The lectin-like domain of thrombomodulin interferes with complement activation and protects against arthritis. J Thromb Haemost. 2006;4:1813–24. doi: 10.1111/j.1538-7836.2006.02033.x. [DOI] [PubMed] [Google Scholar]
- 14.Blackburn JS, Brinckerhoff CE. Matrix metalloproteinase-1 and thrombin differentially activate gene expression in endothelial cells via PAR-1 and promote angiogenesis. Am J Pathol. 2008;173:1736–46. doi: 10.2353/ajpath.2008.080512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Presanis JS, Hajela K, Ambrus G, Gal P, Sim RB. Differential substrate and inhibitor profiles for human MASP-1 and MASP-2. Mol Immunol. 2004;40:921–9. doi: 10.1016/j.molimm.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 16.Takahashi K, Chang WC, Takahashi M, Pavlov V, Ishida Y, La Bonte L, et al. Mannose-binding lectin and its associated proteases (MASPs) mediate coagulation and its deficiency is a risk factor in developing complications from infection, including disseminated intravascular coagulation. Immunobiol. 2011;216:96–102. doi: 10.1016/j.imbio.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polley MJ, Nachman RL. Human complement in thrombin-mediated platelet function: uptake of the C5b-9 complex. J Exp Med. 1979;150:633–45. doi: 10.1084/jem.150.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang CJ, Hsu LA, Ko YH, Chen PL, Chuang YT, Lin CY, et al. Thrombin regulates matrix metalloproteinase-9 expression in human monocytes. Biochem Biophy Res Commun. 2009;385:241–6. doi: 10.1016/j.bbrc.2009.05.049. [DOI] [PubMed] [Google Scholar]
- 19.Rosen T, Schatz F, Kuczynski E, Lam H, Koo AB, Lockwood CJ. Thrombin-enhanced matrix metalloproteinase-1 expression: a mechanism linking placental abruption with premature rupture of the membranes. J Maternal-Fetal Neonat Med. 2002;11:11–7. doi: 10.1080/jmf.11.1.11.17. [DOI] [PubMed] [Google Scholar]
- 20.Hiller O, Lichte A, Oberpichler A, Kocourek A, Tschesche H. Matrix metalloproteinases collagenase-2, macrophage elastase, collagenase-3, and membrane type 1-matrix metalloproteinase impair clotting by degradation of fibrinogen and factor XII. J Biol Chem. 2000;275:33008–13. doi: 10.1074/jbc.M001836200. [DOI] [PubMed] [Google Scholar]
- 21.Hart ML, Ceonzo KA, Shaffer LA, Takahashi K, Rother RP, Reenstra WR, et al. Gastrointestinal ischemia–reperfusion injury is lectin complement pathway dependent without involving C1q. J Immunol. 2005;174:6373–80. doi: 10.4049/jimmunol.174.10.6373. [DOI] [PubMed] [Google Scholar]
- 22.Bolger MS, Ross DS, Jiang H, Frank MM, Ghio AJ, Schwartz DA, et al. Complement levels and activity in the normal and LPS-injured lung. Am J Physiol Lung Cell Mol Physiol. 2007;292:L748–59. doi: 10.1152/ajplung.00127.2006. [DOI] [PubMed] [Google Scholar]
- 23.Gorsuch WB, Guikema BJ, Fritzinger DC, Vogel CW, Stahl GL. Humanized cobra venom factor decreases myocardial ischemia–reperfusion injury. Mol Immunol. 2009;47:506–10. doi: 10.1016/j.molimm.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choudhury S, Wilson MR, Goddard ME, O’Dea KP, Takata M. Mechanisms of early pulmonary neutrophil sequestration in ventilator-induced lung injury in mice. Am J Physiol Lung Cell Mol Physiol. 2004;287:L902–10. doi: 10.1152/ajplung.00187.2004. [DOI] [PubMed] [Google Scholar]
- 25.Belperio JA, Keane MP, Burdick MD, Londhe V, Xue YY, Li K, et al. Critical role for CXCR2 and CXCR2 ligands during the pathogenesis of ventilator-induced lung injury. J Clin Invest. 2002;110:1703–16. doi: 10.1172/JCI15849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prodeus AP, Zhou X, Maurer M, Galli SJ, Carroll MC. Impaired mast cell-dependent natural immunity in complement C3-deficient mice. Nature. 1997;390:172–5. doi: 10.1038/36586. [DOI] [PubMed] [Google Scholar]
- 27.Moller-Kristensen M, Ip WK, Shi L, Gowda LD, Hamblin MR, Thiel S, et al. Deficiency of mannose-binding lectin greatly increases susceptibility to postburn infection with Pseudomonas aeruginosa. J Immunol. 2006;176:1769–75. doi: 10.4049/jimmunol.176.3.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wilson MR, Choudhury S, Goddard ME, O’Dea KP, Nicholson AG, Takata M. High tidal volume upregulates intrapulmonary cytokines in an in vivo mouse model of ventilator-induced lung injury. J Appl Physiol. 2003;95:1385–93. doi: 10.1152/japplphysiol.00213.2003. [DOI] [PubMed] [Google Scholar]
- 29.Banda NK, Levitt B, Wood AK, Takahashi K, Stahl GL, Holers VM, et al. Complement activation pathways in murine immune complex-induced arthritis and in C3a and C5a generation in vitro. Clin Exp Immunol. 2010;159:100–8. doi: 10.1111/j.1365-2249.2009.04035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banda NK, Takahashi K, Wood AK, Holers VM, Arend WP. Pathogenic complement activation in collagen antibody-induced arthritis in mice requires amplification by the alternative pathway. J Immunol. 2007;179:4101–9. doi: 10.4049/jimmunol.179.6.4101. [DOI] [PubMed] [Google Scholar]
- 31.van oud Alblas AB, van Furth R. Origin, kinetics, and characteristics of pulmonary macrophages in the normal steady state. J Exp Med. 1979;149:1504–18. doi: 10.1084/jem.149.6.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ward PA, Johnson KJ, Till GO. Animal models of oxidant lung injury. Respiration. 1986;50(Suppl 1):5–12. doi: 10.1159/000195082. [DOI] [PubMed] [Google Scholar]
- 33.Till GO, Johnson KJ, Kunkel R, Ward PA. Intravascular activation of complement and acute lung injury. Dependency on neutrophils and toxic oxygen metabolites. J Clin Invest. 1982;69:1126–35. doi: 10.1172/JCI110548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang H, Downey GP, Suter PM, Slutsky AS, Ranieri VM. Conventional mechanical ventilation is associated with bronchoalveolar lavage-induced activation of polymorphonuclear leukocytes: a possible mechanism to explain the systemic consequences of ventilator-induced lung injury in patients with ARDS. Anesthesiol. 2002;97:1426–33. doi: 10.1097/00000542-200212000-00014. [DOI] [PubMed] [Google Scholar]
- 35.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–7. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 36.Miyao N, Suzuki Y, Takeshita K, Kudo H, Ishii M, Hiraoka R, et al. Various adhesion molecules impair microvascular leukocyte kinetics in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2006;290:L1059–68. doi: 10.1152/ajplung.00365.2005. [DOI] [PubMed] [Google Scholar]
- 37.Mulligan MS, Till GO, Smith CW, Anderson DC, Miyasaka M, Tamatani T, et al. Role of leukocyte adhesion molecules in lung and dermal vascular injury after thermal trauma of skin. Am J Pathol. 1994;144:1008–15. [PMC free article] [PubMed] [Google Scholar]
- 38.Kipnis E, Guery BP, Tournoys A, Leroy X, Robriquet L, Fialdes P, et al. Massive alveolar thrombin activation in Pseudomonas aeruginosa-induced acute lung injury. Shock. 2004;21:444–51. doi: 10.1097/00024382-200405000-00008. [DOI] [PubMed] [Google Scholar]
- 39.Fligiel SE, Standiford T, Fligiel HM, Tashkin D, Strieter RM, Warner RL, et al. Matrix metalloproteinases and matrix metalloproteinase inhibitors in acute lung injury. Hum Pathol. 2006;37:422–30. doi: 10.1016/j.humpath.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 40.Petrucci N, Iacovelli W. Ventilation with lower tidal volumes versus traditional tidal volumes in adults for acute lung injury and acute respiratory distress syndrome. Cochrane Database Syst Rev. 2004:CD003844. doi: 10.1002/14651858.CD003844.pub2. [DOI] [PubMed] [Google Scholar]
- 41.Collard CD, Montalto MC, Reenstra WR, Buras JA, Stahl GL. Endothelial oxidative stress activates the lectin complement pathway: role of cytokeratin 1. Am J Pathol. 2001;159:1045–54. doi: 10.1016/S0002-9440(10)61779-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang M, Alicot EM, Chiu I, Li J, Verna N, Vorup-Jensen T, et al. Identification of the target self-antigens in reperfusion injury. J Exp Med. 2006;203:141–52. doi: 10.1084/jem.20050390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vogel CW, Fritzinger DC. Humanized cobra venom factor: experimental therapeutics for targeted complement activation and complement depletion. Curr Pharm Des. 2007;13:2916–26. doi: 10.2174/138161207782023748. [DOI] [PubMed] [Google Scholar]
- 44.Hill LD, Sun L, Leuschen MP, Zach TL. C3 synthesis by A549 alveolar epithelial cells is increased by interferon-gamma and dexamethasone. Immunol. 1993;79:236–40. [PMC free article] [PubMed] [Google Scholar]
- 45.Strunk RC, Eidlen DM, Mason RJ. Pulmonary alveolar type II epithelial cells synthesize and secrete proteins of the classical and alternative complement pathways. J Clin Invest. 1988;81:1419–26. doi: 10.1172/JCI113472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walsh MC, Bourcier T, Takahashi K, Shi L, Busche MN, Rother RP, et al. Mannose-binding lectin is a regulator of inflammation that accompanies myocardial ischemia and reperfusion injury. J Immunol. 2005;175:541–6. doi: 10.4049/jimmunol.175.1.541. [DOI] [PubMed] [Google Scholar]
- 47.Sun S, Wang H, Zhao G, An Y, Guo Y, Du L, et al. Complement inhibition alleviates paraquat-induced acute lung injury. Am J Respir Cell Mol Biol. doi: 10.1165/rcmb.2010-0444OC. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gipson TS, Bless NM, Shanley TP, Crouch LD, Bleavins MR, Younkin EM, et al. Regulatory effects of endogenous protease inhibitors in acute lung inflammatory injury. J Immunol. 1999;162:3653–62. [PubMed] [Google Scholar]
- 49.Banda NK, Thurman JM, Kraus D, Wood A, Carroll MC, Arend WP, et al. Alternative complement pathway activation is essential for inflammation and joint destruction in the passive transfer model of collagen-induced arthritis. J Immunol. 2006;177:1904–12. doi: 10.4049/jimmunol.177.3.1904. [DOI] [PubMed] [Google Scholar]