

Abstract
There are over 150 human proteins that have been categorized as bona fide DNA repair proteins. These DNA repair proteins maintain the integrity of the genome, reducing the onset of cancer, disease and aging phenotypes. Variations in expression and/or function would therefore impact genome integrity as well as the cellular response to genotoxins. Global gene expression analysis is an effective approach to uncover defects in DNA repair gene expression and to discover cellular and/or organismal effects brought about by external stimuli such as environmental genotoxicants, chemotherapeutic regimens, viral infections as well as developmental and age-related stimuli. Given the significance of genome stability in cell survival and response to stimuli, we have hypothesized that cells may undergo transcriptional re-programming to accommodate defects in basal DNA repair capacity to promote survival. As a test of this hypothesis, we have compared the transcriptome in three DNA polymerase ß knockout (Polß-KO) mouse embryonic fibroblasts (MEFs) and the corresponding wild-type (WT) littermate control cell lines. Each Polß-KO cell line was found to have a range of genes up-regulated, when compared to its WT littermate control cell line. Interestingly, six (6) genes were commonly up regulated in all three Polß-KO cell lines, including Sox2, one of several genes associated with the induction of pluripotent stem cells. Herein, we present these findings and suggest that loss of DNA repair and the induction of cellular transcriptional re-programming may, in part, contribute to tumor formation and the cellular response to external stimuli.
Keywords: DNA polymerase ß, mouse embryonic fibroblast, Sox2, gene expression profiling, transcriptional reprogramming
Introduction
Human cells repair thousands of DNA lesions per day to prevent the accumulation of DNA mutations or genome aberrations that can impact cellular survival and genomic integrity [1]. To facilitate the repair of these lesions, cells have multiple DNA repair and DNA damage response mechanisms that signal the presence of lesions and promote DNA repair [2]. Defects in these repair and response pathways can promote tumorigenesis and, indeed, are common in human cancers [3,4]. The overall strategy of most genotoxic chemotherapeutics is to create large amounts of DNA damage that overwhelm DNA repair systems to promote tumor cell death. Hence, the requirement for DNA repair in response to radiation and genotoxic chemotherapeutics implicates DNA repair proteins as prime targets for improving response to currently available anti-cancer regimens. Further, another strategy is to exploit the DNA repair and response defects that are present in cancer cells thereby specifically targeting tumor cells while sparing healthy cells from a high load of unrepaired DNA damage [5,6].
Cancer-specific DNA repair gene defects provide a mechanistic understanding of therapeutic response. For example, recurrent glioma appears to be resistant to alkylator therapy due to somatic mutations in the mismatch repair protein MSH6 [7]. However, cancer-specific DNA repair defects can also offer novel approaches for tumor-selective therapy [8-10]. Since the discovery that cells with defects in the BRCA genes are selectively killed by the inhibition of PARP, PARP inhibitors have rapidly made their way into the clinic [11,12]. In addition, we and others have shown that the Base Excision Repair (BER) pathway is important for the response to the chemotherapeutic agent Temozolomide (TMZ) [13-15] and TMZ potentiation by targeting the essential BER protein DNA polymerase ß (Polß) is enhanced in BRCA-deficient tumor cells [16], suggesting that defects in BER might also influence response to select agents. As we learn more about the changes in the DNA repair status of tumor cells and the effect of these DNA repair mutations on protein function and the cellular response to genotoxins such as radiation and chemotherapy, we become poised to exploit these defects to our advantage. As was described [5], there is a critical need to identify tumor-related DNA repair defects (mutations) and more importantly, evaluate the functional and biological context of these mutations with regard to cellular transformation and the response to chemotherapy.
DNA-damaging agents, either from endogenous or exogenous sources, activate DNA damage responses leading to a reprogramming of gene expression by mechanisms that are not fully elucidated. These include signal transduction processes mediated by cascades of post-translational modifications such as ATM-mediated phosphorylation [17], chromatin-remodeling [18] and poly-ADP-ribose (PAR)-mediated alterations in protein activity and mRNA or microRNA expression [19,20]. Recent evidence suggests that this reprogramming occurs at multiple levels including changes in the synthesis and stability of specific RNAs and alteration in mRNA splicing patterns.
We have hypothesized that DNA repair defects will impact mRNA expression suggestive of DNA repair pathway specific RNA signatures. This is supported by multiple recent findings in addition to the well-established role of NER and response to blocks to RNA polymerase (transcriptional-coupled NER; TC-NER) [21-24]. BER genes have been shown to be involved in Myc and ER-mediated transcription [25-27] and NER proteins have been reported to play a direct role in transcription [28-30] whereas HR defects (e.g., BRCA1, ATM) can give rise to significant alterations in miRNA and mRNA expression profiles, all in support of our hypothesis that RNA expression profiles may be uniquely impacted by the DNA repair status of the cell [31]. As anticipated from our hypothesis, specific DNA repair proteins can have unique transcriptional effects, such as the loss of XPC inducing an up-regulation of the short form of caspase 2 that leads to enhanced UV-induced apoptosis [32].
To evaluate the impact of DNA repair status on transcriptional profiles, we utilized a well-characterized DNA repair deficient cell system, mouse embryonic fibroblast (MEFs) deficient in the BER protein Polß. The BER pathway is responsible for resolving up to 20,000 lesions per cell per day, which include oxidative and alkylation damage. Polß is a member of the X family of DNA polymerases and is an essential protein in the BER pathway [13,15,33]. Polß is the primary polymerase involved in BER, through its bifunctional 5’ deoxyribose phosphate (5’dRP)-lyase and DNA polymerase activities, so defective or error-prone Polß activity leads to aberrant BER and genomic instability which are associated with carcinogenesis. Polß has been implicated in several cellular functions, including genome stability [34], telomere maintenance and meiosis [35]. Defects in Polß have been linked with cancer [36], aging, neurodegeneration and its expression is critical for the cellular response to environmental and chemotherapeutic genotoxins [37]. This latter function involves its primary role as the major DNA polymerase in the BER pathway. Polß is a bi-functional, two-domain, single-polypeptide 39kDa enzyme. Further, cancer specific mutations of Polß have been identified [36,38-43]. In many cases, the proteins are dysfunctional, such as the E295K mutation found in gastric cancer [44,45]. There has been considerable effort put forth to characterize the cellular involvement of Polß in both mouse and human cells with regard to the response to DNA damaging agents. Polß plays a critical role in the repair of genomic base damage [34] and in the absence of Polß, cells are unable to efficiently repair the highly toxic 5’dRP moiety and therefore are hypersensitive to the cytotoxic effect of different types of alkylating agents such as MMS, MNU and N-methyl-N’-nitro-N-nitrosoguanidine (MNNG) [37], the thymidine analog 5-hydroxymethyl-2′-deoxyuridine [46] as well as the therapeutic alkylating agent TMZ [15,33,46].
However, what is not clear is how the loss of this essential DNA repair gene has altered the global cellular state. Although MEFs and human cells appear fully functional in the absence of Polß [15,33,37,47], mice are not viable past birth [37,48,49] and in one case, the response to DNA damaging agents was shown to vary with continued passage of the cells [50]. Given the significance of genome stability in cell survival and response to stimuli, we have hypothesized that cells may undergo transcriptional re-programming to accommodate defects in basal DNA repair capacity to promote survival. As a test of this hypothesis, we have compared the transcriptome in three DNA polymerase ß knockout (Polß-KO) mouse embryonic fibroblasts (MEFs) and the corresponding wild-type (WT) littermate control cell lines. Each Polß-KO cell line was found to have a range of genes up-regulated, when compared to its WT littermate control cell line. Interestingly, six (6) genes were commonly up regulated in all three Polß-KO cell lines, including Sox2, one of several genes associated with the induction of pluripotent stem cells. Herein, we present these findings and suggest that loss of DNA repair and the induction of cellular transcriptional re-programming may, in part, contribute to tumor formation and the cellular response to external stimuli.
Materials and methods
Chemicals and reagents
Cell culture media and supplies where from InVitrogen-Gibco (Carlsbad, CA). Fetal bovine serum (FBS) was from Sigma (St. Louis, MO). We used the following primary antibodies: DNA polymerase beta monoclonal Abs 18S [51] and Clone 61 (mAb clone 61; Thermo Fisher Scientific; Waltham, MA) and PCNA mAb (#sc-56, Santa Cruz Biotechnology, Santa Cruz, CA). Secondary antibodies GAM-HRP and GAR-HRP conjugates and signal generation substrates were from Bio-Rad (Hercules, CA) and Pierce (Rockford, IL), respectively. All electrophoresis reagents were from Bio-Rad (Hercules, CA). The miRNeasy Mini RNA Isolation and purification kit (cat # 217004) was from Qiagen (Valencia, CA). Ambion WT Expression Kit (#4411974) was from Ambion (Foster City, CA). GeneChip WT Terminal labelling and controls kit (#901524) and GeneAtlas Hybridization, Wash, and Stain Kit (#900720) were from Affymetrix (Santa Clara, CA). SuperScript® III First-Strand cDNA Synthesis SuperMix (#18080-400) was from InVitrogen (Carlsbad, CA). TaqMan® Fast Advanced Master Mix and Taqman assay primers were obtained from Life Technologies (Carlsbad, CA).
Cell culture
Transformed Polß-KO mouse embryonic fibroblast (MEF) cell lines (38Δ4, 50TAg and 88TAg) and the corresponding littermate WT control MEF cell lines (36.3, 53TAg and 92TAg) have been described previously [37,52]. The cell lines 92TAg and 88TAg are available from the ATCC (CRL-2816 and CRL-2820, respectively). MEFs were cultured at 37°C in a humidified incubator with 5% CO2 in DMEM supplemented with 10% FBS, penicillin (50 units/mL), streptomycin (50 μg/mL) and Glutamax (4 mmol/L). A375 cells [53,54] were obtained from ATCC (CRL-1619) and cultured at 37°C in a humidified incubator with 5% CO2 in DMEM supplemented with 10% FBS, penicillin (50 units/mL) and streptomycin (50 μg/mL).
Lentiviral transduction of human A375 cells
A375 is a human epithelial cell line derived from malignant melanoma [54]. The A375 cells were modified using a lentiviral vector for the expression of GFP (FIV-CDF1-copGFP) or GFP+shRNA specific to human Polß (FIV-H1(hPolβ)-copGFP), essentially as we have described previously [13-15]. Briefly, lentiviral particles were generated by transfection of three plasmids (the expression plasmid, e.g., pFIV-H1-puro-hPOLB.1; plus pFIV-34N and pVSV-G) into 293-FT cells [55] using FuGene 6. Forty-eight hours after transfection, lentivirus-containing supernatant was collected and passed through 0.45μM filters to isolate the viral particles. Lentiviral transduction was performed as described earlier [13]. Briefly, 6.0 × 104 cells were seeded into a 6-well plate 24 hours before transduction. Cells were transduced for 18 hours at 32°C and then cultured for 72 hours at 37°C. Cells expressing GFP or both GFP and Polß specific shRNA were isolated by fluorescence activated cell sorting (FACS) in the UPCI Cytometry Facility.
Immunoblot analysis
MEF and A375 cell nuclear extracts were prepared and protein concentrations were determined as described by us previously [13,51]. Briefly, nuclear extracts were prepared using the NucBuster nuclear protein extraction reagent (Novagen). Protein concentration was determined using Bio-Rad protein assay reagents according to the manufacturer’s instructions. Nuclear protein (30 μg) was separated by electrophoresis in a 4-12% SDS-polyacrylamide gel and electrotransferred to a 0.45μm nitrocellulose membrane (Trans-Blot, Bio-Rad). Antigens were detected using standard protocols. Primary antibodies: for nuclear extracts from MEFs, Polß was detected using anti-Polß mAb 18S (1:1,000); for nuclear extracts from A375 cells, Polß was detected using anti-Polß mAb Clone 61 (1:500). The horseradish peroxidase (HRP)-conjugated secondary antibodies (GAM-HRP or GAR-HRP; Bio-Rad) were diluted 1:10,000 in TBST/5% milk. Each membrane was stripped and re-probed with anti-PCNA (1:1000) antibodies to correct for differences in protein loading.
RNA Isolation
For each MEF or A375 cell line, cells were cultured in a 100mm cell culture dish (90% confluence) and then split into three 100mm dishes. When cells reached 50~60% confluence, cells in each dish were collected individually and then 1x105 cells of each paired GFP and Polß-KO or Polß-KD cells were seeded into the same 6 well tissue culture plate with 3 wells per cell line. After incubation at 37°C for 24 h, culture medium was removed and then each well was washed twice with 1x cold PBS. Cells were then lysed with 1ml Qiazol Lysis Reagent per three wells. Total RNA was then extracted and purified using the Qiagen miRNeasy Mini kit according to the manufacturer’s instructions. Briefly, after mixing with 200μl chloroform, each cell lysate was centrifuged at 12,000xg to separate the aqueous and organic phases. Each aqueous phase was transferred to a clean 1.5 ml tube and then mixed with 1.5 volumes 100% ethanol. The mixture was then transferred to an RNeasy Mini column and RNA was bound to the capture membrane by a 30 second centrifugation at 12000 rpm. After one wash with buffer RWT and then two washes with buffer RPE, RNA was eluted from each column with 30μl RNase-free water. For each pair of GFP and Polß-KO or Polß-KD cell lines, three independent pairs of RNAs were prepared. RNA quality was determined using an Agilent 2100 Bioanalyzer in the UPCI Cancer Biomarkers Facility. In all cases, the RNA integrity number (RIN) was greater than 9.5. RNA concentration was determined using a Nanodrop 2000.
Microarray analysis
Comparative analysis of mRNA expression for the WT and Polß-KO MEFs was determined using the Mouse Gene 1.1 ST Array and the Affymetrix GeneAtlas system, as per the manufacturer’s instructions. Microarray analysis for each of the MEF cell lines (GFP or Polß KO) was accomplished with 100ng purified total RNA (described above) and the corresponding cRNA using an Ambion WT Expression Kit (Ambion #4411974), as described by the manufacturer. The resulting cDNA was fragmented and end-labelled, as described by the manufacturer. The labeled cDNAs were then mixed with hybridization master mix and the hybridization cocktails were then denatured at 95°C for 5 minutes, followed by 45°C for 5 minutes then kept at 45°C until applied to the hybridization tray (GeneAtlas System; 120μl hybridization cocktail of a cell line was transferred into a well of a 4 well hybridization tray). The array strip was immerse into hybridization cocktail and incubated in the Hybridization Station at 48°C for 20 hours. After hybridization, the strip was washed and stained in the GeneAtlas Fluidics Station using the GeneAtlas Hybridization, Wash, and Stain Kit (Affymetrix #900720) and the intensity of each hybridized probe was generated using the GeneAtlas™ Imaging Station. Raw .cel files from the Mouse Gene 1.1 ST Array were analyzed using the ‘oligo’ package in R Bioconductor specifically designed to analyze Gene ST arrays. The raw data was normalized and summarized using robust multichip average (RMA). The data summarized by transcript clusters was used for further analysis. For transcripts represented by multiple clusters, the cluster with the highest IQR (Interquartile range; a descriptive statistic used to summarize the extent of the spread of the data) was selected to represent the transcript’s expression. As a result of the filtering procedure all transcripts are represented by a single cluster for further statistical analysis.
The ‘genefilter” package in R (Bioconductor. org) was used to perform differential gene expression analysis. Genes differentially expressed in Polß-KO versus WT controls were identified using the univariate t test (p < 0.001). The p-values obtained were adjusted for multiple comparisons using the method of Benjamini and Hochberg [56]. A list of genes differentially expressed by more than 2 fold in all 3 experiments was then obtained. The complete list of differentially expressed genes in each Polß-KO MEF cell line (as compared to the corresponding WT littermate matched cell line) is in Supplemental Table S1. Genes that are commonly up regulated in two or three of the Polß-KO MEFs are listed in Table 1, indicating the fold-increase in each, as determined by the microarray analysis and graphically repre-sented in Figure 2.
Table 1.
Up regulated genes in Polß-KO MEFs
| Genes up regulated* in 38Δ4 and 50TAg cells | Genes up regulated* in 50TAg and 88TAg cells | Genes up regulated* in 38Δ4 and 88TAg cells |
|---|---|---|
| 5730469M10Rik (Pamm) | 5730469M10Rik (Pamm) | 5730469M10Rik (Pamm) |
| Ahr | Ahr | 9030418K01Rik |
| Fam110c | Ctsh | Ahr |
| Hoxb2 | Fam110c | Car12 |
| Hunk | Fez1 | Cd80 |
| Kcnip1 | Kcnip1 | Crabp1 |
| Kcnj2 | Ldhb | Cyp1b1 |
| Meis1 | Lrrc7 | Eda2r |
| Msln | Npnt | Ephx1 |
| Pde3b | Ramp3 | Fam110c |
| Plscr2 | Sfrp1 | Kcnip1 |
| Ptgis | Sox2 | Khdrbs3 |
| Ramp3 | Lass4 | |
| Sim2 | Parvb | |
| Sox2 | Ramp3 | |
| Tspan11 | Slc16a13 | |
| Slit3 | ||
| Sox2 | ||
| Tmem74 |
As determined by the microarray analysis.
Bolded genes are up regulated in all three Polß-KO cells.
Figure 2.
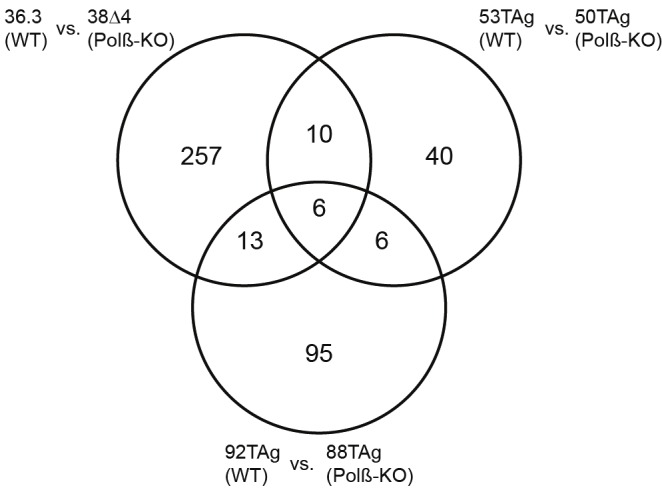
Representation of the number of genes up regulated in Polß-KO MEFs. Venn diagram depicting a graphical representation of the number of genes up regulated in each Polß-KO cell line (as compared to the correspond WT littermate control cell line). The number of identical genes for each comparison and among the three cell line pairs is depicted in the overlapping circles. The names of each gene found to be up regulated in more than one Polß-KO MEF cell line is shown in Table 1.
Quantitative RT-PCR analysis
Validation of the microarray results was determined by quantitative RT-PCR (qRT-PCR) using an Applied Biosystems StepOnePlus system. Briefly, 3μg RNA isolated from the MEF or A375 cells was reverse transcribed to yield first-strand cDNA using InVitrogen Super-Script® III First-Strand cDNA Synthesis SuperMix (#18080-400). Next, 4μl of a 1:50 dilution of the cDNA was used for each Taqman fast RT reaction (20μl). Each sample was analysed in triplicate and the results are an average of all three analyses. Analysis of mRNA expression was conducted as per the manufacturer (ΔΔCT method) using Applied Biosystems TaqMan® Gene Expression Assays (Ahr, Mm00478932_m1; Fam110c, Mm00503576_m1; Kcnip1, Mm01189526; Ramp3, Mm00840142_m1; Sox2 (mouse) Mm03053810_s1; SOX2 (human) Hs01053049_s1; and 5730469-M10Rik, Mm00510430_m1 and normalized to the expression of mouse ß-actin (part #4352663) in the MEFs or human ß-actin (part #4333762T) in the A375 cells.
Results
Global gene expression changes in Polß-KO mouse embryonic fibroblasts
To investigate the global gene expression changes resulting from the loss of expression of Polß, we took advantage of Polß knockout (Polß-KO) mouse embryonic fibroblast (MEF) cell lines and the corresponding WT littermate controls [52]. To achieve reliable transcriptional profile signatures, three different Polß-KO MEF cell lines were used for the analysis and compared to the WT littermate-derived cell lines as controls. The loss of expression of Polß in each cell line was validated by western blot analysis. The WT control MEFs had the expected level of expression of Polß (36.3, 53Tag, or 92TAg cell lines) and the expression of Polß in 38Δ4, 50TAg or 88TAg Polß-KO cell lines was not detectable (Figure 1). Total RNA from each cell line was isolated from cells cultured for 24 hours under the same conditions. We found that the expression of a large number of genes were changed when we compared each Polß-KO cell line to the WT control cell line. To ensure that our findings are reliable, we set a stringent cut off where the expression change of a gene should be above two-fold and the p-value, the indication of statistic significance, should be less than 0.001. 286 genes were more than two-fold up regulated and 196 genes over two-fold down regulated when 38Δ4 (Polß-KO) was compared with 36.3 (WT). There were 64 genes above two-fold up regulated and 48 genes above two-fold down regulated when comparing 50TAg (Polß-KO) with 53TAg (WT). 120 genes were over two-fold up regulated and 153 genes above two-fold down regulated when we compared 88TAg (Polß-KO) with 92TAg (WT). We are most interested in the genes whose expression difference is commonly changed between the cell lines. The common up regulated genes in the Polß-KO cells are shown in Table 1. The Ven diagram in Figure 2 shows the total number of up regulated genes. Though there is no common down regulated gene among those three pairs (except Polß), the expression of the six genes, Sox2, Ramp3, Kcnip1, Fam110c, Ahr and 5730469M10Rik (Pamm) were up regulated in all three Polß-KO MEFs as compared to the corresponding WT cells for each.
Figure 1.
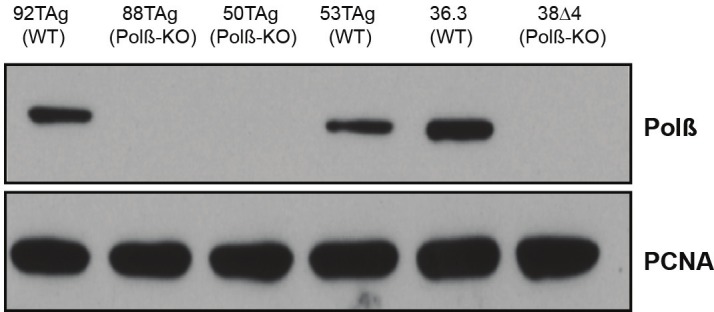
Immunoblot analysis: expression of Polß and PCNA in WT and Polß-KO MEFs. Nuclear lysates from WT MEFs and the corresponding Polß-KO MEFs were probed for expression of Polß and PCNA (loading control) as indicated in the figure. Following separation by SDS-PAGE and transfer to nitrocellulose, the proteins were evaluated for expression as described in the Materials & Methods section. The expected bands for Polß and PCNA are labelled.
Functional network analysis of the genes up regulated in Polß-KO MEFs
Ingenuity Pathway (IPA) analysis was performed on the 6 genes commonly up regulated in each of the comparisons to examine the relationships between them. Significant biological functions and pathways in which these 6 genes are involved include Cell Cycle, Cancer, and importantly, the role of Oct4 in mammalian embryonic stem cell pluripotency. Four of the six genes (Sox2, Ramp3, Kcnip1 and Ahr) are connected into the top-scoring interact network (Figure 5). One gene, JUN and two chemicals, beta-estradiol and Ca2+ were determined by IPA analysis to form the common nodes that connect multiple genes across different pathways. JUN is a transcription factor that recognizes and binds to the enhancer heptamer motif 5’-TGA[CG]TCA-3’ and is involved in numerous cell activities, such as proliferation, apoptosis, survival, tumorigenesis and tissue morphogenesis [57]. It has been reported that JUN promotes ErbB2-induced mammary tumor cell invasion and self-renewal via up regulation of the expression of stem cell factor (SCF) and CCL5 using mouse model [58]. IPA analysis indicates either Sox2 or Ahr directly interacts with JUN. Because the up regulation of both Sox2 and Ahr are involved in the maintenance of the pluripotency of cancer stem cells, it may imply that this interaction may promote the expansion of cancer stem cells. The interaction between JUN and beta-estradiol has been addressed in a rat model. Treating ovariectomized rats with 17 beta-estradiol induced the expression of JUN in all uterine cell types [59]. 17 beta-estradiol also attenuates voltage-dependent Ca2+ current through T- and L-type Ca2+ channels in A7r5 vascular smooth muscle cells [60]. It has also been reported that the application of 17 beta-estradiol to ovariectomized rats dramatically reduced the expression of Ramp3 [61]. Overall, the interaction between 17 beta-estradiol and JUN connects Sox2, Ramp3, Kcnip1, and Ahr in a network and may be related to breast or ovarian malignancy.
Figure 5.

Functional network analysis of the genes up regulated in Polß-KO MEFs. Using Ingenuity Pathways Analysis (IPA), a gene network was defined using the 6 genes found to be up regulated in all 3 Polß-KO MEFs. The 4 (out of 6) genes which are up regulated by more than 2-fold in the 38Δ4, 50TAg and 88TAg cell lines are shown in grey.
Validation of microarray analysis by quantitative RT-PCR of the genes up regulated in all three Polß-KO cell lines
Quantitative RT-PCR (qRT-PCR) analysis of the six common up regulated genes among the three Polß-KO MEF cell lines was performed to validate our microarray results. cDNAs were synthesized from the total RNA of Polß-KO and WT MEF cell lines and used as the templates for qRT-PCR using Taqman expression probes for the Sox2, Ramp3, Kcnip1, Fam110c, Ahr, and Pamm genes, respectively. The results of the qRT-PCR indicated that the expression of all six genes were up regulated in each Polß-KO MEF cell line, which corresponded with the microarray data (Figure 3 and Table 2). The fold change in qRT-PCR was not exactly the same as in the microarray analysis, which might reflect the different sensitivity of each method. The expression of the Sox2 gene in the 38Δ4 cell line was dramatically up regulated to 175-fold when compared with the WT control cell line 36.3.
Figure 3.

Validation of gene up regulation by qRT-PCR. The relative level of expression for (A) Sox2, (B) Kcnip1, (C) Ramp3, (D) Fam110c, (E) Ahr and (F) 5730469M10Rik (Pamm) was determined in each Polß-KO MEF cell line by qRT-PCR via the ΔΔCT method, normalizing the level of expression to the corresponding WT control cell line and to mouse ß-actin within each sample as described in the Materials & Methods section. Results are reported as the mean ± SE of three independent qRT-PCR experiments. The fold increase in mRNA expression for each as determined by qRT-PCR analysis in the 38Δ4, 50TAg and 88TAg cells is shown in Table 2.
Table 2.
Genes of up regulated in all 3 Polß-KO MEFs
| Gene Name | Fold up regulation in 38Δ4 cells* | Fold up regulation in 50TAg cells* | Fold up regulation in 88TAg cells* |
|---|---|---|---|
| 5730469M10Rik (Pamm) | 7.3 | 4.0 | 4.0 |
| Ahr | 5.5 | 2.8 | 2.6 |
| Fam110c | 3.8 | 7.6 | 4.4 |
| Kcnip1 | 106.4 | 6.0 | 12.6 |
| Ramp3 | 44.1 | 6.5 | 4.1 |
| Sox2 | 175.4 | 4.3 | 10.9 |
As determined by the qRT-PCR analysis.
Analysis of SOX2 expression in A375 cells following Polß knockdown
After validating that the loss of expression of Polß results in the up regulation of Sox2 in MEFs, we were interested to know if a deficiency in Polß can also affect the expression of the SOX2 gene in human cell lines. We then developed a Polß deificient human tumor cell line (A375/Polß-KD) by lentiviral mediated expression of Polß-specific shRNA, as we have described [13-15]. The A375-GFP cell line was used as a Polß WT control, which was transduced with the same viral vector expressing only GFP instead of the Polß-shRNA+GFP. Immunoblotting analysis confirmed the deficiency of Polß in the A375/Polß-KD cell line (Figure 4A). A qRT-PCR analysis was performed to compare the expression of SOX2 in the A375/Polß-KD cell line to that of the WT control (A375-GFP). As we predicted, the results show that the expression of SOX2 was more than 2 fold up regulated in the A375/Polß-KD cell line (Figure 4B).
Figure 4.
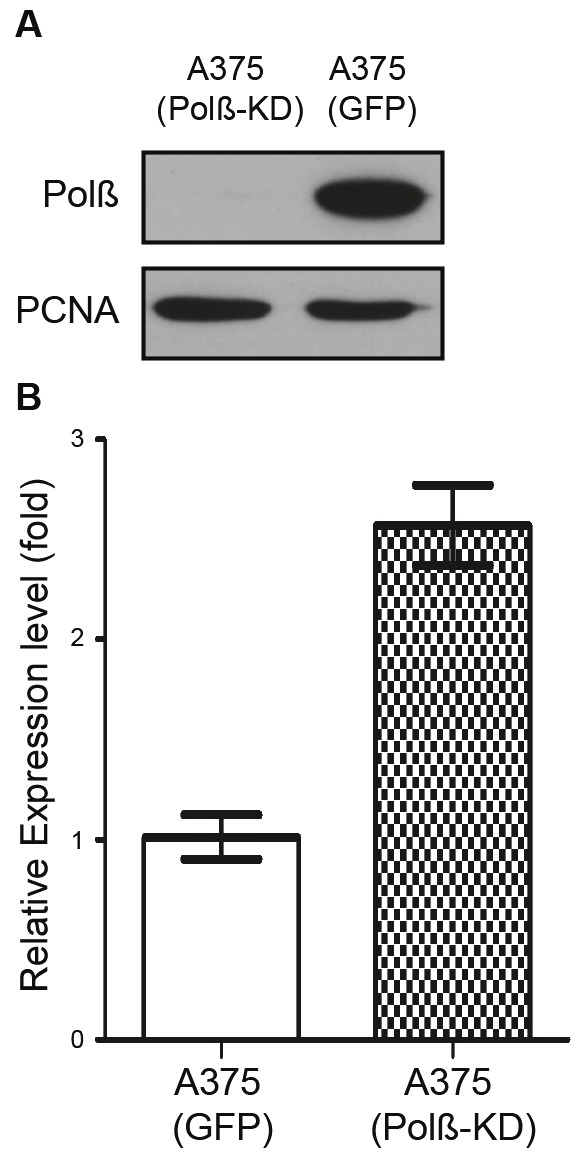
Lentiviral(shRNA)-mediated Polß knockdown and the impact on SOX2 mRNA expression. A. Nuclear lysates from the A375/GFP and A375/Polß-KD cell lines were probed for expression of Polß and PCNA (loading control) as indicated in the figure. Following separation by SDS-PAGE and transfer to nitrocellulose, the proteins were evaluated for expression as described in the Materials & Methods section. The expected bands for Polß and PCNA are labelled. B. The relative level of expression for human SOX2 in the A375/GFP and A375/Polß-KD cell lines were determined by qRT-PCR via the ΔΔCT method, normalizing the level of expression to the corresponding A375/GFP control cell line and to human ß-actin within each sample as described in the Materials & Methods section. Results are reported as the mean ± SE of three independent qRT-PCR experiments.
Discussion
In this study we present a transcriptional profiling analysis detailing the global transcription alterations that result from the loss of expression of the essential base excision repair (BER) protein Polß. Because Polß is a critical protein in BER, the deficiency in Polß will severely impair cellular BER and the response to genotoxic stress from both endogenous and exogenous sources [37,47,62]. Besides its major function of repairing DNA single-strand breaks and deamination, oxidation and alkylation-induced DNA base damage, the BER pathway was recently reported to participate in epigenetic regulation to facilitate gene expression modulation by altering the state of DNA methylation or via a reaction coupled to histone modification [63,64]. Thus, we can speculate that a deficiency in the expression of Polß may also affect the transcriptional profiles of these cell lines via a defect in methylation or epigenetic regulation. The results from our microarray analysis demonstrated a global expression change in each Polß-KO MEF cell line. For each cell line, more than one hundred genes were altered even under a stringent cut off (two-fold expression variation and p-value less than 0.001).
The majority of genes whose expression changed were cell line specific. For example, in the 38Δ4 cell line, the expression of Ifi202b is the most up regulated, which is more than 20-fold higher in the 38Δ4 cell line compared to the 36.3 cell line in our microarray analysis. The increase in expression of the lfi202b gene was linked to an increase of susceptibility for the development of lupus in a mouse model [65]. Due to the strong up-regulation of the expression of the lfi202b gene after loss of function of Polß, our result implies that it is worth to investigate if the deficiency of Polß is also involved in the development of lupus. Sim2 is more than 10-fold up regulated in the 38Δ4 cell line. A recent report indicated that Sim2 can be used as a novel marker of aggressive prostate cancer because the expression of Sim2 gene was highly up regulated in prostate cancer patient samples [66].
Even though the cell line specific expression alterations are interesting, we are more interested in the alterations that are present in all Polß KO MEFs. A total of 6 genes were commonly up regulated in these three Polß-KO MEFs as determined by microarray analysis, and were confirmed with qRT-PCR analysis. These genes include Sox2, Ramp3, Kcnip1, Fam110c, Ahr, and 5730469M10Rik (Pamm). The IPA pathway analysis indicates 4 out of the 6 genes, including Sox2, Ramp3, Kcnip1, and Ahr, are directly or indirectly involving in the protein interaction network centered on JUN and beta estraldiol. This suggests that a deficiency of Polß may play an important role in breast cancer [67].
Sox2 is a transcription factor that is essential for maintaining self-renewal, or pluripotency, of undifferentiated embryonic stem cells. Sox2 is also a marker of cancer stem cells (CSCs), which are highly tumorigenic compared to other subsets and characterized as having the potential for self-renewal, the formation of tumor spheres in low-adherence cultures, and multidrug resistance. CSCs may play a major role in cancer relapse due to their resistance to conventional cancer therapies. The expression of Sox2 is often up regulated to maintain the pluripotency of CSCs. In our Polß-KO MEF cell line, loss of Polß expression resulted in a 175, 4.3 and 10.9 fold up-regulation of Sox2 in 38Δ4, 50Tag and 88Tag cell lines; respectively as validated by qRT-PCR analysis. Therefore, one might then infer that a deficiency in Polß may promote the maintenance of pluripotency in cancer stem cells.
It has been reported that Ahr is involved in the maintenance of the pluripotency of cancer stem cell populations in breast cancer [68]. The inhibition of CXCR4 reduced the growth of tamoxifen-resistant tumors in vivo, which have a larger cancer progenitor population compared with wild-type breast cancer. The inhibition of CXCR4 altered the Ahr signalling network. Moreover, the direct inhibition of the function of Ahr with small molecule antagonists selectively delayed the growth of tumors derived from a subcutaneous inoculation of tamoxifen-resistant MCF7 cells (progenitor cancer cells) into nude mice but not the growth of tumors derived from the tamoxifen-sensitive MCF7 cells (wild type cancer cells) [68]. The loss of Polß expression resulted in 5.5, 2.8 and 2.6 fold up regulated expression of the Ahr gene in 38Δ4, 50Tag and 88Tag cell lines respectively by qRT-PCR analysis. This also implies that a deficiency of Polß expression may facilitate the maintenance of a cancer stem cell population.
Ramp3 is a single-pass membrane-spanning protein with very short intracellular domains. Ramp3 forms complexes with the calcitonin receptor like receptor (CRLR), resulting in the formation of functional receptors for the proangiogenic and protumorigenic peptide adrenomedullin [69]. It has been reported that the expression of Ramp3 is strongly up regulated in human colon, breast, and gastric carcinomas but not in normal colon or gastric epithelial cells. Inhibition of Ramp3 expression in MDA-MB-231 and LM2-4 cells, using a specific shRNA, strongly inhibited their invasiveness and their tumor-forming abilities. Knocking down the expression of Ramp3 dramatically reduced p38 mitogen-activated kinase (p38) phosphorylation as well as ß1-integrin and vimentin expression, each linked to tumor metastasis [69]. Our data suggest loss of Polß expression may also be related to tumor progression via the up-regulation of Ramp3.
Kcnip1 is a member of the family of voltagegated potassium (Kv) channel-interacting proteins. The function Kcnip1 is related to the regulation of A-type currents, and neuronal excitability, in response to changes in intracellular calcium. There is only one report linking Kcnip1 to cancer. Sequencing Kcnip1 cDNA samples from 12 specimens of breast cancer and 12 specimens of normal mammary tissues indicated a new splicing variant of the Kcnip1 gene in cancer samples, which has an insert (162 bp) between exon 1 and exon 2 in the Kcnip1 gene [70]. Recent reports have shown that the expression of Kcnip1 was altered in multiple cancer cell lines. Microarray analysis in the NCI 60 Reference panel indicated that the expression of the Kcnip1 gene has the highest level of up regulation in ACHN renal cancer and OVCAR4 ovarian cancer cell lines [71]. Our ongoing microarray analysis of glioblastoma stem cell (GSC) also indicates that the expression of Kcnip1 was more than 4 fold up regulated in proneurol GSCs compared to normal human actrocytes (not shown). However, both mesenchymal GSC and glioblastoma cell lines such as LN428 and T98G showed more than 1 fold down regulation (unpublished data). Our data suggest that loss of Polß expression may also relate to some special types of tumors such as brain cancer stem cells via the up-regulation of Kcnip1 expression.
Fam110c, which is not in the beta estraldiol related protein network, may also be related to malignant transformation. Fam110c belongs to a gene family consisting of three members, Fam110a, Fam110b, and Fam110c. It has been reported that the Fam110c protein localizes to centrosomes and accumulates at the microtubule organization center in interphase and at spindle poles in mitosis. Overexpression of Fam110c resulted in microtubule aberrancies [72]. Moreover, the depletion of FAM110C by shRNA reduced integrin-mediated filopodia formation, hepatocyte growth factor-induced migration, and phosphorylation of the Akt1 kinase in the epithelial cell line HepG2 [73]. This implies that the Polß-KO induced overexpression of Fam110c may impact the metastasis of tumor cells.
The other gene not in the beta estraldiol related protein network is 5730469M10Rik (Pamm). Pamm is involved in redox regulation of the cell and serves as an antioxidant. It has been reported that the expression of PAMM in RAW264.7 monocytes protected cells under oxidative stress induced by hydrogen peroxide. The authors also indicated the overexpression of PAMM in RAW 264.7 cells prevented an increase in ROS induced by RANKL and inhibited RANKL-induced NFKB1 and JUN activation [74]. Given that one major function of BER is to repair DNA lesions resulting from ROS, the upregulation of the expression of Pamm can be considered as a compensation of dysfunction of BER to reduce oxidative stress.
Taken together, our results show that the loss of function of Polß results in a global transcriptional alteration in each Polß-KO MEF cell line. The up regulation of six genes present in all Polß-KO MEF cell lines results in a transcriptional signature that may be representative of loss of expression of Polß and by inference, loss of function. The over-expression of Sox2, Ramp3, Kcnip1, Fam110c and Ahr are related to either maintaining the pluripotency of CSCs or promoting tumor metastasis. With the observation that 30% of human tumor (from multiple tumor types) present with defects in the expression or function of Polß [36,38,41], and the recent discovery of the participation of BER in epigenetic regulation [63,64], our results suggest that a deficiency in Polß not only results in a loss of BER capacity but may also directly affect the expression of genes critical for malignant transformation and tumor progression.
Acknowledgements
We would like to thank Andrea Braganza for proofreading. This work was supported by grants from the National Institutes of Health (NIH) [GM087798; CA148629; ES019498] to RWS. Support for the UPCI Lentiviral Facility was provided to RWS by the Cancer Center Support Grant from the National Institutes of Health [P30 CA047904]. Support for the UPCI Cancer Biomarkers Facility and the UPCI Cytometry Facility was provided by the Cancer Center Support Grant from the National Institutes of Health [P30 CA047904].
Disclosure of potential conflicts of interest
RWS is a Scientific Consultant for Trevigen, Inc.
Supporting Information
References
- 1.Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–715. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- 2.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Harper JW, Elledge SJ. The DNA damage response: ten years after. Molecular Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 5.Alberts B. Redefining cancer research. Science. 2009;325:1319. doi: 10.1126/science.1181224. [DOI] [PubMed] [Google Scholar]
- 6.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 7.Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB, Batchelor TT, Futreal PA, Stratton MR, Curry WT, Iafrate AJ, Louis DN. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res. 2007;13:2038–2045. doi: 10.1158/1078-0432.CCR-06-2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Javle M, Curtin NJ. The role of PARP in DNA repair and its therapeutic exploitation. British Journal of Cancer. 2011;105:1114–1122. doi: 10.1038/bjc.2011.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sandhu SK, Yap TA, de Bono JS. Poly(ADP-ribose) polymerase inhibitors in cancer treatment: a clinical perspective. European Journal of Cancer. 2010;46:9–20. doi: 10.1016/j.ejca.2009.10.021. [DOI] [PubMed] [Google Scholar]
- 10.Peralta-Leal A, Rodriguez MI, Oliver FJ. Poly(ADP-ribose)polymerase-1 (PARP-1) in carcinogenesis: potential role of PARP inhibitors in cancer treatment. Clin Transl Oncol. 2008;10:318–323. doi: 10.1007/s12094-008-0207-8. [DOI] [PubMed] [Google Scholar]
- 11.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 12.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, Martin NM, Jackson SP, Smith GC, Ashworth A. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 13.Tang JB, Goellner EM, Wang XH, Trivedi RN, St Croix CM, Jelezcova E, Svilar D, Brown AR, Sobol RW. Bioenergetic Metabolites Regulate Base Excision Repair-Dependent Cell Death in Response to DNA Damage. Molecular Cancer Research. 2010;8:67–79. doi: 10.1158/1541-7786.MCR-09-0411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang JB, Svilar D, Trivedi RN, Wang XH, Goellner EM, Moore B, Hamilton RL, Banze LA, Brown AR, Sobol RW. N-methylpurine DNA glycosylase and DNA polymerase beta modulate BER inhibitor potentiation of glioma cells to temozolomide. Neuro Oncol. 2011;13:471–486. doi: 10.1093/neuonc/nor011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trivedi RN, Wang XH, Jelezcova E, Goellner EM, Tang J, Sobol RW. Human methyl purine DNA glycosylase and DNA polymerase ß expression collectively predict sensitivity to temozolomide. Molecular Pharmacology. 2008;74:505–516. doi: 10.1124/mol.108.045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stachelek GC, Dalal S, Donigan KA, Campisi Hegan D, Sweasy JB, Glazer PM. Potentiation of temozolomide cytotoxicity by inhibition of DNA polymerase beta is accentuated by BRCA2 mutation. Cancer Research. 2010;70:409–417. doi: 10.1158/0008-5472.CAN-09-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuoka S, Ballif BA, Smogorzewska A, Mc- Donald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 18.Lukas J, Lukas C, Bartek J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol. 2011;13:1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 19.Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leung AK, Vyas S, Rood JE, Bhutkar A, Sharp PA, Chang P. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Molecular Cell. 2011;42:489–499. doi: 10.1016/j.molcel.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 22.de Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13:768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- 23.Wood RD. DNA repair in eukaryotes. Annu Rev Biochem. 1996;65:135–167. doi: 10.1146/annurev.bi.65.070196.001031. [DOI] [PubMed] [Google Scholar]
- 24.Shuck SC, Short EA, Turchi JJ. Eukaryotic nucleotide excision repair: from understanding mechanisms to influencing biology. Cell Res. 2008;18:64–72. doi: 10.1038/cr.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amente S, Bertoni A, Morano A, Lania L, Avvedimento EV, Majello B. LSD1-mediated demethylation of histone H3 lysine 4 triggers Myc-induced transcription. Oncogene. 2010;29:3691–3702. doi: 10.1038/onc.2010.120. [DOI] [PubMed] [Google Scholar]
- 26.Amente S, Lania L, Avvedimento EV, Majello B. DNA oxidation drives Myc mediated transcription. Cell Cycle. 2010;9:3002–3004. doi: 10.4161/cc.9.15.12499. [DOI] [PubMed] [Google Scholar]
- 27.Perillo B, Ombra MN, Bertoni A, Cuozzo C, Sacchetti S, Sasso A, Chiariotti L, Malorni A, Abbondanza C, Avvedimento EV. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science. 2008;319:202–206. doi: 10.1126/science.1147674. [DOI] [PubMed] [Google Scholar]
- 28.Fuss JO, Tainer JA. XPB and XPD helicases in TFIIH orchestrate DNA duplex opening and damage verification to coordinate repair with transcription and cell cycle via CAK kinase. DNA Repair (Amst) 2011;10:697–713. doi: 10.1016/j.dnarep.2011.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehmann AR. Nucleotide excision repair and the link with transcription. Trends Biochem Sci. 1995;20:402–405. doi: 10.1016/s0968-0004(00)89088-x. [DOI] [PubMed] [Google Scholar]
- 30.Lagerwerf S, Vrouwe MG, Overmeer RM, Fousteri MI, Mullenders LH. DNA damage response and transcription. DNA Repair (Amst) 2011;10:743–750. doi: 10.1016/j.dnarep.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 31.Svejstrup JQ. The interface between transcription and mechanisms maintaining genome integrity. Trends Biochem Sci. 2010;35:333–338. doi: 10.1016/j.tibs.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 32.Wang QE, Han C, Zhang B, Sabapathy K, Wani AA. Nucleotide Excision Repair Factor XPC Enhances DNA Damage-Induced Apoptosis by Downregulating the Antiapoptotic Short Isoform of Caspase-2. Cancer Research. 2012 doi: 10.1158/0008-5472.CAN-11-2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trivedi RN, Almeida KH, Fornsaglio JL, Schamus S, Sobol RW. The Role of Base Excision Repair in the Sensitivity and Resistance to Temozolomide Mediated Cell Death. Cancer Research. 2005;65:6394–6400. doi: 10.1158/0008-5472.CAN-05-0715. [DOI] [PubMed] [Google Scholar]
- 34.Almeida KH, Sobol RW. A unified view of base excision repair: lesion-dependent protein complexes regulated by post-translational modification. DNA Repair. 2007;6:695–711. doi: 10.1016/j.dnarep.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kidane D, Jonason AS, Gorton TS, Mihaylov I, Pan J, Keeney S, de Rooij DG, Ashley T, Keh A, Liu Y, Banerjee U, Zelterman D, Sweasy JB. DNA polymerase beta is critical for mouse meiotic synapsis. Embo J. 2010;29:410–423. doi: 10.1038/emboj.2009.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Starcevic D, Dalal S, Sweasy JB. Is there a link between DNA polymerase beta and cancer? Cell Cycle. 2004;3:998–1001. [PubMed] [Google Scholar]
- 37.Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Requirement of mammalian DNA polymerase-ß in base-excision repair. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 38.Sweasy JB, Lang T, DiMaio D. Is base excision repair a tumor suppressor mechanism? Cell Cycle. 2006;5:250–259. doi: 10.4161/cc.5.3.2414. [DOI] [PubMed] [Google Scholar]
- 39.Sweasy JB, Lauper JM, Eckert KA. DNA polymerases and human diseases. Radiat Res. 2006;166:693–714. doi: 10.1667/RR0706.1. [DOI] [PubMed] [Google Scholar]
- 40.Donigan KA, Hile SE, Eckert KA, Sweasy JB. The human gastric cancer-associated DNA polymerase beta variant D160N is a mutator that induces cellular transformation. DNA Repair (Amst) 2012;11:381–390. doi: 10.1016/j.dnarep.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Donigan KA, Sun KW, Nemec AA, Murphy DL, Cong X, Northrup V, Zelterman D, Sweasy JB. Human POLB Gene Is Mutated in High Percentage of Colorectal Tumors. Journal of Biological Chemistry. 2012;287:23830–23839. doi: 10.1074/jbc.M111.324947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Murphy DL, Donigan KA, Jaeger J, Sweasy JB. The E288K Colon Tumor Variant of DNA Polymerase beta Is a Sequence Specific Mutator. Biochemistry. 2012 doi: 10.1021/bi3003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nemec AA, Donigan KA, Murphy DL, Jaeger J, Sweasy JB. Colon Cancer-associated DNA Polymerase beta Variant Induces Genomic Instability and Cellular Transformation. Journal of Biological Chemistry. 2012;287:23840–23849. doi: 10.1074/jbc.M112.362111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lang T, Dalal S, Chikova A, Dimaio D, Sweasy JB. The E295K DNA polymerase beta gastric cancer-associated variant interferes with base excision repair and induces cellular transformation. Mol Cell Biol. 2007;27:5587–5596. doi: 10.1128/MCB.01883-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iwanaga A, Ouchida M, Miyazaki K, Hori K, Mukai T. Functional mutation of DNA polymerase ß found in human gastric cancer: inability of the base excision repair in vitro. Mutation Research. 1999;435:121–128. doi: 10.1016/s0921-8777(99)00036-1. [DOI] [PubMed] [Google Scholar]
- 46.Horton JK, Joyce-Gray DF, Pachkowski BF, Swenberg JA, Wilson SH. Hypersensitivity of DNA polymerase beta null mouse fibroblasts reflects accumulation of cytotoxic repair intermediates from site-specific alkyl DNA lesions. DNA Repair (Amst) 2003;2:27–48. doi: 10.1016/s1568-7864(02)00184-2. [DOI] [PubMed] [Google Scholar]
- 47.Sobol RW, Prasad R, Evenski A, Baker A, Yang XP, Horton JK, Wilson SH. The lyase activity of the DNA repair protein ß-polymerase protects from DNA-damage-induced cytotoxicity. Nature. 2000;405:807–810. doi: 10.1038/35015598. [DOI] [PubMed] [Google Scholar]
- 48.Sugo N, Aratani Y, Nagashima Y, Kubota Y, Koyama H. Neonatal lethality with abnormal neurogenesis in mice deficient in DNA polymerase ß. EMBO Journal. 2000;19:1397–1404. doi: 10.1093/emboj/19.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase ß gene segment in T cells using cell type-specific gene targeting. Science. 1994;265:103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- 50.Horton JK, Baker A, Berg BJ, Sobol RW, Wilson SH. Involvement of DNA polymerase ß in protection against the cytotoxicity of oxidative DNA damage. DNA Repair (Amst) 2002;1:317–333. doi: 10.1016/s1568-7864(02)00008-3. [DOI] [PubMed] [Google Scholar]
- 51.Jelezcova E, Trivedi RN, Wang XH, Tang JB, Brown AR, Goellner EM, Schamus S, Fornsaglio JL, Sobol RW. Parp1 activation in mouse embryonic fibroblasts promotes Pol beta-dependent cellular hypersensitivity to alkylation damage. Mutat Res. 2010;686:57–67. doi: 10.1016/j.mrfmmm.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sobol RW. DNA polymerase ß null mouse embryonic fibroblasts harbor a homozygous null mutation in DNA polymerase iota. DNA Repair (Amst) 2007;6:3–7. doi: 10.1016/j.dnarep.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ruddon RW, Anderson C, Meade KS, Aldenderfer PH, Neuwald PD. Content of gonadotropins in cultured human malignant cells and effects of sodium butyrate treatment on gonadotropin secretion by HeLa cells. Cancer Research. 1979;39:3885–3892. [PubMed] [Google Scholar]
- 54.Giard DJ, Aaronson SA, Todaro GJ, Arnstein P, Kersey JH, Dosik H, Parks WP. In vitro cultivation of human tumors: establishment of cell lines derived from a series of solid tumors. Journal of the National Cancer Institute. 1973;51:1417–1423. doi: 10.1093/jnci/51.5.1417. [DOI] [PubMed] [Google Scholar]
- 55.Poeschla EM, Wong-Staal F, Looney DJ. Efficient transduction of nondividing human cells by feline immunodeficiency virus lentiviral vectors. Nature Medicine. 1998;4:354–357. doi: 10.1038/nm0398-354. [DOI] [PubMed] [Google Scholar]
- 56.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B-Methodological. 1995;57:289–300. [Google Scholar]
- 57.Meng Q, Xia Y. c-Jun, at the crossroad of the signaling network. Protein Cell. 2011;2:889–898. doi: 10.1007/s13238-011-1113-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiao X, Katiyar S, Willmarth NE, Liu M, Ma X, Flomenberg N, Lisanti MP, Pestell RG. c-Jun induces mammary epithelial cellular invasion and breast cancer stem cell expansion. Journal of Biological Chemistry. 2010;285:8218–8226. doi: 10.1074/jbc.M110.100792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Webb DK, Moulton BC, Khan SA. Estrogen induces expression of c-jun and jun-B protooncogenes in specific rat uterine cells. Endocrinology. 1993;133:20–28. doi: 10.1210/endo.133.1.8319568. [DOI] [PubMed] [Google Scholar]
- 60.Zhang F, Ram JL, Standley PR, Sowers JR. 17 beta-Estradiol attenuates voltage-dependent Ca2+ currents in A7r5 vascular smooth muscle cell line. Am J Physiol. 1994;266:C975–980. doi: 10.1152/ajpcell.1994.266.4.C975. [DOI] [PubMed] [Google Scholar]
- 61.Thota C, Gangula PR, Dong YL, Yallampalli C. Changes in the expression of calcitonin receptor-like receptor, receptor activity-modifying protein (RAMP) 1, RAMP2, and RAMP3 in rat uterus during pregnancy, labor, and by steroid hormone treatments. Biol Reprod. 2003;69:1432–1437. doi: 10.1095/biolreprod.103.015628. [DOI] [PubMed] [Google Scholar]
- 62.Sobol RW, Kartalou M, Almeida KH, Joyce DF, Engelward BP, Horton JK, Prasad R, Samson LD, Wilson SH. Base Excision Repair Intermediates Induce p53-independent Cytotoxic and Genotoxic Responses. Journal of Biological Chemistry. 2003;278:39951–39959. doi: 10.1074/jbc.M306592200. [DOI] [PubMed] [Google Scholar]
- 63.Hajkova P, Jeffries SJ, Lee C, Miller N, Jackson SP, Surani MA. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science. 2010;329:78–82. doi: 10.1126/science.1187945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, Le Coz M, Devarajan K, Wessels A, Soprano D, Abramowitz LK, Bartolomei MS, Rambow F, Bassi MR, Bruno T, Fanciulli M, Renner C, Klein-Szanto AJ, Matsumoto Y, Kobi D, Davidson I, Alberti C, Larue L, Bellacosa A. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xin H, D’Souza S, Jorgensen TN, Vaughan AT, Lengyel P, Kotzin BL, Choubey D. Increased expression of Ifi202, an IFN-activatable gene, in B6. Nba2 lupus susceptible mice inhibits p53-mediated apoptosis. J Immunol. 2006;176:5863–5870. doi: 10.4049/jimmunol.176.10.5863. [DOI] [PubMed] [Google Scholar]
- 66.Halvorsen OJ, Rostad K, Oyan AM, Puntervoll H, Bo TH, Stordrange L, Olsen S, Haukaas SA, Hood L, Jonassen I, Kalland KH, Akslen LA. Increased expression of SIM2-s protein is a novel marker of aggressive prostate cancer. Clin Cancer Res. 2007;13:892–897. doi: 10.1158/1078-0432.CCR-06-1207. [DOI] [PubMed] [Google Scholar]
- 67.Okobia MN, Bunker CH. Molecular epidemiology of breast cancer: a review. Afr J Reprod Health. 2003;7:17–28. [PubMed] [Google Scholar]
- 68.Dubrovska A, Hartung A, Bouchez LC, Walker JR, Reddy VA, Cho CY, Schultz PG. CXCR4 activation maintains a stem cell population in tamoxifen-resistant breast cancer cells through AhR signalling. British Journal of Cancer. 2012;107:43–52. doi: 10.1038/bjc.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brekhman V, Lugassie J, Zaffryar-Eilot S, Sabo E, Kessler O, Smith V, Golding H, Neufeld G. Receptor activity modifying protein-3 mediates the protumorigenic activity of lysyl oxidase-like protein-2. Faseb J. 2011;25:55–65. doi: 10.1096/fj.10-162677. [DOI] [PubMed] [Google Scholar]
- 70.Liu Z, Xiao XJ, Fan FY, Sun YM, Li YM, Yang FJ. [Discovery of a new splicing type of KCHIP1 gene] . Ai Zheng. 2005;24:755–756. [PubMed] [Google Scholar]
- 71.Reinhold WC, Sunshine M, Liu H, Varma S, Kohn KW, Morris J, Doroshow J, Pommier Y. CellMiner: a web-based suite of genomic and pharmacologic tools to explore transcript and drug patterns in the NCI-60 cell line set. Cancer Research. 2012;72:3499–3511. doi: 10.1158/0008-5472.CAN-12-1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hauge H, Patzke S, Aasheim HC. Characterization of the FAM110 gene family. Genomics. 2007;90:14–27. doi: 10.1016/j.ygeno.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 73.Hauge H, Fjelland KE, Sioud M, Aasheim HC. Evidence for the involvement of FAM110C protein in cell spreading and migration. Cell Signal. 2009;21:1866–1873. doi: 10.1016/j.cellsig.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 74.Xu Y, Morse LR, da Silva RA, Odgren PR, Sasaki H, Stashenko P, Battaglino RA. PAMM: a redox regulatory protein that modulates osteoclast differentiation. Antioxid Redox Signal. 2010;13:27–37. doi: 10.1089/ars.2009.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.