

Abstract
The cytokine ciliary neurotrophic factor (CNTF) promotes the growth of neural processes from many kinds of neurons in the developing and regenerating adult nervous system, but the intracellular signaling mechanisms mediating this important function of CNTF are poorly understood. Here, we show that CNTF activates the nuclear factor-κB (NF-κB) transcriptional system in neonatal sensory neurons and that blocking NF-κB-dependent transcription inhibits CNTF-promoted neurite growth. Selectively blocking NF-κB activation by the noncanonical pathway that requires tyrosine phosphorylation of inhibitor κB-α (IκB-α), but not by the canonical pathway that requires serine phosphorylation of IκB-α, also effectively inhibits CNTF-promoted neurite growth. CNTF treatment activates spleen tyrosine kinase (SYK) whose substrates include IκB-α. CNTF-induced SYK phosphorylation is rapidly followed by increased tyrosine phosphorylation of IκB-α, and blocking SYK activation or tyrosine phosphorylation of IκB-α prevents CNTF-induced NF-κB activation and CNTF-promoted neurite growth. These findings demonstrate that NF-κB signaling by an unusual activation mechanism is essential for the ability of CNTF to promote the growth of neural processes in the developing nervous system.
Keywords: NF-κB, neurite, CNTF, SYK, development, sensory neuron
Introduction
Nuclear factor-κB (NF-κB) is a ubiquitously expressed transcription factor system that consists of homodimers or heterodimers of five structurally related proteins: p65, RelB, c-Rel, p50, and p52, of which the p50/p65 heterodimer is the most abundant and widely expressed (Hayden and Ghosh, 2004). NF-κB is held in an inactive form in the cytosol by interaction with a member of the IκB family of inhibitory proteins, IκBα, IκBβ, IκBε, IκBγ, Bcl-3, p100 and p105, of which IκBα is the predominantly expressed inhibitor. In the canonical NF-κB signaling pathway, NF-κB is activated by phosphorylation of IκBα on serine residues 32 and 36 by an IκB kinase complex. This leads to ubiquitination and proteasome-mediated degradation of IκBα and translocation of the liberated NF-κB to the nucleus where it binds to consensus κB sequences in the promoter and enhancer regions of responsive genes (Hayden and Ghosh, 2004). NF-κB can also be activated by several alternative mechanisms including one in which IκBα is phosphorylated on tyrosine 42, which results in its dissociation from NF-κB without proteasome-mediated degradation (Koong et al., 1994; Imbert et al., 1996; Bui et al., 2001; Takada et al., 2003).
Classically, NF-κB has been shown to regulate the expression of genes involved in innate and adaptive immune responses, stress responses, cell survival, and cell proliferation (Liang et al., 2004). In the nervous system, NF-κB is activated by a variety of neurotrophic factors, cytokines, and neurotransmitters, and can promote neuronal survival or bring about neuronal death. NF-κB signaling also regulates synaptic function, plays a role learning and memory and participates in peripheral nerve myelination (Kaltschmidt et al., 2005).
CNTF promotes the survival of a variety of neurons (Horton et al., 1998; Nishimune et al., 2000) and stimulates neurite growth and axon regeneration in several in vitro and in vivo experimental paradigms in the developing and mature nervous system (Hartnick et al., 1996; Cui and Harvey, 2000; Siegel et al., 2000). Binding of CNTF to a receptor complex consisting of gp130, leukemia inhibitory factor receptor β, and CNTF receptor α (Stahl and Yancopoulos, 1994) leads to the activation of several signal transduction pathways including JAK (Janus kinase)/STAT (signal transducer and activator of transcription), MEK [extracellular signal-regulated kinase (ERK) kinase]/MAPK (mitogen-activated protein kinase), phosphoinositide 3-kinase (PI3-K)/Akt, and NF-κB (Nishimune et al., 2000; Rane and Reddy, 2000). Because NF-κB signaling via the canonical activation pathway partially contributes to the neurite growth-promoting effects of the neurotrophins NGF and BDNF (Sole et al., 2004; Gutierrez et al., 2005), we investigated whether NF-κB signaling plays any role in the neurite growth-promoting effects of CNTF. Using neonatal mouse nodose ganglion sensory neurons, which are supported by CNTF (Horton et al., 1998), we show by a variety of complementary experimental approaches that NF-κB signaling is essential for CNTF-promoted neurite growth and that the NF-κB activation mechanism used by CNTF for promoting growth is distinct from that used by the neurotrophins.
Materials and Methods
Neuron culture and transfection.
Dissociated cultures of nodose ganglion neurons from newborn mice were grown in polyornithine/laminin-coated 35 mm dishes in defined medium and were transfected 3 h after plating with plasmid-coated or oligonucleotide-coated (κB decoy dsDNA, 5′-GAGGGGACTTTCCCT-3′; scrambled control dsDNA, 5′-GATGCGTCTGTCGCA-3′) gold microcarriers using the Bio-Rad (Hercules, CA) Gene-gun (Gutierrez et al., 2005). Immunocytochemistry was used to confirm that nodose neurons transfected with expression plasmids for Tyr IκBα, Ser IκBα, and Syk dominant-negative mutants have increased expression of the corresponding protein (data not shown). The survival of transfected neurons was quantified by counting the numbers of yellow fluorescent protein (YFP)-labeled neurons 12 h after plating and at later time points and expressing the latter as a percentage of the former. Survival in nontransfected cultures was estimated by counting the number of neurons in a grid 3 and 24 h after plating and expressing the latter as a percentage of the former.
Quantification of fluorescence.
To estimate the relative level of NF-κB activation, neurons were transfected with a plasmid expressing green fluorescent protein (GFP) under the control of an NF-κB promoter. Neurons were imaged with a Zeiss (Oberkochen, Germany) Axioplan confocal microscope and mean soma fluorescence intensity was obtained using LSM510 software.
Analysis of neuritic arbors.
YFP-labeled neurons were visualized and digitally acquired using an Axioplan Zeiss confocal microscope. In nontransfection experiments, neurons were fluorescently labeled with the vital dye calcein-AM (Invitrogen, Eugene, OR). Neuritic arbors were traced using LSM510 software from which total neurite length and number of branch points was calculated and Sholl analysis was performed (Gutierrez et al., 2005).
Western blots.
Neurons were plated at high density in polyornithine/laminin-coated 96-well plates (5000 neurons per well). Four hours after plating, 50 ng/ml CNTF was added for the indicated times. The cells were lysed in radioimmunoprecipitation assay buffer and insoluble debris was removed by centrifugation. Samples were transferred to polyvinylidene difluoride membranes using the Bio-Rad TransBlot. Membranes were blocked with 5% dried milk in PBS with 0.1% Tween 20. Membranes were then incubated with anti-phospho-Tyr IκB-α antibody (1:200; Abcam, Cambridge, UK), anti-phospho-Syk antibody (1:1000; Cell Signaling Technology, Beverly, MA), anti-IκB-α antibody (1:1000; Cell Signaling Technology), or anti-β-III tubulin antibody (1:10000; Promega, Hawthorne, Australia), which were detected with peroxidase-linked secondary antibody (GE Healthcare Bio-Sciences, Piscataway, NJ) and ECL-plus (GE Healthcare Bio-Sciences). Densitometry was performed using Adobe (San Jose, CA) Photoshop.
Results
Blocking NF-κB-dependent transcription inhibits CNTF-promoted neurite growth
To investigate the importance of NF-κB in mediating the response of neurons to CNTF, we studied the consequences of specifically inhibiting a key step in NF-κB signaling, the binding of activated NF-κB to regulatory elements in target genes. This was blocked by transfecting neurons with double stranded DNA oligonucleotides containing the κB consensus binding sequence. This κB decoy DNA has been successfully used in vitro and in vivo to inhibit NF-κB transcriptional activity by sequestering transcriptionally active NF-κB complexes (Morishita et al., 1997; Tomita et al., 2000; Gutierrez et al., 2005). Postnatal day 0 (P0) nodose ganglion neurons were transfected with gold particles coated with either the κB decoy DNA or a control oligonucleotide of scrambled sequence 3 h after plating. The cultures received CNTF after transfection and neuronal survival and neurite arbor size and complexity were quantified 24 h after plating.
The κB decoy DNA, but not the control oligonucleotide caused a marked threefold reduction in neurite length and branching and a substantial reduction in neurite growth as quantified by Sholl analysis (Fig. 1). Despite the dramatic effect on neurite growth, the cell bodies of neurons transfected with κB decoy DNA retained a normal appearance (Fig. 1E) and continued to survive just as well as control-transfected neurons in the presence of CNTF, with ∼70% of the neurons surviving up to 72 h after plating (Fig. 1C). Accordingly, NFκB reporter activity was significantly reduced in neurons transfected with κB decoy DNA and not in neurons transfected with control DNA (see Fig. 4D). These results suggest that NF-κB-dependent transcription is required for CNTF-promoted neurite growth from newborn mouse nodose ganglion neurons, but is not needed for the survival of these neurons with CNTF. Similar decreases in the size and complexity of neurite arbors without any neuronal loss were observed in CNTF-supported nodose neurons treated with SN50, a cell-permeable inhibitor of NF-κB nuclear translocation (data not shown). Conversely, combined overexpression of p65/p50 in CNTF-supported neurons resulted in a significant increase in neurite length and branching (data not shown).
Figure 1.

Effect of κB decoy DNA on CNTF-promoted neurite growth. Three hours after plating, P0 nodose neurons were transfected with a YFP expression plasmid together with either κB decoy DNA or a scrambled control DNA. A–D, After 24 h incubation with 50 ng/ml CNTF, neurite arbor length (A), branch point number (B), Sholl analysis (D), and neuronal survival (C) were ascertained. Means ± SEs of a typical experiment are shown (50–90 neurons per condition). Very similar data were obtained in three independent experiments. **p < 0.001. E, Photomicrographs show typical control transfected and κB decoy DNA-transfected neurons. Scale bars, 30 μm.
Figure 4.
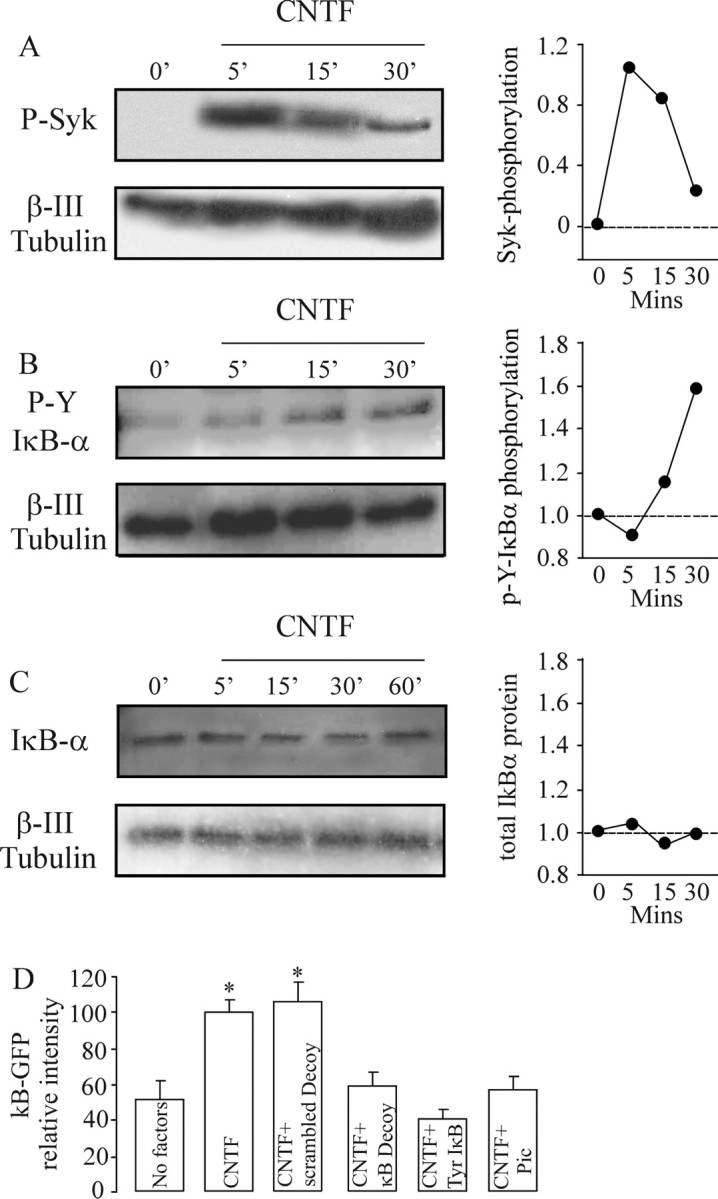
CNTF promotes tyrosine phosphorylation of SYK and IκB-α and enhances NF-κB transcriptional activity without IκB-α degradation. A–C, Representative Western blots showing phosphotyrosine-SYK (A), phosphotyrosine-IκB-α (B), and total IκB-α protein (C) relative to β-III tubulin protein in cultured P0 nodose neurons exposed to 50 ng/ml CNTF for the times indicated. Graphs of densitometric estimates of the relative phosphotyrosine-SYK and phosphotyrosine-IκB-α levels in blots A–C are shown (similar data were obtained in three independent experiments). D, Relative level of NF-κB-driven GFP fluorescence in P0 nodose neurons 24 h after transfection with the NF-κB reporter plasmid together with either the Y42F IκB-α plasmid, an empty control plasmid, κB decoy DNA, scrambled control oligonucleotide, or treatment with 10 μm piceatannol (Syk inhibitor). Means ± SEs of fluorescence measurements on 40–60 neurons per experiment are shown. Statistical comparisons are with respect to controls; *p < 0.05.
CNTF does not promote neurite growth via the canonical NF-κB signaling pathway
The canonical NF-κB signaling pathway can be selectively inhibited by an IκB-α protein possessing serine to alanine substitutions at residues 32 and 36 that prevent its phosphorylation by the IκB kinase complex, but do not affect its association with NF-κB dimers (Roff et al., 1996). We showed previously that expression of this 32/36-SS/AA IκB-α mutant in nodose neurons grown with BDNF causes a 30% reduction in neurite arbor size (Gutierrez et al., 2005). In marked contrast to nodose neurons grown with BDNF, this 32/36-SS/AA IκB-α had no effect whatsoever on the neurite arbors of nodose neurons grown with CNTF, as shown by Sholl analysis (Fig. 2A), suggesting that CNTF does not mediate its effects on neurite growth via the canonical NF-κB pathway.
Figure 2.

CNTF does not promote neurite growth by canonical NF-κB signaling. A–C, Three hours after plating, P0 nodose neurons were transfected with a YFP expression plasmid together with either a plasmid expressing the 32/36-SS/AA IκB-α mutant (Ser IκB-α) or an empty control plasmid (A) were treated with either 4 μm ALLN or vehicle control (B) or were treated with 20 nm MG132 or vehicle control (C). After 24 h incubation with 50 ng/ml CNTF, Sholl analysis was performed. Means ± SEs of a typical experiment are shown (50–90 neurons per condition). Very similar data were obtained in three independent experiments. Statistical comparisons are with respect to the empty plasmid-transfected neurons or vehicle-treated neurons.
Additional confirmation of the lack of involvement of canonical NF-κB signaling in CNTF-promoted neurite growth was obtained by inhibiting another key step in this pathway, proteasome-mediated degradation of ubiquitinated phosphoserine IκB-α (Hayden and Ghosh, 2004). We showed previously that the proteasome inhibitor N-acetyl-Leu-Leu-norleucinal (ALLN) reduces the size of the neurite arbors of nodose neurons grown with BDNF (Gutierrez et al., 2005). Neither ALLN (Fig. 2B) nor another proteasome inhibitor MG132 (Fig. 2C) affected neurite arbor size in CNTF-supplemented cultures, suggesting that proteasome function is not required for CNTF-promoted growth. In contrast, we confirmed that both MG132 and ALLN significantly reduced BDNF-promoted neurite growth (data not shown). Western blot analysis of the level IκB-α protein in nodose neurons at intervals after CNTF treatment also failed to show any evidence of IκB-α degradation (see Fig. 4C).
Tyrosine phosphorylation of IκB-α is required for CNTF-promoted growth
The noncanonical NF-κB activation mechanism that involves tyrosine phosphorylation of IκBα can be effectively and specifically blocked with an IκB-α protein that has a tyrosine to phenylalanine substitution at residue 42 (Imbert et al., 1996). Nodose neurons transfected with a plasmid expressing this Y42F IκB-α mutant had markedly smaller and less branched neurite arbors than control-transfected neurons when grown with CNTF (Fig. 3 A–C). The survival of newborn nodose neurons grown with CNTF was not effected by expression of the Y42F IκB-α mutant with 70–80% of neurons surviving after 24 h, confirming the above data that NF-κB signaling does not mediate the survival response of these neurons to CNTF. Although expression of the Y42F IκB-α mutant markedly inhibited CNTF-promoted neurite growth, its expression had no significant effect on the size and complexity of neurite arbors of nodose neurons grown with BDNF (data not shown). These results suggest that phosphotyrosine IκB-α-dependent activation of NF-κB is essential for CNTF-promoted neurite-growth but not for BDNF-promoted growth
Figure 3.
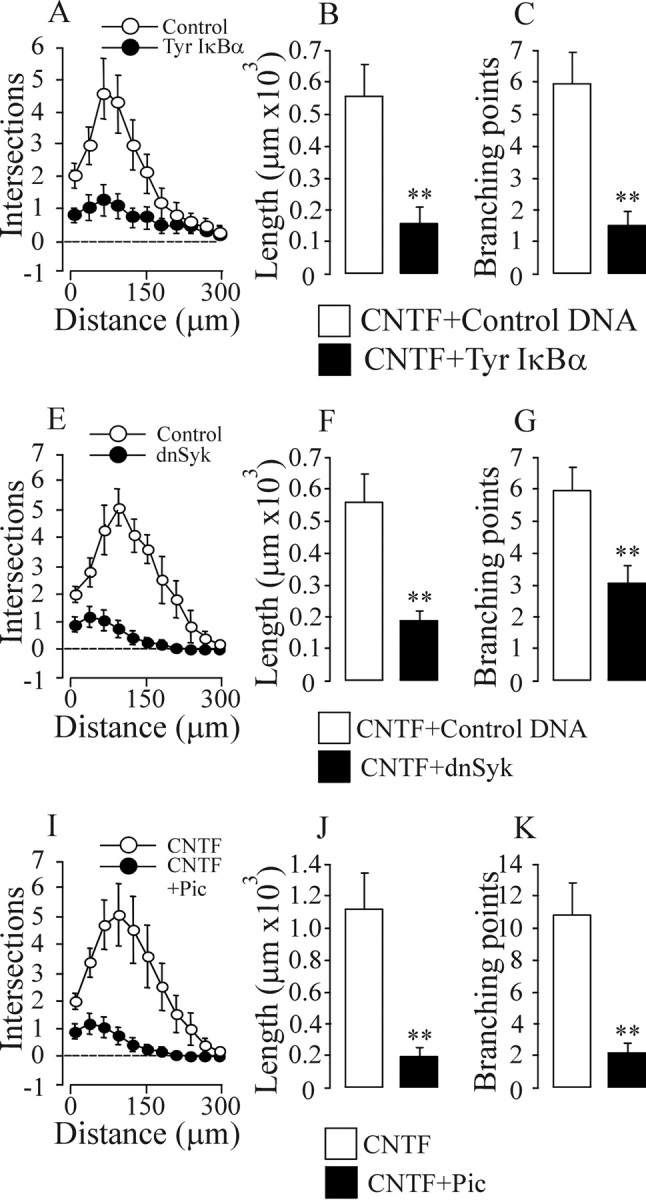
Tyrosine phosphorylation of IκBα and activation of SYK kinase are required for CNTF-promoted neurite growth. A–K, Three hours after plating, P0 nodose neurons were transfected with a YFP expression plasmid together with either a plasmid expressing the Y42F IκB-α mutant (Tyr IκB-α) or an empty control plasmid (A–C), or were transfected with a YFP expression plasmid together with either a plasmid expressing a dominant-negative SYK protein (dnSyk) or an empty control plasmid (E–G), or treated with either 10 μm piceatannol (Pic) or vehicle control (I–K). After 24 h incubation with 50 ng/ml CNTF, Sholl analysis (A, E, I), neurite arbor length (B, F, G), and branch point number (C, G, K) were ascertained. Means ± SEs of a typical experiment are shown (50–90 neurons per condition). Very similar data were obtained in three independent experiments. Statistical comparisons are with respect to either control-transfected neurons or vehicle-treated neurons. **p < 0.001.
SYK is required for CNTF-promoted neurite growth
We investigated the potential involvement of the spleen tyrosine kinase (SYK) in CNTF-promoted neurite growth because this protein-tyrosine kinase has been shown previously to phosphorylate IκB-α on tyrosine 42 in myeloid cells (Takada et al., 2003). Because the enzymatic activity of SYK is regulated by tyrosine phosphorylation (Berton et al., 2005), we quantified the level of phospho-SYK in nodose neurons at intervals after CNTF treatment to see whether CNTF is capable of activating SYK. Western blotting using an anti-phospho-SYK antibody failed to detect phospho-SYK in untreated neurons, but within 5 min of CNTF treatment, a very strong signal was evident which decreased markedly by 30 min (Fig. 4A). In contrast, BDNF treatment did not induce SYK phosphorylation (data not shown). Western blot analysis using a specific anti-phosphotyrosine IκBα antibody revealed that the rapid rise and peak in phospho-SYK 5 min after CNTF treatment was followed by a rise in phosphotyrosine IκB-α that was first evident after 15 min treatment with CNTF and was further elevated after 30 min (Fig. 4B).
To determine whether the sequential tyrosine phosphorylation of SYK and IκBα in response to CNTF is necessary for CNTF-promoted NF-κB activation, we transfected nodose neurons with a reporter construct in which GFP is under the control of an NF-κB promoter and studied the effects of inhibiting SYK activation and blocking tyrosine phosphorylation of IκBα on reporter signal intensity (Fig. 4D). Whereas CNTF treatment promoted a twofold increase in reporter signal compared with untreated controls, this rise was completely blocked in neurons treated with the potent selective SYK inhibitor piceatannol (Oliver et al., 1994) and in neurons transfected with the Y42F IκB-α plasmid.
To test the role of SYK in mediating the neurite growth-promoting effects of CNTF, we studied the neurite arbors of CNTF-supported nodose neurons that were either treated with piceatannol or transfected with a plasmid expressing dominant-negative SYK. Both approaches caused a substantial reduction in CNTF-promoted neurite growth (Fig. 3 E–K) and NF-κB activation, but not BDNF-promoted neurite growth (data not shown), and had no effect on survival with either factor (data not shown). These findings indicate that CNTF-dependent activation of SYK and the subsequent activation of NF-κB by tyrosine phosphorylation of IκB-α are essential steps in the neurite growth response of neurons to CNTF, but not BDNF.
Discussion
Using a variety of complementary experimental approaches, we have demonstrated that NF-κB signaling by an uncommon noncanonical activation mechanism is essential for the ability of CNTF to promote neurite growth. Inhibiting NF-κB-dependent transcription, blocking tyrosine phosphorylation of IκBα and inhibiting SYK activation each caused very substantial reductions in the size and complexity of the neurite arbors of CNTF-stimulated neurons. Although NF-κB signaling makes a small contribution to the neurite growth-promoting effects of the neurotrophins NGF and BDNF (Sole et al., 2004; Gutierrez et al., 2005), the essential role of NF-κB signaling in CNTF-promoted growth is a striking, novel finding. Although CNTF is known to activate signaling pathways implicated in regulating neurite growth such as MEK/ERK and PI3-K/Akt (Alonzi et al., 2001), there have been no direct studies of the involvement of these or other signaling pathways in mediating the neurite growth promoting effects of CNTF and related neurotrophic cytokines in primary neurons. Our results therefore provide an important insight into the intracellular signaling mechanisms that are crucial for the neurite growth-promoting actions of cytokines like CNTF.
A striking finding of our study is that CNTF uses a distinctive noncanonical NF-κB activation mechanism involving tyrosine phosphorylation of IκBα to promote neurite growth. Previous work has shown that the contribution of NF-κB signaling to neurotrophin-promoted neurite growth occurs via the canonical pathway that involves serine phosphorylation of IκBα and proteasome mediated degradation of IκBα (Sole et al., 2004; Gutierrez et al., 2005). Our demonstration that neither the 32/36-SS/AA IκB-α mutant nor proteasome inhibition affect CNTF-promoted neurite growth indicate that the canonical NF-κB activation mechanism plays no part in the neurite growth-promoting actions of CNTF. Rather, we show that CNTF promotes tyrosine phosphorylation of IκB-α and selectively blocking this with a Y42F IκB-α mutant completely inhibits CNTF-enhanced NF-κB transcriptional activity and blocks CNTF-promoted neurite growth. Because the Y42F IκB-α mutant does not affect BDNF-promoted neurite growth, our findings imply that different NF-κB activation mechanisms are exclusively required for the neurite growth-promoting effects of CNTF and neurotrophins.
Our work has implicated the SYK protein-tyrosine kinase in CNTF-promoted NF-κB activation and neurite growth. We have shown that CNTF promotes rapid sequential tyrosine phosphorylation of SYK and IκB-α. Pharmacological blockade of SYK and expression of a dominant-negative SYK protein inhibit CNTF-promoted neurite growth. SYK is a widely expressed protein-tyrosine kinase (Yanagi et al., 2001) that has been extensively studied from the standpoint of its role in immunoreceptor signaling in leukocytes (Berton et al., 2005). Although overexpression of SYK has been reported to promote the differentiation of embryonal carcinoma P19 cells into neuron-like cells (Tsujimura et al., 2001), we provide the first clear evidence in primary neurons that SYK plays a crucial role in regulating the growth of neural processes. In future work it will be interesting to ascertain the link between the CNTF receptor complex and SYK and to establish whether other signaling pathways activated by CNTF cooperate with NF-κB in mediating the effects of this cytokine on neurite growth.
Investigation of the key role of NF-κB signaling in CNTF-promoted neurite growth has been facilitated in newborn nodose neurons because CNTF supports the survival of the majority of these neurons in culture and NF-κB inhibition has no detectable effect on their survival. Previous work has established a neuroprotective role for NF-κB for a variety of CNS neurons (Kaltschmidt et al., 2005) and NF-κB signaling has been shown to contribute to the survival response of embryonic sympathetic and sensory to NGF (Maggirwar et al., 1998; Hamanoue et al., 1999) and adult DRG neurons to tumor necrosis factor-α (Fernyhough et al., 2005). Although it has been reported that NF-κB plays a role in mediating the survival response of cultured fetal nodose neurons to CNTF (Middleton et al., 2000), our more extensive investigation in which NF-kB activation has been blocked by variety of complementary approaches contradicts this general conclusion. Whereas inhibiting NF-κB signaling leaves the cell bodies of newborn nodose neurons without processes, these cell bodies retained a normal appearance and were stained with the vital dye calcein-AM with no significant loss in number up to 48 h in vitro. Likewise, inhibiting NF-κB activation in fetal nodose neurons by the same diverse approaches virtually eliminated neurite growth while leaving cell bodies intact and viable (data not shown). Our findings therefore clearly demonstrate that NF-κB signaling is crucial for the growth of neurites from developing nodose neurons stimulated with CNTF, but is not required for the survival response of these neurons to CNTF.
Studying primary neurons is a very powerful approach for elucidating growth factor physiology and signaling in the appropriate cellular and developmental context. Our studies of newborn nodose neurons have demonstrated a striking, essential role for NF-κB signaling in CNTF-promoted neurite growth and revealed that the NF-κB activation mechanism mediating this response of neurons to CNTF is distinct from the NF-κB activation mechanism mediating the much smaller contribution of NF-κB signaling to neurotrophin-promoted neurite growth. Although the very limited availability of primary neurons presents considerable technical challenges for biochemical studies of signaling networks, future progress on unraveling how cytokines and neurotrophins influence NF-κB signaling in distinctive ways and how this in turn has such marked effects on neurite growth will be important in understanding the emerging role of NF-κB signaling in regulating neuronal morphology in development.
Footnotes
This work was supported by The Wellcome Trust. N.G. was supported by the Beatriu de Pinós Fellowship (Generalitat de Catalunya, Spain). We thank Yumi Tohyama for the dominant-negative SYK construct.
References
- Alonzi T, Middleton G, Wyatt S, Buchman V, Betz UA, Muller W, Musiani P, Poli V, Davies AM. Role of STAT3 and PI 3-kinase/Akt in mediating the survival actions of cytokines on sensory neurons. Mol Cell Neurosci. 2001;18:270–282. doi: 10.1006/mcne.2001.1018. [DOI] [PubMed] [Google Scholar]
- Berton G, Mocsai A, Lowell CA. Src and Syk kinases: key regulators of phagocytic cell activation. Trends Immunol. 2005;26:208–214. doi: 10.1016/j.it.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Bui NT, Livolsi A, Peyron JF, Prehn JH. Activation of nuclear factor κB and Bcl-x survival gene expression by nerve growth factor requires tyrosine phosphorylation of IκBα. J Cell Biol. 2001;152:753–764. doi: 10.1083/jcb.152.4.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Harvey AR. CNTF promotes the regrowth of retinal ganglion cell axons into murine peripheral nerve grafts. NeuroReport. 2000;11:3999–4002. doi: 10.1097/00001756-200012180-00019. [DOI] [PubMed] [Google Scholar]
- Fernyhough P, Smith DR, Schapansky J, Van Der Ploeg R, Gardiner NJ, Tweed CW, Kontos A, Freeman L, Purves-Tyson TD, Glazner GW. Activation of nuclear factor-κB via endogenous tumor necrosis factor α regulates survival of axotomized adult sensory neurons. J Neurosci. 2005;25:1682–1690. doi: 10.1523/JNEUROSCI.3127-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez H, Hale V, Dolcet X, Davies A. NF-κB signalling regulates the growth of neural processes in the developing peripheral and central nervous systems. Development. 2005;132:1713–1726. doi: 10.1242/dev.01702. [DOI] [PubMed] [Google Scholar]
- Hamanoue M, Middleton G, Wyatt S, Jaffray E, Hay RT, Davies AM. p75-mediated NF-κB activation enhances the survival response of developing sensory neurons to nerve growth factor. Mol Cell Neurosci. 1999;14:28–40. doi: 10.1006/mcne.1999.0770. [DOI] [PubMed] [Google Scholar]
- Hartnick CJ, Staecker H, Malgrange B, Lefebvre PP, Liu W, Moonen G, Van de Water TR. Neurotrophic effects of BDNF and CNTF, alone and in combination, on postnatal day 5 rat acoustic ganglion neurons. J Neurobiol. 1996;30:246–254. doi: 10.1002/(SICI)1097-4695(199606)30:2<246::AID-NEU6>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Signaling to NF-κB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- Horton AR, Barlett PF, Pennica D, Davies AM. Cytokines promote the survival of mouse cranial sensory neurones at different developmental stages. Eur J Neurosci. 1998;10:673–679. doi: 10.1046/j.1460-9568.1998.00079.x. [DOI] [PubMed] [Google Scholar]
- Imbert V, Rupec RA, Livolsi A, Pahl HL, Traenckner EB, Mueller-Dieckmann C, Farahifar D, Rossi B, Auberger P, Baeuerle PA, Peyron JF. Tyrosine phosphorylation of IκB-α activates NF-κB without proteolytic degradation of IκB-α. Cell. 1996;86:787–798. doi: 10.1016/s0092-8674(00)80153-1. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Widera D, Kaltschmidt C. Signaling via NF-κB in the nervous system. Biochim Biophys Acta. 2005;1745:287–299. doi: 10.1016/j.bbamcr.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Koong AC, Chen EY, Giaccia AJ. Hypoxia causes the activation of nuclear factor kappa B through the phosphorylation of IκBα on tyrosine residues. Cancer Res. 1994;54:1425–1430. [PubMed] [Google Scholar]
- Liang Y, Zhou Y, Shen P. NF-κB and its regulation on the immune system. Cell Mol Immunol. 2004;1:343–350. [PubMed] [Google Scholar]
- Maggirwar SB, Sarmiere PD, Dewhurst S, Freeman RS. Nerve growth factor-dependent activation of NF-κB contributes to survival of sympathetic neurons. J Neurosci. 1998;18:10356–10365. doi: 10.1523/JNEUROSCI.18-24-10356.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton G, Hamanoue M, Enokido Y, Wyatt S, Pennica D, Jaffray E, Hay RT, Davies AM. Cytokine-induced nuclear factor kappa B activation promotes the survival of developing neurons. J Cell Biol. 2000;148:325–332. doi: 10.1083/jcb.148.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita R, Sugimoto T, Aoki M, Kida I, Tomita N, Moriguchi A, Maeda K, Sawa Y, Kaneda Y, Higaki J, Ogihara T. In vivo transfection of cis element “decoy” against nuclear factor-κB binding site prevents myocardial infarction. Nat Med. 1997;3:894–899. doi: 10.1038/nm0897-894. [DOI] [PubMed] [Google Scholar]
- Nishimune H, Vasseur S, Wiese S, Birling MC, Holtmann B, Sendtner M, Iovanna JL, Henderson CE. Reg-2 is a motoneuron neurotrophic factor and a signalling intermediate in the CNTF survival pathway. Nature Cell Biol. 2000;2:906–914. doi: 10.1038/35046558. [DOI] [PubMed] [Google Scholar]
- Oliver JM, Burg DL, Wilson BS, McLaughlin JL, Geahlen RL. Inhibition of mast cell Fc epsilon R1-mediated signaling and effector function by the Syk-selective inhibitor, piceatannol. J Biol Chem. 1994;269:29697–29703. [PubMed] [Google Scholar]
- Rane SG, Reddy EP. Janus kinases: components of multiple signaling pathways. Oncogene. 2000;19:5662–5679. doi: 10.1038/sj.onc.1203925. [DOI] [PubMed] [Google Scholar]
- Roff M, Thompson J, Rodriguez MS, Jacque JM, Baleux F, Arenzana-Seisdedos F, Hay RT. Role of IκBα ubiquitination in signal-induced activation of NFκB in vivo. J Biol Chem. 1996;271:7844–7850. doi: 10.1074/jbc.271.13.7844. [DOI] [PubMed] [Google Scholar]
- Siegel SG, Patton B, English AW. Ciliary neurotrophic factor is required for motoneuron sprouting. Exp Neurol. 2000;166:205–212. doi: 10.1006/exnr.2000.7528. [DOI] [PubMed] [Google Scholar]
- Sole C, Dolcet X, Segura MF, Gutierrez H, Diaz-Meco MT, Gozzelino R, Sanchis D, Bayascas JR, Gallego C, Moscat J, Davies AM, Comella JX. The death receptor antagonist FAIM promotes neurite outgrowth by a mechanism that depends on ERK and NF-κB signaling. J Cell Biol. 2004;167:479–492. doi: 10.1083/jcb.200403093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl N, Yancopoulos GD. The tripartite CNTF receptor complex: activation and signalling involves components shared with other cytokines. J Neurobiol. 1994;25:1454–1466. doi: 10.1002/neu.480251111. [DOI] [PubMed] [Google Scholar]
- Takada Y, Mukhopadhyay A, Kundu GC, Mahabeleshwar GH, Singh S, Aggarwal BB. Hydrogen peroxide activates NF-κB through tyrosine phosphorylation of IκBα and serine phosphorylation of p65: evidence for the involvement of IκBα kinase and Syk protein-tyrosine kinase. J Biol Chem. 2003;278:24233–24241. doi: 10.1074/jbc.M212389200. [DOI] [PubMed] [Google Scholar]
- Tomita N, Morishita R, Tomita S, Gibbons GH, Zhang L, Horiuchi M, Kaneda Y, Higaki J, Ogihara T, Dzau VJ. Transcription factor decoy for NFκB inhibits TNF-α-induced cytokine and adhesion molecule expression in vivo. Gene Ther. 2000;7:1326–1332. doi: 10.1038/sj.gt.3301243. [DOI] [PubMed] [Google Scholar]
- Tsujimura T, Yanagi S, Inatome R, Takano T, Ishihara I, Mitsui N, Takahashi S, Yamamura H. Syk protein-tyrosine kinase is involved in neuron-like differentiation of embryonal carcinoma P19 cells. FEBS Lett. 2001;489:129–133. doi: 10.1016/s0014-5793(01)02097-x. [DOI] [PubMed] [Google Scholar]
- Yanagi S, Inatome R, Takano T, Yamamura H. Syk expression and novel function in a wide variety of tissues. Biochem Biophys Res Commun. 2001;288:495–498. doi: 10.1006/bbrc.2001.5788. [DOI] [PubMed] [Google Scholar]