

Abstract
Nuclear factor κB (NF-κB) signaling is known to promote neurite growth from developing sensory neurons and to enhance the size and complexity of pyramidal neuron dendritic arbors in the developing cerebral cortex. In marked contrast, here we show that NF-κB signaling can also exert a potent inhibitory influence on neurite growth in certain neurons, and can either promote or inhibit neurite growth in the same neurons depending on the mechanism of NF-κB activation. In neonatal superior cervical ganglion sympathetic neurons, enhancing NF-κB transcriptional activity by overexpressing either the p65 NF-κB subunit or the IκB kinase-β (IKKβ) subunit of the IκB kinase complex, or by tumor necrosis factor α (TNFα) treatment, strongly inhibits neurite growth. Paradoxically in neonatal nodose ganglion sensory neurons, enhancing NF-κB transcriptional activity by p65/p50 overexpression increases neurite growth, whereas enhancing NF-κB transcriptional activity by IKKβ overexpression inhibits neurite growth. In addition to activating NF-κB, IKKβ overexpression leads to phosphorylation of p65 on serine 536. Blockade of serine 536 phosphorylation by a S536A-p65 mutant protein prevents the growth-inhibitory effects of IKKβ overexpression in both sensory and sympathetic neurons and the growth-inhibitory effects of TNFα on sympathetic neurons. Furthermore, expression of a p65 S536D phosphomimetic mutant inhibits neurite growth from sensory neurons. These results demonstrate that NF-κB can either stimulate or inhibit neurite growth in developing neurons depending on the phosphorylation status of p65.
Keywords: axon, brain development, growth, phosphorylation, signal transduction, transcription factor
Introduction
Nuclear factor κB (NF-κB) is a ubiquitously expressed transcription factor system that consists of homodimers or heterodimers of five structurally related proteins: p65 (RelA), RelB, c-Rel, p50, and p52. NF-κB dimers are held in an inactive form in the cytosol by interaction with a member of the IκB family of inhibitory proteins: IκBα, IκBβ, IκBε, IκBγ, Bcl-3, p100, and p105. The p65/p50 heterodimer is the most abundant and widely expressed form of NF-κB, and IκBα is the predominantly expressed inhibitor. In the classical NF-κB signaling pathway, NF-κB is activated by phosphorylation of IκBα on Ser32 and Ser36 by the IκB kinase-β (IKKβ) catalytic subunit of an IκB kinase (IKK) complex that consists of IKKβ together with another catalytic subunit IKKα and a regulatory subunit IKKγ. This leads to ubiquitination and proteasome-mediated degradation of IκBα and translocation of the liberated NF-κB to the nucleus where it binds to κB elements in the promoter and enhancer regions of responsive genes, resulting in gene induction or gene repression (Perkins, 2007).
NF-κB has been studied most extensively in the immune system in which it plays a key role in regulating the expression of genes involved in innate and adaptive immune responses, inflammatory responses, cell survival, and cell proliferation (Liang et al., 2004). In the nervous system, NF-κB is activated by a variety of neurotrophic factors, cytokines, and neurotransmitters, and has been implicated in regulating neuronal survival and death, peripheral nerve myelination, and synaptic function (Mémet, 2006). NF-κB is also emerging as an important positive regulator of axonal and dendritic growth in developing peripheral and central neurons. NF-κB signaling contributes to cortical pyramidal dendrite growth and to the neurite growth-promoting effects of NGF and brain-derived neurotrophic factor (BDNF) in PC12 cells and sensory neurons, respectively (Sole et al., 2004; Gutierrez et al., 2005), and is critical for ciliary neurotrophic factor (CNTF)-promoted neurite growth from sensory neurons (Gallagher et al., 2007).
In the present study, we investigated the roles of NF-κB signaling in two well characterized populations of peripheral neurons of newborn mice. Sympathetic neurons of the superior cervical ganglion (SCG) survive and extend neurites in response to NGF (Glebova and Ginty, 2005), whereas sensory neurons of the nodose ganglion survive and extend neurites in response to BDNF (Gutierrez et al., 2005). We report that enhanced NF-κB signaling inhibits neurite growth from sympathetic neurons, in contrast to its growth-promoting effects in sensory neurons. Furthermore, we show that this functional reversal in the role of NF-κB is determined by the phosphorylation status of a single residue in p65 that can be brought about by differential engagement of the IKK complex in these two neuronal populations.
Materials and Methods
Neuron culture and transfection.
Dissociated cultures of nodose ganglion or SCG neurons from newborn mice were grown in polyornithine/laminin-coated 35 mm tissue culture dishes in defined medium and were transfected 3 h after plating with plasmid-coated gold microcarriers using the Bio-Rad Gene-gun (Gutierrez et al., 2005). In all experiments, except those involving SN50, the neurons were transfected with a yellow fluorescent protein (YFP) expression plasmid to identify transfected neurons and visualize their neurite arbors for analysis by confocal microscopy. The microcarriers were also coated with pcDNA3.1 plasmid(s) expressing the required protein(s), as specified for each set of experiments. Control-transfected neurons received pcDNA3.1 containing a short sequence of noncoding DNA. For experiments using κB decoy DNA, neurons were transfected with the YFP plasmid together with decoy or control oligonucleotides as described previously (Gutierrez et al., 2005).
SCG neurons were grown in medium containing 10 ng/ml NGF (R&D Systems), and nodose neurons were grown in medium containing 10 ng/ml BDNF (R&D Systems). Where indicated, 10 ng/ml tumor necrosis factor α (TNFα) (R&D Systems) or 20 μm of either SN50 or the SM control peptide (Calbiochem) were added to the medium. Neuronal survival was estimated after 24 h in culture as described previously (Gutierrez et al., 2005).
Quantification of NF-κB activity.
To estimate the relative level of NF-κB activation, neurons were transfected with a plasmid expressing green fluorescent protein (GFP) under the control of an NF-κB promoter. Neurons were imaged with a Carl Zeiss Axioplan confocal microscope, and mean soma fluorescence intensity was obtained using LSM510 software (Gutierrez et al., 2005).
Analysis of neuritic arbors.
YFP-labeled neurons, or in the case of experiments using SN50 in which the neurons were fluorescently labeled with the vital dye calcein-AM (Invitrogen), were visualized and digitally acquired using an Axioplan Carl Zeiss laser-scanning confocal microscope. Neuritic arbors were traced using LSM510 software from which total neurite length and number of branch points were calculated and Sholl analysis performed (Gutierrez and Davies, 2007).
Western blots.
In all experiments, neurons were plated at high density in polyornithine/laminin-coated 96-well plates (5000 neurons per well). Three hours after plating, 10 ng/ml NGF for SCG neurons or 10 ng/ml BDNF for nodose neurons was added to the wells for the indicated times. TNFα (10 ng/ml) was added where indicated. The cells were lysed in radioimmunoprecipitation buffer, and insoluble debris was removed by centrifugation. Samples were transferred to polyvinylidene difluoride membranes using the Bio-Rad TransBlot (Bio-Rad). The membranes were blocked with 5% dried milk in PBS with 0.1% Tween 20 and were incubated with either anti-phospho-Ser IκB-α (1:1000; Abcam), anti-IκBα (1:1000; Abcam), anti-phospho-S536-p65 (1:1000; Cell Signaling Technology), anti-phospho-IKK (1:1000; Cell Signaling Technology), anti-IKK (1:1000; Cell Signaling Technology), anti-p65 (1:1000; Cell Signaling Technology), or anti-βIII-tubulin (1:10,000; Promega) antibodies that were detected with peroxidase-linked secondary antibodies (1:2000; Promega) and ECL-plus (GE Healthcare). Densitometry was performed using Adobe Photoshop.
Immunocytochemistry.
Cultured neurons were fixed in ice-cold methanol for 5 min, washed with PBS, blocked with 5% BSA in PBS, and incubated with anti-phospho-S536-p65 antibody (1:50; Cell Signaling Technology) at 4°C for 18 h. After washing, neurons were incubated with anti-rabbit-Cy3 secondary antibody (Alexa Fluor; Invitrogen; 1:500) and counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) (Millipore Bioscience Research Reagents). Neurons were imaged with a Carl Zeiss Axioplan confocal microscope and mean soma fluorescence intensity was obtained using LSM510 software (n ≥40 neurons per experimental condition taken from three independent experiments).
Results
Blockade of NF-κB signaling does not affect neurite growth from SCG neurons
To assess the requirement for NF-κB signaling in the growth of neurites from developing sympathetic neurons, we inhibited several different steps in the NF-κB signaling network in SCG neurons cultured from newborn mice and quantified the size and complexity of neurite arbors after 24 h incubation with NGF. In all cases, we assessed the effectiveness of NF-κB inhibition by transfecting the neurons with a plasmid expressing GFP under the control of an NF-κB promoter and compared fluorescence intensity in experimental and control neurons (Gutierrez et al., 2005; Gallagher et al., 2007). We also quantified neuronal survival in all experiments to ascertain whether any of the treatments adversely affected survival. We previously showed that BDNF does not influence NF-κB activation in postnatal nodose ganglion sensory neurons (Gutierrez et al., 2005). We repeated the same experiment in postnatal SCG neurons and found that NGF likewise does not affect NF-κB activation in these neurons (data not shown).
We reported in nodose neurons that constitutive activation of NF-κB significantly contributes to BDNF-promoted neurite growth (Gutierrez et al., 2005). Here, we selectively inhibited NF-κB signaling by the classic pathway in postnatal SCG neurons by transfecting these neurons with a plasmid that expresses a mutated IκBα protein with serine-to-alanine substitutions at residues 32 and 36 (Roff et al., 1996). Although neurons expressing this S32A/S36A IκBα mutant displayed a marked reduction in NF-κB reporter signal compared with control-transfected neurons (Fig. 1 A), this protein had no effect on the neurite arbor size and complexity, as shown by Sholl analysis, which provides a graphic representation of neurite branching with distance from the cell body (Fig. 1 B). Measurements of total neurite length and branch point number and counts of the number of surviving neurons at the end of the experiment (>80% of the number plated) revealed no significant differences between S32A/S36A IκBα-transfected and control-transfected neurons (data not shown).
Figure 1.
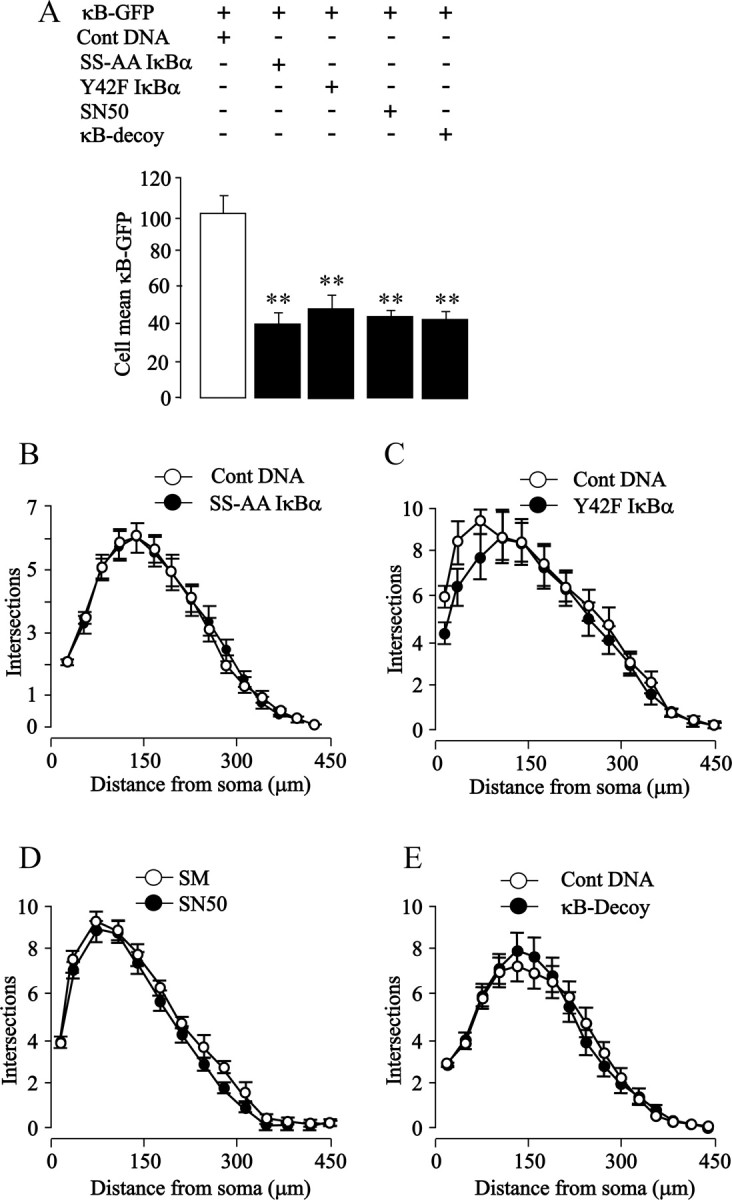
Blockade of NF-κB signaling does not prevent neurite growth from SCG neurons. A–E, Shown are bar charts of NF-κB-dependent transcriptional activity (A) and Sholl plots of the neurite arbors of P0 SCG neurons in which different steps in NF-κB signaling were inhibited by transfection with an S32A/336A IκBα plasmid (B), transfection with a Y42A IκBα plasmid (C), treatment with 20 μm SN50 (D), or transfection with κB decoy DNA (E). Controls were transfected with a pcDNA3.1 plasmid with a noncoding insert (B, C), treated with 20 μm SM control peptide (D), or transfected with scrambled sequence oligonucleotide (E). Neurons were cotransfected with the κB-GFP reporter for measuring transcriptional activity and with the YFP plasmid for Sholl analysis (except for the SN50/SM experiments in which the neurite arbors were labeled with calcein-AM). All data were obtained 24 h after transfection (performed 3 h after plating) or SN50/SM treatment (added 1 h after plating). The combined results of three separate experiments are plotted (means and SEs of the data from >150 neurons for each condition). Statistically significant differences from controls are indicated (**p < 0.001).
We also reported in nodose neurons that NF-κB activation by phosphorylation of IκBα at tyrosine 42 is essential for CNTF-promoted neurite growth (Gallagher et al., 2007), and it has been shown that NGF activates NF-κB in PC12 cells via tyrosine phosphorylation of IκBα (Bui et al., 2001). This NF-κB signaling pathway was selectively inhibited in postnatal SCG neurons by transfecting the neurons with a plasmid that expresses a Y42F IκBα mutant (Imbert et al., 1996; Gallagher et al., 2007). Expression of this mutant protein caused a marked decrease in NF-κB reporter signal compared with control-transfected neurons (Fig. 1 A) but did not affect the shape of the Sholl profile (Fig. 1 C). Total neurite length, branch point number, and neuronal survival were also not significantly different between Y42F IκBα-transfected and control-transfected neurons (data not shown).
Translocation of activated NF-κB from the cytoplasm to the nucleus is inhibited by SN50, a cell-permeable peptide that blocks the nuclear localization sequence of p50 (Lin et al., 1995). Although SN50 caused a clear reduction in NF-κB reporter signal (Fig. 1 A), there were no significant differences in the Sholl profiles of neurons grown with an inactive control peptide (SM) (Fig. 1 D). There were also no significant differences in total neurite length, branch point number, and neuronal survival between SN50-treated and SM-treated cultures (data not shown).
Finally, we investigated the consequences of blocking binding of activated NF-κB to regulatory elements in target genes by transfecting SCG neurons with double-stranded DNA oligonucleotides containing the κB consensus binding sequence (Morishita et al., 1997). This κB decoy DNA, but not control oligonucleotides with a scrambled sequence, caused a marked reduction in NF-κB reporter signal (Fig. 1 A) but did not affect the Sholl profile (Fig. 1 E). There were also no significant differences in total neurite length, branch point number, and neuronal survival between neurons transfected with κB decoy DNA and control DNA (data not shown).
Because neurite growth from nodose sensory neurons depends on NF-κB signaling during a brief developmental window during the immediate perinatal period (Gutierrez et al., 2005), we studied the effect of inhibiting NF-κB signaling in SCG neurons at two additional postnatal ages to see whether neurite growth from these neurons might also depend on NF-κB signaling during an equivalent developmental window. Blockade of NF-κB signaling by either SN50 or κB decoy DNA in postnatal day 3 (P3) and P6 SCG neurons had no effect on neurite growth (data not shown). Finally, although neurite growth from postnatal SCG cultured requires NGF, we investigated the effects of inhibiting NF-κB signaling in SCG neurons grown without NGF. In cultures supplemented with caspase inhibitors to prevent apoptosis in the absence of NGF, inhibiting NF-κB signaling by any of the above approaches had no effect on neurite growth (data not shown). Together, the above findings demonstrate that SCG neurons cultured from newborn mice have clearly detectable NF-κB transcriptional activity that can be substantially reduced by interfering with IκBα activation by either ser32/ser36 or tyr42 phosphorylation, by preventing nuclear translocation of activated NF-κB, and by sequestering transcriptionally active NF-κB complexes. None of these manipulations affects neuronal survival, and in marked contrast to hippocampal pyramidal neurons, sensory neurons, and PC12 cells (Sole et al., 2004; Gutierrez et al., 2005; Gallagher et al., 2007), none affects neurite growth, indicating that NF-κB signaling does not promote neurite growth in sympathetic neurons under these experimental conditions.
Enhancing NF-κB signaling inhibits neurite growth from SCG neurons
Given the surprising finding that neurite growth from SCG neurons is unaffected by inhibiting NF-κB signaling, we investigated whether experimentally enhancing NF-κB signaling in these neurons has any effect on neurite arbor size. To this end, SCG neurons were transfected with plasmids expressing the p65 and p50 NF-κB subunits together with the NF-κB reporter plasmid to assess the level of NF-κB-dependent transcription. Overexpression of p65 and p50 caused a marked elevation in NF-κB reporter signal (Fig. 2 A) and highly significant reductions in total neurite length and branching (Fig. 2 B,C). These dramatic effects of increased NF-κB-dependent transcription on neurite growth are also clearly illustrated by the Sholl profile of transfected neurons (Fig. 2 D) and by images of representative neurite arbors (Fig. 2 F). Despite the marked reductions in neurite arbor size and complexity brought about by overexpressing NF-κB subunits, there were no significant differences in the numbers of control-transfected and NF-κB-transfected neurons 48 h after transfection, indicating that overexpression of these proteins does not adversely affect sympathetic neuron survival (data not shown). These data show, in marked contrast to sensory neurons, that enhancing NF-κB signaling in newborn sympathetic neurons inhibits neurite growth in proportion to the increase in NF-κB-dependent transcription.
Figure 2.

Enhancing NF-κB signaling inhibits neurite growth from SCG neurons. P0 SCG neurons were transfected 3 h after plating with the YFP plasmid together with one or more of the following plasmids as specified in the figure: a control pcDNA3.1 plasmid with a noncoding insert, the κB-GFP reporter for measuring transcriptional activity, and plasmids expressing p65, p50, or wild-type IKKβ (wt IKKβ). A–E, Twenty-four hours after transfection, NF-κB-dependent transcriptional activity was quantified (A), and neurite arbors were analyzed to measure total neurite length (B), count the number of branch points (C), and obtain Sholl profiles (D, E). The combined results of three separate experiments are plotted (means and SEs of the data from >150 neurons for each condition). Statistically significant differences are indicated (*p < 0.05; **p < 0.001). F, Photomicrographs of a representative control-transfected neuron and neurons overexpressing the proteins indicated. Scale bar, 50 μm.
To confirm that the growth-inhibitory effect of overexpressing NF-κB subunits in sympathetic neurons results from enhanced NF-κB-dependent transcription rather than some nonspecific consequence of overexpressing these particular proteins, we overexpressed IKKβ to promote phosphorylation of IκBα, a key substrate of IKKβ and an essential step in activating the classical NF-κB pathway. Overexpression of IKKβ significantly increased NF-κB-dependent transcription (Fig. 2 A) and greatly reduced neurite length and branching compared with control-transfected neurons (Fig. 2 B,C,E,F). These data again show that enhancing NF-κB signaling in sympathetic neurons significantly reduces neurite growth.
Furthermore, to ascertain whether NF-κB signaling in cultured SCG neurons is normally dependent on the catalytic activity of IKKβ, we transfected newborn SCG neurons with a plasmid expressing a dominant-negative K44A IKKβ protein. Quantification of the NF-κB reporter signal in these neurons revealed a significant reduction compared with control-transfected neurons, indicating that IKKβ makes a significant contribution to the basal level of NF-κB signaling in cultured newborn SCG neurons. As with blocking NF-κB activation with the S32A/336A IκBα mutant, expression of the K44A IKKβ mutant did not significantly affect neurite length and branching and had no significant effect on survival (data not shown). This provides additional confirmation that blockade of NF-κB signaling does not interfere with neurite growth from newborn SCG neurons.
IKKβ and p65/p50 overexpression have opposite effects in sensory neurons
The neurite growth-inhibitory effect of enhancing NF-κB signaling in sympathetic neurons was particularly surprising given the neurite growth-promoting effects of NF-κB signaling observed in sensory neurons and cortical pyramidal neurons (Gutierrez et al., 2005; Gallagher et al., 2007). This finding and our demonstration that there are no significant differences in the basal levels of endogenous NF-κB reporter activity between sensory and sympathetic neurons (data not shown) indicate that NF-κB can exert opposing effects on the growth of neural processes in different neuronal populations. To ascertain whether these opposing effects of NF-κB signaling reflect neuron-specific differences, we explored the effects of activating NF-κB by alternative mechanisms in sensory neurons. To this end, P0 nodose ganglion neurons were transfected with either an IKKβ plasmid or both p65 and p50 plasmids. Quantification of the GFP signal from the cotransfected NF-κB reporter revealed that overexpression of IKKβ and p65/p50 each caused marked and similar increases in NF-κB transcriptional activity (Fig. 3 A). Analysis of neurite arbor size and complexity 24 h after transfection revealed that p65/p50 overexpression caused significant increases in neurite length and branching (Fig. 3 B–D,F). Surprisingly, IKKβ overexpression had a very marked inhibitory effect on neurite growth (Fig. 3 B,C,E,F). Neither p65/p50 overexpression nor IKKβ overexpression affected neuronal survival (data not shown). These results show that activating NF-κB by different mechanisms can have opposing effects on neurite growth in the same neurons, suggesting the existence of a functional switch in the NF-κB signaling network.
Figure 3.

Opposite effects of IKKβ overexpression and p65/p50 overexpression on neurite growth from nodose neurons. P0 nodose neurons were transfected 3 h after plating with the YFP plasmid together with one or more of the following plasmids as specified in the figure: a control pcDNA3.1 plasmid with a noncoding insert, the κB-GFP reporter for measuring transcriptional activity, and plasmids expressing p65, p50, or wild-type IKKβ (wt IKKβ). A–E, Twenty-four hours after transfection, NF-κB-dependent transcriptional activity was quantified (A), and neurite arbors were analyzed to measure total neurite length (B), count the number of branch points (C), and obtain Sholl profiles (D, E). All treatments were performed in the same experiments, but are shown separately for clarity. The combined results of three separate experiments are plotted (means and SEs of the data from >150 neurons for each condition). Statistically significant differences from controls are indicated (*p < 0.05; **p < 0.001). F, Photomicrographs of a representative control-transfected neuron and neurons overexpressing the proteins indicated. Scale bar, 50 μm.
Selective IKKβ activation and p65 phosphorylation on Ser536 in SCG neurons
Our finding that IKKβ overexpression in nodose sensory neurons switches NF-κB signaling from growth-promoting to growth-inhibitory prompted us to examine potential differences in the basal level of activation of the IKK complex in sympathetic and nodose neurons. For these experiments, protein extracts of cultured newborn SCG and nodose neurons were probed with an anti-phospho-IKK antibody. Whereas SCG neurons had clearly detectable levels of phospho-IKK proteins, these phosphoproteins were barely detectable in nodose neurons. These striking differences in basal activation were observed after 24 h in culture (Fig. 4 A,C) and 1 h after plating (data not shown). This suggests much higher, sustained activation of the IKK complex in sympathetic neurons compared with nodose ganglion sensory neurons.
Figure 4.

IKKβ and p65 phosphorylation in SCG and nodose neurons. A, B, Representative Western blots comparing the levels of phospho-IKK and total IKK (A) and phospho-S536-p65 and total p65 (B) in the same quantity of total protein extracted from P0 SCG and nodose neurons 24 h after plating. C, Quantification of the relative levels of these proteins from densitometric scans of three separate experiments of each type are plotted. D, Photomicrograph showing immunostainings of phospho-S536-p65 in cultured P0 nodose and SCG neurons 24 h after transfection with a YFP plasmid together with an IKKβ plasmid. DAPI nuclear staining, YFP labeling, and phospho-S536-p65 immunostaining in the same fields are shown from left to right. The bar chart shows the relative intensity of phospho-S536-p65 immunostaining in IKKβ-overexpressing nodose and SCG neurons (black bars) normalized to the level in the respective control-transfected nodose and SCG neurons (pcDNA3.1 plasmid, white bars). E, Photomicrograph of immunostainings for total p65 in cultured P0 nodose and SCG neurons 24 h after transfection with a YFP plasmid together with a p65 plasmid. DAPI staining, YFP labeling, and p65 immunostaining in the same fields are shown from left to right. The bar chart shows the relative intensity of p65 immunostaining in p65-overexpressing nodose and SCG neurons (black bars) normalized to the level in the respective control-transfected nodose and SCG neurons (pcDNA3.1 plasmid, white bars). F, Photomicrograph of immunostainings for phospho-S536-p65 in cultured P0 nodose and SCG neurons 24 h after transfection with a YFP plasmid together with a p65 plasmid. DAPI staining, YFP labeling, and phospho-S536-p65 immunostaining in the same fields are shown from left to right. The bar chart shows the relative intensity of phospho-S536-p65 immunostaining in p65-overexpressing nodose and SCG neurons (black bars) normalized to the level in the respective control-transfected nodose and SCG neurons (white bars). Means and SEs of measurements from >150 neurons for each condition are shown in D–F. Statistically significant difference from control-transfected neurons is indicated (**p < 0.001).
Among the substrates of the IKK complex is the p65 NF-κB subunit, which it phosphorylates on Ser536 (Sakurai et al., 1999; Yang et al., 2003; Jeong et al., 2005; Sasaki et al., 2005). Given the high constitutive activation of the IKK complex in sympathetic compared with nodose neurons, we asked whether there are corresponding differences in the level of phospho-S536-p65 in these neurons. Protein extracts of sympathetic, but not sensory neurons, displayed strong immunoreactivity for phospho-S536-p65 after 24 h in culture (Fig. 4 B,C) and 1 h after plating (data not shown), also demonstrating sustained levels of phospho-S536-p65 in SCG neurons. To confirm that this difference was not attributable to endogenous differences in the basal level of NF-κB activation between sympathetic and sensory neurons, we compared directly the NF-κB reporter signal in these two neuronal populations. These measurements revealed no significant differences in signal intensity (SCG, 44.3 ± 2.4 SEM; nodose, 40.8 ± 1.56 SEM). Furthermore, no significant differences were observed when the reporter signal was normalized to the fluorescence intensity of a cotransfected constitutively expressed RFP to control for potential differences in transfection efficiency between these two neuronal populations (SCG, 0.723 ± 0.04 SEM; nodose, 0.848 ± 0.12 SEM).
Activation of IKKβ (but not IKKα) promotes phosphorylation of p65 on Ser536
Because phospho-S536-p65 has been proposed to regulate transcription of a distinctive set of target genes (Okazaki et al., 2003; Buss et al., 2004; Sasaki et al., 2005), our demonstration that sympathetic neurons possess substantially higher levels of this form of p65 than sensory neurons raised the possibility that S536 phosphorylation might be responsible for switching NF-κB signaling from growth-promoting to growth-inhibiting. Given that IKKβ overexpression in nodose neurons reverses the effects of NF-κB activation on neurite growth, we investigated whether IKKβ overexpression leads to an elevation in the level of phospho-S536-p65 in these neurons. Western blot analysis could not be used to assess the level of phospho-S536-p65 in neurons overexpressing IKKβ because of the low efficiency of ballistic transfection. We therefore used immunofluorescent detection to visualize phospho-S536-p65 in transfected neurons using an anti-phospho-S536-p65 antibody. Digital quantification of labeling intensity by confocal microscopy revealed that IKKβ overexpression caused marked increases in phospho-S536-p65 immunofluorescence in both nodose neurons and SCG neurons relative to the respective control-transfected neurons (Fig. 4 D). Interestingly, nodose and SCG neurons transfected with an IKKα plasmid in place of the IKKβ plasmid showed no increase in phospho-S536-p65 immunofluorescence compared with control-transfected neurons (data not shown). These findings show that overexpression of IKKβ, but not IKKα, enhances phosphorylation of p65 on Ser536 in both sensory and sympathetic neurons.
Because overexpression of p65/p50 in nodose and SCG neurons has opposite effects on neurite growth, we asked whether the overexpressed dimer is differentially phosphorylated in these two neuronal populations. To this end, cultures of newborn nodose and SCG neurons were cotransfected with both p65 and p50 plasmids together with a YFP plasmid to identify transfected neurons. After 24 h, cultures were fixed and divided into two groups, one of which was labeled for total p65 immunofluorescence and the other for phospho-S536-p65. Both SCG and nodose neurons overexpressing p65/p50 exhibited intense labeling for total p65 relative to nontransfected neurons and control-transfected neurons (Fig. 4 E). In contrast, an increase in phospho-S536-p65 immunofluorescence was only observed in SCG neurons overexpressing p65/p50. The level of phospho-S536-p65 immunofluorescence in nodose neurons overexpressing p65/p50 was not significantly different from nontransfected and control-transfected neurons (Fig. 4 F).
TNFα inhibits neurite growth from SCG neurons by an NF-κB-dependent mechanism
To further explore the implications of the negative regulation of neurite growth by NF-κB signaling in postnatal sympathetic neurons, we looked at the potential effects of TNFα on NF-κB signaling and neurite growth in these neurons. TNFα is a potent activator of NF-κB in a variety of cells (Hehlgans and Pfeffer, 2005), and TNFα has been shown to act on embryonic SCG neurons to accelerate the onset of apoptosis after NGF deprivation (Barker et al., 2001). Because TNFα can activate several different signaling pathways depending on the cellular context (Hehlgans and Pfeffer, 2005), we first examined whether TNFα activates the classical NF-κB pathway in postnatal SCG neurons. To ascertain whether TNFα increases IκBα phosphorylation, Western blots of proteins extracted from SCG neurons treated with TNFα for times ranging from 5 to 120 min were probed with an IκBα phosphoserine antibody. To avoid proteosome-mediated degradation of IκBα complicating the analysis of these blots, the proteosome inhibitor N-acetyl-Leu-Leu-norleucinal (ALLN) was included in the medium. TNFα promoted a marked increase in phosphoserine IκBα within 5 min followed by a gradual decline to near-prestimulation levels by 120 min (Fig. 5 A). In the absence of ALLN, Western blots showed a clear decrease in total IκBα protein after 60 min followed by a characteristic NF-κB-dependent resynthesis of IκBα (Fig. 5 B). These findings together with a substantial increase in the NF-κB reporter signal in neurons treated with TNFα (Fig. 5 D) demonstrate that TNFα activates NF-κB in newborn SCG neurons.
Figure 5.

TNFα inhibits neurite growth from SCG neurons by an NF-κB-dependent mechanism. A, B, Representative Western blots for phosphoserine-IκB-α (A) and total IκB-α (B) in protein extracts of P0 SCG neurons treated with 10 ng/ml TNFα from 5 to 120 min (0′, untreated neurons) 24 h after plating. The cultures used for phosphoserine-IκB-α detection received 10 mm ALLN 30 min before TNFα treatment. C, Representative Western blots for phospho-IKK and phospho-S536-p65 in protein extracts of P0 SCG neurons treated 24 h after plating with 10 ng/ml TNFα for 15–30 min (0′, untreated neurons). D, NF-κB-dependent transcriptional activity in P0 SCG neurons transfected 3 h after plating with the YFP plasmid and κB-GFP reporter plus either the S32A/336A IκBα plasmid or pcDNA3.1 control plasmid and cultured for an additional 24 h with or without 10 ng/ml TNFα. E–G, Total neurite length (E), number of branch points (F), and Sholl profiles (G) of P0 SCG neurons transfected 3 h after plating with the YFP plasmid and either the S32A/336A IκBα plasmid or pcDNA3.1 control plasmid and cultured for an additional 24 h with or without 10 ng/ml TNFα. The combined results of three separate experiments are plotted (means and SEs of the data from >150 neurons for each condition). Statistically significant differences from controls are indicated (**p < 0.001).
Analysis of the neurite arbors of postnatal SCG neurons incubated with TNFα for 24 h revealed marked reductions in overall length (45% shorter than control neurons) (Fig. 5 E), in branching (61% fewer branch points) (Fig. 5 F), and in the Sholl profile (Fig. 5 G). Despite its marked effect on axon growth, TNFα did not significantly affect the number of SCG neurons surviving with NGF at this stage of development (data not shown). To confirm that the suppression of neurite growth by TNFα was attributable to NF-κB activation, postnatal SCG neurons were transfected with the S32A/336A IκBα plasmid before TNFα treatment. Expression of this superrepressor IκBα not only prevented the TNFα-induced increase in NF-κB-reporter signal (Fig. 5 D) but completely eliminated the suppression of neurite growth by TNFα (Fig. 5 E–G). These results show that TNFα inhibits neurite growth from postnatal sympathetic neurons by an NF-κB-dependent mechanism and confirm that NF-κB signaling negatively regulates neurite growth from these neurons.
Because TNFα activates IKKβ during the induction of NF-κB signaling, we investigated whether treating newborn SCG neurons with TNFα increases the level of phospho-S536-p65. Western blot analysis of proteins extracted from cultured SCG neurons 15 and 30 min after TNFα treatment revealed clear increases in both phospho-IKK (Fig. 5 C) and phospho-S536-p65 (Fig. 5 C), confirming that TNFα activates IKK and enhances S536 phosphorylation in these neurons. Similar experiments with TNFα were not performed on nodose neurons because these neurons do not respond to this cytokine.
Blocking phosphorylation of p65 at S536 abolishes neurite growth inhibition
To test directly whether enhanced phosphorylation of p65 on S536 is responsible for the growth-inhibitory effect of NF-κB activation in SCG and nodose neurons, we transfected these neurons with a plasmid that expresses a p65 protein with a serine-to-alanine substitution at residue 536 that prevents phosphorylation at this site but does not abolish the transcriptional activity of p65 (Okazaki et al., 2003; Buss et al., 2004; Sasaki et al., 2005). Expression of this S536A-p65 mutant had no effect on the survival of either nodose or SCG neurons (data not shown) and caused a significant, more than twofold increase in κB reporter signal in both kinds of neurons (p < 0.0001) (data not shown). Despite enhancing NF-κB transcriptional activity in sympathetic neurons, expression of this S536A-p65 mutant, unlike overexpressed wild-type p65, failed to inhibit neurite growth (Fig. 6 A–C) and completely abolished the neurite growth-inhibitory effects of TNFα (Fig. 6 D–F). It also completely abolished the neurite growth-inhibitory effects of overexpressed IKKβ in both SCG (see Fig. 8 A–C) and nodose neurons (Fig. 7 A–C). Expression of the S536A-p65 mutant did not promote neurite growth from either SCG or nodose neurons cultured without neurotrophins (data not shown). Together, these findings suggest that phosphorylation of p65 at S536 is required for the neurite growth-inhibitory effects of enhancing NF-κB signaling with either TNFα or IKKβ overexpression.
Figure 6.

Blocking phosphorylation of p65 at S536 in SCG neurons abolishes neurite growth inhibition induced by TNFα and p65 overexpression. A–C, Total neurite length (A), number of branch points (B), and Sholl profiles (C) of the neurite arbors of P0 SCG neurons transfected 3 h after plating with the YFP plasmid together with either a pcDNA3.1 control plasmid, p65 plasmid, or a S536A-p65 plasmid and cultured for an additional 24 h. D–F, Total neurite length (D), number of branch points (E), and Sholl profiles (F) of the neurite arbors of P0 SCG neurons transfected 3 h after plating with the YFP plasmid and either the pcDNA3.1 control plasmid or S536A p65 plasmid and cultured for an additional 24 h with or without 10 ng/ml TNFα. The combined results of three separate experiments are plotted (means and SEs of the data from >150 neurons for each condition). Statistically significant difference from control-transfected neurons is indicated (**p < 0.001).
Figure 8.

S536A-p65 abolishes IKKβ-induced neurite growth inhibition, and S536D-p65 has a similar inhibitory effect to wild-type p65 on neurite growth from SCG neurons. A–C, Total neurite length (A), number of branch points (B), and Sholl profiles (C) of the neurite arbors of P0 SCG neurons transfected 3 h after plating with the YFP plasmid and either the pcDNA3.1 control plasmid, IKKβ plasmid, or the IKKβ plasmid plus the S536A-p65 plasmid and cultured for an additional 24 h. D–F, Total neurite length (D), number of branch points (E), and Sholl profiles (F) of the neurite arbors of P0 SCG neurons transfected 3 h after plating with the YFP plasmid and either the pcDNA3.1 control plasmid, the p50 plasmid plus the p65 plasmid, or the p50 plasmid plus the S536D-p65 plasmid and cultured for an additional 24 h. The combined results of three separate experiments of each kind are plotted (means and SEs of the data from >150 neurons for each condition). Statistically significant difference from control-transfected neurons is indicated (**p < 0.001).
Figure 7.

S536A-p65 abolishes IKKβ-induced neurite growth inhibition, and S536D-p65 inhibits neurite growth from nodose neurons. A–C, Total neurite length (A), number of branch points (B), and Sholl profiles (C) of the neurite arbors of P0 nodose neurons transfected 3 h after plating with the YFP plasmid and either the pcDNA3.1 control plasmid, IKKβ plasmid, or the IKKβ plasmid plus the S536A-p65 plasmid and cultured for an additional 24 h. D–F, Total neurite length (D), number of branch points (E), and Sholl profiles (F) of the neurite arbors of P0 nodose neurons transfected 3 h after plating with the YFP plasmid and either the pcDNA3.1 control plasmid, the p50 plasmid plus the p65 plasmid, or the p50 plasmid plus the S536D-p65 plasmid and cultured for an additional 24 h. The combined results of three separate experiments of each kind are plotted (means and SEs of the data from >150 neurons for each condition). Statistically significant differences from control-transfected neurons are indicated (*p < 0.05; **p < 0.001).
Phosphomimetic S536D-p65 mutant inhibits neurite growth from SCG and nodose neurons
As a final test of the importance of S536 phosphorylation in neurite growth inhibition, we transfected nodose neurons with a plasmid expressing a dominant-positive phosphomimetic p65 mutant (S536D) that behaves like constitutively active phospho-S536-p65 (Sasaki et al., 2005). Expression of this mutant had no effect on neuronal survival (data not shown) and also caused a more than twofold increase in κB reporter signal (p < 0.0001) (data not shown). Whereas overexpression of wild-type p65 together with p50 promoted significant increases in neurite length and branching of 65 and 30%, respectively, expression of S536D-p65 mutant together with p50 led to significant decreases of 38 and 47%, respectively, relative to control-transfected neurons (Fig. 7 D,E). These marked changes in neurite growth were also clearly apparent in the corresponding Sholl plots (Fig. 7 F). In SCG neurons, neurite growth inhibition was observed after overexpression of either wild-type p65 and p50 or S536D-p65 and p50 relative to control-transfected neurons (Fig. 8 D–F). Together, these results demonstrate that phosphomimetic modification of p65 on S536 is sufficient to reverse the functional role of NF-κB in sensory neurons.
Discussion
Recent work has established a key role for NF-κB signaling in promoting the growth of neural processes in the developing PNS and CNS. Blocking NF-κB signaling impairs pyramidal dendrite growth in the hippocampus and reduces NGF-promoted neurite growth from PC12 cells and BDNF-promoted neurite growth from nodose ganglion sensory neurons (Sole et al., 2004; Gutierrez et al., 2005). Most strikingly, CNTF-promoted neurite growth from nodose neurons is virtually eliminated by inhibiting NF-κB signaling (Gallagher et al., 2007). In marked contrast to these studies, we show that blocking multiple steps in NF-κB signaling in neonatal sympathetic neurons does not affect neurite growth. Dominant-negative IKKβ, S32/36A, and Y42F IκBα mutants, nuclear translocation blockade with SN50 and sequestration of activated NF-κB by decoy κB DNA bring about large decreases in NF-κB transcriptional activity in these neurons, but have no effect on neurite arbor size and complexity. These findings together with our demonstration that the constitutive level of NF-κB signaling in sympathetic neurons is no different to that in nodose sensory neurons (data not shown) clearly indicate that constitutive NF-κB signaling in sympathetic neurons does not promote neurite growth from these neurons. Moreover, enhancing NF-κB signaling in these neurons by overexpressing IKKβ, p65, or p65/p50 or by TNFα treatment greatly reduces neurite growth, indicating that NF-κB signaling can exert a potent inhibitory effect on neurite growth in these neurons.
In neonatal nodose neurons, our finding that overexpressing p65/p50 increases neurite growth, whereas overexpressing IKKβ strongly inhibits neurite growth, clearly demonstrates that NF-κB can promote or inhibit neurite growth in the same neurons depending on the mechanism of activation. The much lower level of phosphorylated IKK in protein extracts of BDNF-supported neonatal nodose neurons suggests that IKK activation plays little or no role in the regulation of neurite growth by constitutive NF-κB activity in these neurons. This agrees with our observation that expression of either dominant-negative IKKα or dominant-negative IKKβ has no effect on BDNF-promoted neurite growth from these neurons (data not shown). Furthermore, CNTF-promoted neurite growth from neonatal nodose neurons engages an NF-κB signaling pathway that, crucially, does not require activation of the IKK complex (Gallagher et al., 2007). Together, these data show that the positive regulation of neurite growth by either an inducible or constitutive NF-κB signal in nodose neurons occurs through IKK-independent mechanisms.
In contrast to nodose neurons, Western blot analysis revealed that sympathetic neurons possess high constitutively active IKK. Accordingly, p65, a known substrate of IKKβ (Sakurai et al., 1999), is constitutively phosphorylated at serine 536 in sympathetic neurons, whereas nodose neurons contain barely detectable levels of phospho-S536-p65. The level of phospho-S536-p65 is markedly elevated in sympathetic neurons after TNFα treatment and IKKβ overexpression, and phospho-S536-p65 becomes abundantly detectable in nodose neurons overexpressing IKKβ. This correlation between elevated phospho-S536-p65 and neurite growth inhibition is causative because expressing a p65 protein with an alanine substitution at serine 536 prevents its neurite growth-inhibitory effect as well as that of TNFα and IKKβ overexpression in sympathetic neurons and IKKβ overexpression in nodose neurons. Furthermore, expression of a dominant-positive S536D-p65 mutant that has been shown to behave like phospho-S536-p65 (Sakurai et al., 1999; Buss et al., 2004), drastically reduces neurite growth, strongly suggesting that phosphorylation of p65 at serine 536 is in itself sufficient to trigger the reversal of NF-κB function in the regulation of neurite growth.
The importance of phospho-S536-p65 in switching NF-κB signaling from growth promoting to growth inhibitory explains why p65/p50 overexpression in sympathetic and nodose neurons has opposite effects on neurite growth. In sympathetic neurons, constitutive activation of IKKβ would be expected to bring about this posttranslational modification in overexpressed p65, whereas in nodose neurons overexpressed p65 remains unphosphorylated on this residue (Fig. 9). This raises the question of the actual role played by the basal level of phospho-S536-p65 in sympathetic neurons. Although our data demonstrate that augmenting NF-κB activity in sympathetic neurons inhibits growth, its blockade failed to enhance axonal growth. This might be because the capacity of neurite elongation machinery is rate limiting in sympathetic neurons under the present experimental conditions. In this regard, we recently reported that NGF-promoted axon growth and target innervation requires an autocrine signaling loop in sympathetic neurons involving a member of the TNF superfamily, glucocorticoid-induced TNF receptor related gene (GITR), and its ligand, GITR ligand (GITRL) (O'Keeffe et al., 2008). The level of GITRL–GITR signaling in culture could therefore place a limit on the ability of these neurons to extend neurites. Alternatively, it is possible that phospho-S536-p65 has to be elevated above a certain threshold before it exerts an inhibitory effect, and only when the level of phospho-S536-p65 is elevated in these neurons is growth retardation observed. This latter possibility is consistent with the fact that the endogenous visible levels of phospho-S536-p65 seem not to negatively influence NGF-promoted growth in sympathetic neurons and that NGF neither induces nor inhibits NF-κB-dependent transcriptional activity in neonate sympathetic neurons (data not shown).
Figure 9.

Schematic model showing the proposed mechanism underlying the opposing effects of NF-κB signaling on neurite growth. In nodose sensory neurons, IKKβ-independent constitutive NF-κB signaling in BDNF-supplemented cultures leads to a transcriptionally active form of p65 that is not phosphorylated on S536 that promotes neurite growth. In SCG sympathetic neurons, IKKβ-dependent enhanced NF-κB signaling after TNFα treatment leads to a transcriptionally active form of p65 that is phosphorylated on S536 that inhibits neurite growth.
We found that IKKβ but not IKKα promoted p65 phosphorylation and growth inhibition in SCG and nodose neurons. Although both IKKβ and IKKα have been shown to be equally capable of phosphorylating p65 at serine 536, differences in the relative importance of either IKK protein in promoting p65 phosphorylation have also been described in different cellular systems (Jeong et al., 2005; Lawrence et al., 2005). Although we focused on the role of IKKβ in p65 phosphorylation, we cannot rule out the potential involvement of other serine kinases modifying p65 in developing neurons. Serine 536 is known to be targeted by several other kinases including RSK1, IKKε, and TBK1 (Mettouchi et al., 1997; Sakurai et al., 1999, 2003; Fujita et al., 2003; Jiang et al., 2003; Yang et al., 2003; Bohuslav et al., 2004; Buss et al., 2004; Mattioli et al., 2004; Jeong et al., 2005; Adli and Baldwin, 2006). This raises the possibility that other signaling pathways might influence the phosphorylation status of p65 in particular neurons and hence modulate the function of NF-κB on axonal and dendritic growth. The schematic model shown in Figure 9 summarizes the proposed mechanism underlying the functional switch for NF-κB signaling based on the phosphorylation status of p65 at S536.
The events after NF-κB activation that inhibit or promote growth of neural processes are unclear. NF-κB activation induces or represses the expression of many genes in different cellular contexts (Hoffmann et al., 2006). Some NF-κB-regulated genes are potentially relevant in the control of neural process growth, such as the neural cell adhesion molecule, tenascin-C, and β1 integrin (Mettouchi et al., 1997; Simpson and Morris, 2000; Wang et al., 2003). It is possible that the opposing effects on neurite growth of phosphorylated and nonphosphorylated S536 p65 could result from the regulation of different sets of genes. Serine 536 is located within a C-terminal transactivation domain of p65 (Viatour et al., 2005), and the consequences of mutation at this residue for p65 transcriptional activity differ according to the promoter being analyzed. For example, S536A substitution substantially reduced the κB-luciferase reporter signal in fibroblasts in which IKKα and/or IKKβ were activated in a variety of ways (Madrid et al., 2000; Jiang et al., 2003; Yang et al., 2003; O'Mahony et al., 2004). In contrast, S536A substitution did not reduce p65 transcriptional activity in fibroblasts with respect to TNF-mediated interleukin-6 (IL-6) induction (Okazaki et al., 2003) and IL-1-mediated IL-8 induction (Buss et al., 2004). Moreover, the S536D phosphomimetic p65 mutant induced IL-8 transcription in the Jurkat T-cell line, whereas the S536A p65 mutant did not (Sasaki et al., 2005). These observations have led to the proposal that phosphorylated S536 p65 and nonphosphorylated S536 p65 regulate the transcription of different sets of genes (Sasaki et al., 2005). For this reason, it will be informative to study the expression of NF-κB-regulated genes in neurons under conditions in which NF-κB signaling has opposing actions on neurite growth.
In summary, our results demonstrate that NF-κB can both stimulate and inhibit neurite growth in developing neurons depending on the phosphorylation status of p65. Although opposite effects of NF-κB signaling have been described in the regulation of cell survival and cell death in different cellular systems (Mémet, 2006; Schwabe and Brenner, 2007), to our knowledge we provide the first demonstration of a reversal in NF-κB function resulting from the phosphorylation of a single residue in an active NF-κB subunit. Our results will help to explain conflicting data regarding seemingly diverging roles played by NF-κB signaling in the nervous system and provide additional support for the importance of this signaling pathway in neural plasticity and development.
Footnotes
This work was supported by a grant from The Wellcome Trust. N.G. was supported by the Beatriu de Pinós Fellowship (Generalitat de Catalunya, Spain). Our thanks to Ron Hay (University of Dundee, Dundee, UK) for kindly providing the IKKα, IKKβ, IκBα, p50, and p65 plasmids and to Carl Sasaki (National Institute on Aging, Baltimore, MD) for kindly providing the p65 mutant plasmids.
References
- Adli M, Baldwin AS. IKK-i/IKKepsilon controls constitutive, cancer cell-associated NF-kappaB activity via regulation of Ser-536 p65/RelA phosphorylation. J Biol Chem. 2006;281:26976–26984. doi: 10.1074/jbc.M603133200. [DOI] [PubMed] [Google Scholar]
- Barker V, Middleton G, Davey F, Davies AM. TNFalpha contributes to the death of NGF-dependent neurons during development. Nat Neurosci. 2001;4:1194–1198. doi: 10.1038/nn755. [DOI] [PubMed] [Google Scholar]
- Bohuslav J, Chen LF, Kwon H, Mu Y, Greene WC. p53 induces NF-kappaB activation by an IkappaB kinase-independent mechanism involving phosphorylation of p65 by ribosomal S6 kinase 1. J Biol Chem. 2004;279:26115–26125. doi: 10.1074/jbc.M313509200. [DOI] [PubMed] [Google Scholar]
- Bui NT, Livolsi A, Peyron JF, Prehn JH. Activation of nuclear factor kappaB and Bcl-x survival gene expression by nerve growth factor requires tyrosine phosphorylation of IkappaBalpha. J Cell Biol. 2001;152:753–764. doi: 10.1083/jcb.152.4.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss H, Dörrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-κB at serine 536 is mediated by multiple protein kinases including IκB kinase (IKK)-α, IKKβ, IKKε, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J Biol Chem. 2004;279:55633–55643. doi: 10.1074/jbc.M409825200. [DOI] [PubMed] [Google Scholar]
- Fujita F, Taniguchi Y, Kato T, Narita Y, Furuya A, Ogawa T, Sakurai H, Joh T, Itoh M, Delhase M, Karin M, Nakanishi M. Identification of NAP1, a regulatory subunit of IkappaB kinase-related kinases that potentiates NF-kappaB signaling. Mol Cell Biol. 2003;23:7780–7793. doi: 10.1128/MCB.23.21.7780-7793.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher D, Gutierrez H, Gavalda N, O'Keeffe G, Hay R, Davies AM. Nuclear factor-κB activation via tyrosine phosphorylation of inhibitor κB-α is crucial for ciliary neurotrophic factor-promoted neurite growth from developing neurons. J Neurosci. 2007;27:9664–9669. doi: 10.1523/JNEUROSCI.0608-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glebova NO, Ginty DD. Growth and survival signals controlling sympathetic nervous system development. Annu Rev Neurosci. 2005;28:191–222. doi: 10.1146/annurev.neuro.28.061604.135659. [DOI] [PubMed] [Google Scholar]
- Gutierrez H, Davies AM. A fast and accurate procedure for deriving the Sholl profile in quantitative studies of neuronal morphology. J Neurosci Methods. 2007;163:24–30. doi: 10.1016/j.jneumeth.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Gutierrez H, Hale VA, Dolcet X, Davies A. NF-κB signalling regulates the growth of neural processes in the developing peripheral and central nervous systems. Development. 2005;132:1713–1726. doi: 10.1242/dev.01702. [DOI] [PubMed] [Google Scholar]
- Hehlgans T, Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology. 2005;115:1–20. doi: 10.1111/j.1365-2567.2005.02143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene. 2006;25:6706–6716. doi: 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- Imbert V, Rupec RA, Livolsi A, Pahl HL, Traenckner EB, Mueller-Dieckmann C, Farahifar D, Rossi B, Auberger P, Baeuerle PA, Peyron JF. Tyrosine phosphorylation of I kappa B-alpha activates NF-kappa B without proteolytic degradation of I kappa B-alpha. Cell. 1996;86:787–798. doi: 10.1016/s0092-8674(00)80153-1. [DOI] [PubMed] [Google Scholar]
- Jeong SJ, Pise-Masison CA, Radonovich MF, Park HU, Brady JN. A novel NF-kappaB pathway involving IKKbeta and p65/RelA Ser-536 phosphorylation results in p53 Inhibition in the absence of NF-kappaB transcriptional activity. J Biol Chem. 2005;280:10326–10332. doi: 10.1074/jbc.M412643200. [DOI] [PubMed] [Google Scholar]
- Jiang X, Takahashi N, Matsui N, Tetsuka T, Okamoto T. The NF-kappa B activation in lymphotoxin beta receptor signaling depends on the phosphorylation of p65 at serine 536. J Biol Chem. 2003;278:919–926. doi: 10.1074/jbc.M208696200. [DOI] [PubMed] [Google Scholar]
- Lawrence T, Bebien M, Liu GY, Nizet V, Karin M. IKKalpha limits macrophage NF-kappaB activation and contributes to the resolution of inflammation. Nature. 2005;434:1138–1143. doi: 10.1038/nature03491. [DOI] [PubMed] [Google Scholar]
- Liang Y, Zhou Y, Shen P. NF-kappaB and its regulation on the immune system. Cell Mol Immunol. 2004;1:343–350. [PubMed] [Google Scholar]
- Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-kappa B by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem. 1995;270:14255–14258. doi: 10.1074/jbc.270.24.14255. [DOI] [PubMed] [Google Scholar]
- Madrid LV, Wang CY, Guttridge DC, Schottelius AJ, Baldwin AS, Jr, Mayo MW. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-kappaB. Mol Cell Biol. 2000;20:1626–1638. doi: 10.1128/mcb.20.5.1626-1638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattioli I, Sebald A, Bucher C, Charles RP, Nakano H, Doi T, Kracht M, Schmitz ML. Transient and selective NF-κB p65 serine 536 phosphorylation induced by T cell costimulation is mediated by IκB kinase β and controls the kinetics of p65 nuclear import. J Immunol. 2004;172:6336–6344. doi: 10.4049/jimmunol.172.10.6336. [DOI] [PubMed] [Google Scholar]
- Mémet S. NF-kappaB functions in the nervous system: from development to disease. Biochem Pharmacol. 2006;72:1180–1195. doi: 10.1016/j.bcp.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Mettouchi A, Cabon F, Montreau N, Dejong V, Vernier P, Gherzi R, Mercier G, Binétruy B. The c-Jun-induced transformation process involves complex regulation of tenascin-C expression. Mol Cell Biol. 1997;17:3202–3209. doi: 10.1128/mcb.17.6.3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita R, Sugimoto T, Aoki M, Kida I, Tomita N, Moriguchi A, Maeda K, Sawa Y, Kaneda Y, Higaki J, Ogihara T. In vivo transfection of cis element “decoy” against nuclear factor-kappaB binding site prevents myocardial infarction. Nat Med. 1997;3:894–899. doi: 10.1038/nm0897-894. [DOI] [PubMed] [Google Scholar]
- Okazaki T, Sakon S, Sasazuki T, Sakurai H, Doi T, Yagita H, Okumura K, Nakano H. Phosphorylation of serine 276 is essential for p65 NF-kappaB subunit-dependent cellular responses. Biochem Biophys Res Commun. 2003;300:807–812. doi: 10.1016/s0006-291x(02)02932-7. [DOI] [PubMed] [Google Scholar]
- O'Keeffe GW, Gutierrez H, Pandolfi PP, Riccardi C, Davies AM. NGF-promoted axon growth and target innervation requires GITRL-GITR signaling. Nat Neurosci. 2008;11:135–142. doi: 10.1038/nn2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Mahony AM, Montano M, Van Beneden K, Chen LF, Greene WC. Human T-cell lymphotropic virus type 1 tax induction of biologically active NF-kappaB requires IkappaB kinase-1-mediated phosphorylation of RelA/p65. J Biol Chem. 2004;279:18137–18145. doi: 10.1074/jbc.M401397200. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8:49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Roff M, Thompson J, Rodriguez MS, Jacque JM, Baleux F, Arenzana-Seisdedos F, Hay RT. Role of IkappaBalpha ubiquitination in signal-induced activation of NFkappaB in vivo. J Biol Chem. 1996;271:7844–7850. doi: 10.1074/jbc.271.13.7844. [DOI] [PubMed] [Google Scholar]
- Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W. IkappaB kinases phosphorylate NF-kappaB p65 subunit on serine 536 in the transactivation domain. J Biol Chem. 1999;274:30353–30356. doi: 10.1074/jbc.274.43.30353. [DOI] [PubMed] [Google Scholar]
- Sakurai H, Suzuki S, Kawasaki N, Nakano H, Okazaki T, Chino A, Doi T, Saiki I. Tumor necrosis factor-alpha-induced IKK phosphorylation of NF-kappaB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J Biol Chem. 2003;278:36916–36923. doi: 10.1074/jbc.M301598200. [DOI] [PubMed] [Google Scholar]
- Sasaki CY, Barberi TJ, Ghosh P, Longo DL. Phosphorylation of RelA/p65 on serine 536 defines an IκBα-independent NF-κB pathway. J Biol Chem. 2005;280:34538–34547. doi: 10.1074/jbc.M504943200. [DOI] [PubMed] [Google Scholar]
- Schwabe RF, Brenner DA. Nuclear factor-kappaB in the liver: friend or foe? Gastroenterology. 2007;132:2601–2604. doi: 10.1053/j.gastro.2007.04.058. [DOI] [PubMed] [Google Scholar]
- Simpson CS, Morris BJ. Regulation of neuronal cell adhesion molecule expression by NF-kappa B. J Biol Chem. 2000;275:16879–16884. doi: 10.1074/jbc.275.22.16879. [DOI] [PubMed] [Google Scholar]
- Sole C, Dolcet X, Segura MF, Gutierrez H, Diaz-Meco MT, Gozzelino R, Sanchis D, Bayascas JR, Gallego C, Moscat J, Davies AM, Comella JX. The death receptor antagonist FAIM promotes neurite outgrowth by a mechanism that depends on ERK and NF-kappa B signaling. J Cell Biol. 2004;167:479–492. doi: 10.1083/jcb.200403093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Wang JH, Manning BJ, Wu QD, Blankson S, Bouchier-Hayes D, Redmond HP. Endotoxin/lipopolysaccharide activates NF-κB and enhances tumor cell adhesion and invasion through a β1 integrin-dependent mechanism. J Immunol. 2003;170:795–804. doi: 10.4049/jimmunol.170.2.795. [DOI] [PubMed] [Google Scholar]
- Yang F, Tang E, Guan K, Wang CY. IKKβ plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J Immunol. 2003;170:5630–5635. doi: 10.4049/jimmunol.170.11.5630. [DOI] [PubMed] [Google Scholar]