

Abstract
Objectives
Macrophage endothelial lipase (EL) is associated with increased atherosclerosis and inflammation. Because of their anti-inflammatory properties we hypothesized that n-3 fatty acids (FA), in contrast to saturated FA, would lower macrophages and arterial EL and inflammatory markers.
Methods and Results
Murine J774 and peritoneal macrophages were incubated with eicosapentaenoic acid (EPA) or palmitic acid (PA) in the presence or absence of lipopolysaccaride (LPS). LPS increased EL mRNA and protein. PA alone or with LPS dose-dependently increased EL mRNA and protein. In contrast, EPA dose-dependently abrogated effects of LPS or PA on increasing EL expression. EL expression closely linked to PPARγ expression. EPA blocked rosiglitazone (a PPARγ agonist)-mediated EL activation and GW9662 (a PPARγ antagonist) blocked PA-mediated EL stimulation. EPA alone or with LPS blunted LPS-mediated stimulation of macrophage pro-inflammatory IL-6, IL-12p40, TLR4 mRNA and increased anti-inflammatory IL-10 and mannose receptor mRNA. In vivo studies in LDL receptor knockout mice showed that high saturated fat rich diets, but not n-3 diets, increased arterial EL, PPARγ and pro-inflammatory cytokine mRNA.
Conclusions
n-3 FA, in contrast to saturated FA, decrease EL in parallel with modulating pro- and anti-inflammatory markers, and these effects on EL link to PPARγ.
Keywords: n-3 fatty acids, endothelial lipase, inflammation, PPARγ, atherosclerosis
Inflammation adversely affects arterial wall biology.1,2 Much evidence supports a pro-atherogenic and pro-inflammatory effect of saturated fatty acids (FA).3,4 In contrast, protective actions with respect to the arterial wall have been attributed to n-3 FA.5-7 n-3 FA delivered from dietary fish oil are incorporated into atherosclerotic plaques, enhancing stability, whereas n-6 FA do not have these effects.5 Recent reviews indicate that increased consumption of long-chain n-3 FA, eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), but not of α-linolenic acid (their n-3 essential fatty acid precursor), reduced the rates of all-cause mortality, and cardiac and sudden death.6 n-3 FA have been shown to reduce the macrophage infiltration into the vessel wall and secretion of pro-atherogenic and pro-inflammatory growth factors and cytokines by monocytes and macrophages.7
Macrophages play a pivotal role in the development and progression of atherosclerosis. Endothelial lipase (EL) is one of several lipases synthesized and secreted by macrophages. High levels of EL expression have been observed in macrophages present in human atherosclerotic plaques.8 EL deficiency is associated with a 70% decrease in atherosclerotic lesions in apoE KO mice.9 A pro-atherogenic effect of EL in macrophages has also been ascribed to bridging functions, which plays a role in the uptake of lipoproteins10 or recruitment of monocytes by blood vessel walls.11 Moreover, EL expression is upregulated in macrophages by pro-inflammatory cytokines.10 Upregulation of macrophage EL by toll-like receptor (TLR) 4 and 3 negatively modulates IL-10 and positively modulates IL-12 production, potentially influencing atherosclerosis.12 Accordingly, EL might be considered to be an attractive pharmacological target in the prevention of atherosclerosis.
Peroxisome proliferator activated receptor (PPAR)γ also has been implicated in atherogenesis. PPARγ is highly expressed in macrophages-derived foam cells in atherosclerotic lesions13 and activation of PPARγ has been shown to induce macrophage lipid accumulation by increasing the expression of the oxidized LDL scavenger receptor CD36 and lipoprotein lipase (LpL).13-15 In contrast to the proposed potentially pro-atherogenic effects of PPARγ, other limited evidence suggests PPARγ may mediate anti-inflammatory effects by negatively regulating pro-inflammatory cytokine expression.16 Also, macrophage PPARγ deficiency increases atherosclerosis in C57BL/6 and LDL receptor knockout (LDL-R KO) mice,17 indicating an anti-atherogenic role for PPARγ. However, at present, no definitive studies support the premise that PPARγ is required for anti-inflammatory effects in macrophages.18-22 In fact, recent clinical trials have raised concerns on increased risk of myocardial infarction and cardiovascular death in diabetic patients treated with rosiglitazone,23, 24a strong PPARγ agonist.
We previously demonstrated that high-saturated fat (SAT) diets increased contributions of LDL selective uptake (SU) to total arterial LDL-CE deposition and that increased SU parallels increased LpL levels and distribution in the arterial wall.25 In contrast, n-3 rich diets decreased arterial total LDL delivery and abrogated LDL SU in parallel with changing arterial wall LpL.26 We now questioned whether FA would regulate EL expression since different dietary FA modulated arterial LpL levels and distribution as described in our previous reports.25-27 Specifically, we asked whether n-3 FA, EPA, in contrast to a saturated FA, palmitic acid (PA), would decrease expression of EL and, if so, would these changes correlate with changes in inflammatory markers and in PPARs which may also modulate lipid metabolism and inflammatory responses.13,14,18
Our results demonstrate that PA increases EL expression and decreases anti-inflammatory IL-10 expression in macrophages. In contrast, EPA decreases EL expression, in parallel with decreasing pro-inflammatory markers and increasing anti-inflammatory markers. The changes in macrophage EL by FA were strongly related to the regulation of PPARγ. Moreover, LDL-R KO mice fed SAT diets, but not n-3 diets, showed the increases in EL, PPARγ and pro-inflammatory responses in the arterial wall.
Methods
Some methods are described in more detail in online Supplemental Materials and Methods (available at http://atvb.ahajournals.org).
Cell Culture
Murine macrophage-like cells, J774 (A2), were grown in Dulbecco’s modified Eagle’s medium containing 10% FBS (v/v), 1% glutamine (v/v) and 1% penicillin/streptomycin (v/v). Thioglycollate-elicited macrophages were obtained from C57BL/6 mice (12-14 weeks old) by peritoneal lavage with PBS at 4 days after injection of 1 mL of 3.8% thioglycollate broth (Sigma-Aldrich). Cells were suspended in RPMI 1640 supplemented with 10% FBS (v/v), 1% glutamine (v/v) and 1% penicillin/streptomycin (v/v), and incubated at 37°C for 3 h. For all experiments, the cells were washed twice with phosphate-buffered saline (PBS) and the FA-containing media were added at different doses for 3h, while the control cells received only BSA medium. Then, cells were incubated with LPS (1 μg/mL), PPARγ agonist or antagonist for 4h (for mRNA) or 21h (for protein).
For experiments requiring PPARγ mRNA knock-down, J774 cells (50-70% confluence) were transfected with optimized concentrations of either mouse PPARγ short hairpin RNA (shRNA) plasmid (sc-29456-sh, Santa Cruz Biotechnology, Inc.), or control nonsense shRNA plasmid (sc-108066) using shRNA transfection reagent (sc-29528), according to the manufacturer’s instructions. Twenty four hours after transfection, cells were treated with specific FA and processed for real-time PCR analyses as described below.
Quantitative real-time PCR
Total RNA was isolated with TRIzol reagent (Invitrogen) and quantitative real-time PCR was carried out on an iCycler real-time machine (BioRad) using the SYBR® Green PCR master kit (Applied Biosystems). Values were normalized to GAPDH levels.
Western Blot
EL protein expression was analyzed by western blot normalized to β-actin protein.
Animals and Diets
Eight-weeks-old male LDL-R KO mice were purchased from Jackson Laboratory. After 1 week acclimatization, mice were fed a semipurified, normal chow (total 5% fat, 0.02% cholesterol, w/w) or a high-fat, semipurified diet (total 19% fat, 0.2% cholesterol, w/w) enriched in either n-3 (91% menhaden fish oil and 9% corn oil; Harlan Teklad; TD. 07500) or saturated fat (SAT; 78% saturated fat from coconut oil, 13% monounsaturated fat from olive oil, and 9% polyunsaturated fat from corn oil; Harlan Teklad; TD. 08081) for 12 weeks similar to our previous report.26 The aorta was dissected and measured for mRNA of EL, PPARγ, and specific inflammatory markers. All procedures were approved by the Institutional Animal Care and Use Committee of Columbia University.
Statistical Analyses
Statistical analyses were determined by one-way ANOVA (for comparing FA), 2-tailed Student’s t-test, or Pearson correlation coefficients. Data are expressed as mean ± SE.
Results
LPS increases EL expression in macrophages
We first examined the effect of LPS, a potent endotoxin, on EL expression in macrophages. J774 cells were incubated with increasing concentration of LPS (0.25-10 μg/mL). LPS increased EL mRNA by a maximum of 2.5 fold at 1 μg/mL (Supplemental Figure 1A). Similarly, LPS dose-dependently increased protein levels of EL with a maximum increase also at 1 μg/mL (Supplemental Figure 1B). We thus chose concentration of LPS of 1 μg /mL for the experiments below.
EPA inhibits the increase of EL mRNA and protein induced by LPS more than other FA
To determine the effects of different FA on macrophage EL mRNA expression, J774 cells were cultured in the presence or absence of LPS, 1 μg/mL, for 4h after preincubation with 150 μM of unsaturated FA (eicosapentaenoic acid, EPA; arachidonic acid, AA; linoleic acid, LA), monounsaturated FA (oleic acid, OA) and saturated FA (palmitic acid, PA) for 3h. Exposure of cells to LA and PA alone significantly increased macrophage EL mRNA levels compared to non-FA control by 2.2 fold and 2.6 fold, respectively (Figure 1A). PA together with LPS induced 1.4 fold higher levels of EL mRNA compared to LPS alone. In contrast, EPA, AA and OA inhibited LPS-induced EL mRNA levels by 64%, 37% and 39%, respectively. Among FA tested, PA induced the strongest response on increasing EL expression, whereas EPA was most potent at inhibiting EL expression induced by LPS.
FIGURE 1. Effects of different FA on EL expression in macrophages.
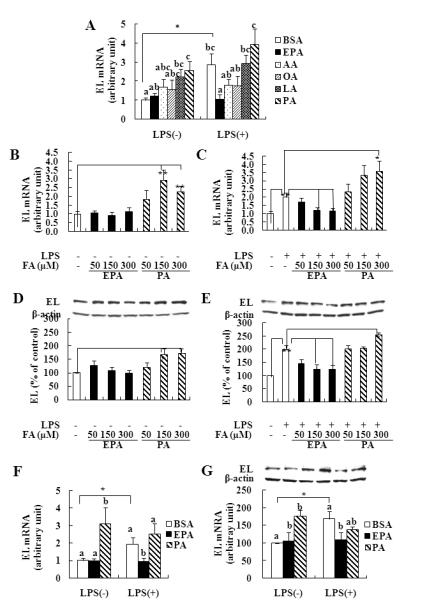
J774 (A-E) or peritoneal macrophages (F, G) were cultured in the presence or absence of LPS, 1 μg/mL, for 4h (mRNA, A-C, F) or 21h (protein, D, E, G) after preincubation with 150 μM of FA for 3h. abcMeans with unlike letters are significantly different at p<0.05 (one-way ANOVA). *p<0.05, **p<0.01 (student’s t-test).
Since EPA and PA appeared to show the greatest differences on EL mRNA expression, we chose EPA and PA for dose-response experiments. J774 cells were cultured in the presence or absence of LPS, 1 μg/mL, for 4 to 21h after preincubation with 50, 150, 300 μM of EPA or PA for 3h. PA alone as well as together with LPS dose-dependently increased EL mRNA (Figure 1B and 1C). In contrast, EPA alone had no effect on EL mRNA, but completely abrogated effects of LPS on increasing EL mRNA. Consistent with mRNA expression, EPA had no significant effect on EL protein expression but inhibited LPS-mediated increase of EL protein (Figure 1D and 1E). PA alone as well as with LPS increased EL protein levels in a dose-dependent manner.
PA and LPS have similar effects on EL expression in peritoneal macrophages as in J774 cells and EPA inhibits LPS-mediated increase of EL expression
To determine whether EPA and PA also regulate EL expression in primary macrophages, we treated EPA and PA with or without LPS in peritoneal macrophages from C57BL/6 mice. Similar to J774 cells, LPS and PA alone significantly increased EL mRNA compared to control by 2.0 and 3.0 fold, respectively, whereas EPA significantly inhibited LPS-induced EL mRNA by ~30% (Figure 1F). EL protein levels were also increased by LPS and PA, but EPA significantly abrogated effects of LPS on increasing EL protein (Figure 1G). Thus, EPA can mitigate the effects of PA-mediated increases of EL in different macrophage lines.
EPA inhibits PA-mediated increases of EL
To determine effects of EPA together with PA on macrophage EL, we incubated cells with different ratios of EPA and PA (Figure 2). EL mRNA was lower in J774 cells treated with the combinations of PA plus EPA compared to the group treated only with PA; these differences became significant when EPA accounted for 1/3 or more of total FA (Figure 2A). EL protein levels showed similar responses to incubations of PA vs PA and EPA (Figure 2B).
FIGURE 2. Effects of different ratios of EPA and PA on EL.

J774 macrophages were incubated with the indicated concentrations of EPA and PA for 7h (mRNA, A) or 24h (protein, B). *p<0.05, **p<0.01 (student’s t-test).
EPA and PA have different effects on regulation of PPARγ expression
Because PPARs are linked to a number of processes important to atherogenesis,14,28 we examined potential associations of PPARγ and PPARα with EL expression. PPARγ expression was much more affected by FA than PPARα expression. In J774 cells, but not peritoneal macrophages, EPA alone or with LPS tended to increase PPARα mRNA whereas PA had no effect (data not shown). In contrast, PA alone as well as with LPS significantly increased PPARγ mRNA expression (Figure 3A and B). In addition, PPARγ mRNA positively correlated with EL mRNA in both macrophages, respectively (r=0.34, p<0.05; r=0.43, p<0.01).
FIGURE 3. Effects of FA on macrophage PPARγ mRNA expression.

J774 (A) or peritoneal macrophages (B) were incubated with 150 μM of EPA or PA as previously described in Figure 1. abMeans with unlike letters are significantly different at p<0.05 (one-way ANOVA).
In order to determine how changes in PPARγ might affect macrophage EL expression, we treated cells with increasing concentrations of rosiglitazone and/or GW9662 in the presence or absence of EPA or PA. Rosiglitazone is a known PPARγ agonist and GW9662 is a PPARγ antagonist. Rosiglitazone (0.1~50 μM) dose-dependently increased EL protein and mRNA, whereas GW9662 markedly blocked rosiglitazone-mediated increases of EL protein and mRNA in J774 cells (Supplemental Figure 2, and Figure 4A and 4B). Moreover, GW9662 significantly inhibited PA-induced EL mRNA and protein. Of note, EPA abrogated rosiglitazone-mediated increases of EL mRNA and protein in J774 cells (Figure 4C). PPARγ mRNA expression was dose-dependently increased by rosiglitazone, whereas GW9662 and EPA abrogate effects of rosiglitazone on increasing PPARγ mRNA in J774 cells (Supplemental Figure 3). We also found that EPA inhibited the increase of mRNA levels of LpL and CD36, well known PPARγ target genes,15,16 after incubation with rosiglitazone (Supplemental Figure 4).
FIGURE 4. Interactions of EPA and PA with a PPARγ agonist/antagonist on EL expression in macrophages.

J774 (A-C) or peritoneal macrophages (D-E) were pretreated with or without FA for 3h and then continuously cultured with PPARγ agonist rosiglitazone (ROSI) and/or PPARγ antagonist GW9662 for 4h (mRNA) or 21h (protein). *p<0.05, **p<0.01 (student’s t-test).
To determine whether the PPARγ agonist and antagonist also regulate EL expression in primary macrophages, we treated peritoneal macrophage with rosiglitazone and GW9662 in the presence of FA. Similar to J774 cells, rosiglitazone increased EL mRNA and protein by 70% and 60%, respectively, and EPA significantly blocked rosiglitazone-induced EL mRNA expression by 86% (Figure 4D). Also, GW9662 markedly blocked the PA and rosiglitazone-mediated increases of EL mRNA by 96% and 78%, respectively, and inhibited the increase in EL protein induced by PA and rosiglitazone (Figure 4D and 4E). Thus, EL in cultured cells in vitro closely linked to PPARγ and was regulated by FA, in part, through modifying macrophage PPARγ expression.
To further support the above association between PPARγ and EL expression we carried out experiments where we used shRNA mediated knock-down of PPARγ expression in J774 cells. We achieved a knock-down efficiency of >80% using PPARγ shRNA as compared to control shRNA controls (n=6, p<0.05). We then compared the effects of PA on increasing EL mRNA in these PPARγ knock-down J774 cells and found a mean 63% decrease in EL mRNA expression after incubation with PA compared to control levels (n=6, p=0.04). Thus, decreases in PPARγ blunt the ability of PA to increase EL expression in macrophages, indicating that EL is modulated, in part, by PPARγ-dependent pathways.
EPA decreases pro-inflammatory markers, but increases anti-inflammatory markers in peritoneal macrophages
LPS is a bacterial endotoxin that is commonly used to stimulate inflammatory responses. Wang et al12 reported that induction of macrophage EL by LPS can modulate macrophage inflammatory responses. To explore the relationships of EL and inflammatory markers, we compared well defined pro-and anti-inflammatory markers in peritoneal macrophages incubated with EPA and PA. LPS significantly increased pro-inflammatory cytokines, IL-6 and IL-12p40 (Figure 5A). However, EPA alone or with LPS blunted the stimulating effects of LPS on IL-6 and IL-12p40 mRNA by 62% and 60%, respectively. Also, EPA markedly attenuated TLR4 mRNA. We also found that in J774 cells, LPS increased IL-6 and IL-12p40 mRNA by 17- and 12-fold, respectively (p<0.001, p<0.001) and that these effects were diminished by EPA (data not shown). PA and EPA effects on TNF-α expression were not similar to other pro-inflammatory markers; PA alone increased TNF-α mRNA level similar to LPS (LPS vs BSA, 56%; PA vs BSA, 55%), whereas EPA alone or with LPS had no effect. In contrast, anti-inflammatory IL-10 and mannose receptor (MR) were increased in EPA-treated cells by 2.1 and 1.5 fold, respectively (Figure 5B). PA had little effect on pro-inflammatory markers but decreased IL-10 mRNA (Figure 5A and B). Interestingly, EL mRNA showed positive correlations with increasing mRNA levels of pro-inflammatory markers such as IL-6, IL-12p40, TLR4 and vascular cell adhesion molecule-1 (VCAM-1) (Supplemental Figure 5). In contrast, there were negative correlations between EL and anti-inflammatory markers, IL-10 and MR, respectively. PPARγ mRNA was also positively correlated with pro-inflammatory IL-6, IL-12p40, TLR4 and VCAM-1 mRNA, whereas it was negatively correlated with anti-inflammatory IL-10 mRNA (Supplemental Figure 5). There were no significant correlations between PPARα and inflammatory markers (data not shown).
FIGURE 5. Effects of FA on pro- (A) and anti-inflammatory markers (B) mRNA expression in murine peritoneal macrophages.

Cells were incubated with 150 μM of EPA or PA as previously described in Figure 1. abMeans with unlike letters are significantly different (p<0.05). *p<0.05 (student’s t-test). MR; mannose receptor.
Dietary saturated vs n-3 diet changes arterial EL, PPARγ and inflammatory markers expression in LDL-R KO mice in vivo
We next investigated the potential effects of 12-week feeding of n-3-rich and SAT-rich diets on arterial EL, PPARγ and inflammatory gene expression in aorta of LDL-R KO mice which are susceptible to atherosclerosis. SAT diets led to 10-fold greater arterial EL mRNA compared to chow (Figure 6A). There was no significant difference in arterial EL mRNA levels between n-3 and chow diets. SAT diets were associated with markedly increased arterial PPARγ mRNA compared to chow diets, whereas the n-3 diets showed a 26% decrease in PPARγ mRNA (Figure 6B). Similar to in vitro data in macrophages, SAT diets increased arterial IL-6 and IL-12p40 mRNA 2.6-fold and 5.8-fold compared to chow, respectively, whereas arterial IL-10 mRNA was lowered in SAT-fed mice compared to chow-fed mice by 22% (Figure 6C-6E). In contrast, n-3 diets reduced both pro-inflammatory cytokine mRNA levels in aorta of LDL-R KO mice compared to chow by 74% and 50%, respectively, but increased IL-10 mRNA compared to SAT diet by 69%. Thus, in vivo effects of diets rich in SAT vs n-3 FA on arterial expression of EL and inflammatory markers paralleled effects observed in cultured macrophages in vitro.
FIGURE 6. Effects of n-3- and saturated FA-rich diets on arterial EL (A), PPARγ (B) and inflammatory markers (C-E) mRNA expression in LDL-R KO mice.

abMeans with unlike letters are significantly different at p<0.05 (one-way ANOVA).
Discussion
Macrophage-derived EL in the arterial wall is associated with increased atherosclerosis and arterial inflammatory markers in mice.9,10,12 Because of their anti-inflammatory properties we hypothesized that n-3 FA, in contrast to saturated FA, would lower expression of EL in vitro and in vivo. Our results show that a saturated FA, PA, increases macrophage EL expression and decreases anti-inflammatory IL-10 expression. In contrast, an n-3 FA, EPA, inhibits the increase of EL expression induced by LPS and PA, and this is accompanied by decreases in pro-inflammatory markers and increases in anti-inflammatory markers in cultured macrophages. Moreover, regulation of macrophage EL in response to FA is closely linked to changes in PPARγ activation. In vivo studies also show that SAT diets, but not n-3 diets, increase EL, PPARγ and pro-inflammatory cytokine expression, but decrease anti-inflammatory cytokine mRNA in aorta of LDL-R KO mice, suggesting that changes in EL by FA have important regulatory roles on atherosclerosis and inflammation in vivo as well as in vitro.
LPS, a major inflammatory stimulus, can play an important role in lipoprotein metabolism and atherosclerosis.29 Yasuda et al10 reported that EL expression was increased by LPS. In our current study, LPS also markedly increased EL protein as well as mRNA expression in J774 and peritoneal macrophages. Interestingly, saturated PA alone and/or with LPS increased EL mRNA and protein levels in both macrophages. In contrast, n-3 EPA had little effects on EL mRNA and protein, but markedly inhibited the increase of macrophage EL expression induced by LPS. Furthermore, EL expression was significantly lower in cells treated with a combination of PA plus EPA compared to PA alone. Based on these results, EPA suppresses the increase of macrophage EL expression induced by EL activators, PA and LPS.
PPARγ is a nuclear transcription factor that regulates numerous genes involved in lipoprotein metabolism and is highly expressed in macrophages, including foam cells of atherosclerotic lesions.14 PPARγ activation increased macrophage LpL mRNA and protein expression,15 and promoted uptake of oxidized LDL through induction of macrophage CD36 expression,13,14 which suggest the potential role of PPARγ in the pathogenesis of atherosclerosis. Furthermore, the PPARγ agonists, rosiglitazone and pioglitazone, enhance macrophage apoptosis via a PPARγ-independent mechanism, and pioglitazone promotes advanced plaque progression in LDL-R KO mice through enhancement of advanced lesion macrophage apoptosis.30 Our data indicate that EL might also contribute to atherogenesis, and that PPARγ activation is associated with this regulation of EL.
In the current study, PPARγ activation using rosiglitazone as well as PA was related to increased EL mRNA and protein, and GW9662, a PPARγ antagonist, EPA and PPARγ-knock-down cells abrogated stimulation by rosiglitazone or PA. Rosiglitazone significantly increased PPARγ mRNA in a dose-dependent manner, whereas pioglitazone, a ~10 times less potent an activator of PPARγ than rosiglitazone,31 had little effects on PPARγ as well as on EL, in our experiments in macrophages (data not shown). Also, GW9662 and EPA blocked rosiglitazone-induced PPARγ mRNA expression. Similar to our results with EPA, n-3 FA-DHA, suppressed CD36 expression induced by PPARγ agonist through the inhibition of transcriptional activity of PPARγ in human monocytes and colon tumor cells, 32 and EPA and DHA reduced the PPARγ response element (called PPRE) reporter activity in colon cancer cells. Edwards et al33 proposed that n-3 FA may directly, or after being metabolized, activates ERK or other pathways that counteracts PPARγ signaling. On the other hand, PA significantly increased PPARγ mRNA or protein expression in several cell types such as cardiomyocytes,34,35 and a high fat diet enriched in PA enhanced PPARγ expression in macrophages.36 PA also stimulated the activity of the PPRE in primary human adipocytes, suggesting activation of this nuclear signaling cascade.37 Taken together, our findings suggest that macrophage EL expression is partially mediated by the upregulation of PPARγ, and that saturated vs n-3 FA affect the expression of EL, at least in part, by regulating PPARγ. It is possible that the transcriptional induction of EL gene might be mediated through binding PPAR-RXR heterodimer, since it is reported that CD36 and the LpL promoter is a direct target of PPAR-RXR heterodimer.14,38 Others have also shown different effects of saturated vs n-3 FA on PPARγ signaling and these differences are also related to the specific tissue or cell analyzed.32-37 To clearly understand whether regulation of macrophage EL is most likely mediated through PPARγ-dependent mechanism, further experiments using ligand binding assays are needed to be performed.
A PPARγ-related mechanism for regulation of arterial wall EL by FA is supported by our in vivo findings. SAT diets, but not n-3 diets, increased arterial EL and PPARγ mRNA in LDL-R KO mice, and EL was positively correlated with PPARγ (p<0.01). Ishida et al9 reported that EL protein was increased in aorta from apoE KO mice and this was accentuated by a high fat diet (0.15% cholesterol, 21% milk fat)10. Also, there was a decrease in atherosclerotic lesions in animals lacking both EL and apoE compared with apoE KO alone.10 Moreover, EL protein39 and PPARγ mRNA40 were increased in the aorta, especially the atherosclerotic lesions in high-cholesterol diet fed animals. Herein we show that n-3 diets do not share the stimulatory effects of SAT diets in vivo.
We also found that, in parallel with the changes in macrophage EL and PPARγ, EPA alone or plus LPS reduced IL-6 and IL-12p40 mRNA in macrophages but increased the mRNA of IL-10 and MR which stimulates anti-inflammatory cytokines production including IL-10.41 Pro-inflammatory cytokines such as IL-6 and IL-12 promote the development of atherosclerotic lesions,42,43 whereas anti-inflammatory IL-10 have anti-atherogenic effects.44 EL as well as PPARγ positively correlated with pro-inflammatory markers but negatively correlated with anti-inflammatory markers. Moreover, similar changes in arterial inflammatory markers were found in LDL-R KO mice, suggesting that FA-regulated inflammatory responses could also occur in vivo. In contrast to other inflammatory cytokines mRNA expression, there was no significant effect of EPA on TNF-α mRNA expression in macrophages. Preliminary data (not shown) on LDL-R KO mice also show that feeding of n-3 diets for 12 weeks did not affect TNF-α mRNA expression in aorta compared to chow or SAT diets. Renier et al.45 reported that macrophages derived from mice fed n-3 diets showed a significant decrease in TNF-α mRNA after 15 weeks, but not 6 weeks, suggesting that the maximum effect of n-3 FA might require a relatively longer observation period than that used in our study.
Several studies have consistently reported the pro-inflammatory effect of EL in macrophages.12,46 On the other hand, there have been contradictory reports on the potency of PPARγ activation on macrophage inflammatory responses. PPARγ agonists reduced pro-inflammatory cytokine production, including TNF-α and IL-6, in human monocytes,16 whereas Thieringer et al19 failed to obtain an inhibitory effect of PPARγ agonists on TNF-α and IL-6 production in human monocytes or macrophages. Furthermore, PPARγ activation appeared to increase plasma cytokine levels in mice after LPS administration.19 Consistent with this finding, rosiglitazone increased LPS-induced TNF-α production in rat peritoneal macrophages.20 In fact, a PPARγ activator exerted anti-inflammatory effects in a PPARγ-independent mechanism via inhibition of NF-κB-dependent transcription,21 and PPAR agonists inhibited cytokine production in PPARγ-deficient macrophages,22 indicating that anti-inflammatory effects may also be mediated by other biological pathways.
Another possible mechanism by which FA affect macrophage-derived EL may be due to regulation of TLR4 and NF-κB, a transcription factor involved in TLR activation. EL expression was induced by inflammatory cytokines and LPS in endothelial cells and macrophages through NF-κB activation.39,47 In vitro treatment with n-3 FA such as EPA diminished TLR4 and NF-κB signaling, while saturated FA enhanced these.44,46,48-50 Consistent with these findings, we found that EPA attenuated TLR4 mRNA, and this correlated with lower EL expression. Thus, it seems possible that FA modulation of EL expression may also occur through interference with TLR4 pathways in macrophages. Indeed, Rader’s group has shown that EL is increased with increased TLR4 expression.12 Experiments in TLR4 knock-down cells would be informative in determining if TLR4 is also directly linked to effects of FA on EL.
Our findings describe mechanisms whereby decreases in EL associated with dietary n-3 FA might be associated not only with higher plasma HDL levels, as described by others51 but also decreases in inflammatory pathways contributory to atherosclerosis. EL was associated with PPARγ expression and the macrophage-derived lipase could be modified by specific FA, in part, through regulating macrophage PPARγ expression. We suggest that similar changes of the EL and inflammation could occur in vivo. The ability of EL to anchor LDL in macrophages could also contribute to atherogenesis.10 We hypothesize that diets rich in n-3 FA (e.g. EPA), in contrast to saturated FA (e.g., PA) decrease progression of atherosclerosis in humans, in part, by down-regulating inflammatory markers and PPARγ, together with decreasing macrophage EL in the arterial wall.
Supplementary Material
Acknowledgments
UJJ, CT, CLC and HH carried out experiments and RJD drafted the outline of the manuscript with UJJ. UJJ wrote the original manuscript and RJD, CLC and TSW performed substantial editing. We also thank Drs. Silke Vogel and Ira Goldberg, Columbia University, for their critical reviews of this manuscript.
Sources of Funding This work was supported by NIH grant HL 40404 (RJD), T32 DK007647 (CLC) T32 HL007343 (CLC) and 5K08AG025833 (TSW), and a fellowship from the International Nutrition Foundation/Ellison Medical Foundation (CT).
Footnotes
Disclosures Richard J. Deckelbaum, MD received an honorarium from the American Society of Nutrition in 2011 for helping organize, chair and speak at a symposium on omega-3 fatty acids titled “Heart Healthy Omega-3s for Food: Stearidonic Acid (SDA) as a Sustainable Choice” at the Experimental Biology 2011 meeting. No other authors had any conflict of interest.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ross R. Atherosclerosis - An inflammatory disease. New Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Hansson GK, Libby P. The immune response in atherosclerosis: a double-edged sword. Nat Rev Immunol. 2006;6:508–519. doi: 10.1038/nri1882. [DOI] [PubMed] [Google Scholar]
- 3.Watts GF, Jackson P, Burke V, Lewis B. Dietary fatty acids and progression of coronary artery disease in men. Am J Clin Nutr. 1996;64:202–209. doi: 10.1093/ajcn/64.2.202. [DOI] [PubMed] [Google Scholar]
- 4.Kromhout D. Fatty acids, antioxidants, and coronary heart disease from an epidemiological perspective. Lipids. 1999;34:S27–S31. doi: 10.1007/BF02562225. [DOI] [PubMed] [Google Scholar]
- 5.Thies F, Garry JMC, Yaqoob P, Rerkasem K, Williams J, Shearman CP, Gallagher PJ, Calder PC, Grimble RF. Association of n-3 polyunsaturated fatty acids with stability of atherosclerotic plaques: a randomised controlled trial. Lancet. 2003;361:477–484. doi: 10.1016/S0140-6736(03)12468-3. [DOI] [PubMed] [Google Scholar]
- 6.Wang C, Harris WS, Chung M, Lichtenstein AH, Balk EM, Kupelnick B. n-3 Fatty acids from fish or fish-oil supplements, but not alpha-linolenic acid, benefit cardiovascular disease outcomes in primary- and secondary-prevention studies: a systematic review. Am J Clin Nutr. 2006;84:5–17. doi: 10.1093/ajcn/84.1.5. [DOI] [PubMed] [Google Scholar]
- 7.Matsumoto M, Sata M, Fukuda D, Tanaka K, Soma M, Hirata Y, Nagai R. Orally administered eicosapentaenoic acid reduces and stabilizes atherosclerotic lesions in ApoE-deficient mice. Atherosclerosis. 2008;197:524–533. doi: 10.1016/j.atherosclerosis.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 8.Azumi H, Hirata K, Ishida T, Kojima Y, Rikitake Y, Takeuchi S, Inoue N, Kawashima S, Hayashi Y, Itoh H, Quertermous T, Yokoyama M. Immunohistochemical localization of endothelial cell-derived lipase in atherosclerotic human coronary arteries. Cardiovasc Res. 2003;58:647–654. doi: 10.1016/s0008-6363(03)00287-6. [DOI] [PubMed] [Google Scholar]
- 9.Ishida T, Choi SY, Kundu RK, Spin J, Yamashita T, Hirata K, Kojima Y, Yokoyama M, Cooper AD, Quertermous T. Endothelial lipase modulates susceptibility to atherosclerosis in apolipoprotein-E-deficient mice. J Biol Chem. 2004;279:45085–45092. doi: 10.1074/jbc.M406360200. [DOI] [PubMed] [Google Scholar]
- 10.Yasuda T, Hirata K, Ishida T, Kojima Y, Tanaka H, Okada T, Quertermous T, Yokoyama M. Endothelial lipase is increased by inflammation and promotes LDL uptake in macrophages. J Atheroscler Thromb. 2007;14:192–201. doi: 10.5551/jat.e502. [DOI] [PubMed] [Google Scholar]
- 11.Kojima Y, Hirata K, Ishida T, Shimokawa Y, Inoue N, Kawashima S, Quertermous T, Yokoyama M. Endothelial lipase modulates monocyte adhesion to the vessel wall. A potential role in inflammation. J Biol Chem. 2004;279:54032–54038. doi: 10.1074/jbc.M411112200. [DOI] [PubMed] [Google Scholar]
- 12.Wang X, Jin W, Rader DJ. Upregulation of macrophage endothelial lipase by toll-like receptors 4 and 3 modulates macrophage interleukin-10 and -12 production. Circ Res. 2007;100:1008–1015. doi: 10.1161/01.RES.0000263011.34709.c5. [DOI] [PubMed] [Google Scholar]
- 13.Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93:229–240. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- 14.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 15.Li L, Beauchamp MC, Renier G. Peroxisome proliferator-activated receptor alpha and gamma agonists upregulate human macrophage lipoprotein lipase expression. Atherosclerosis. 2002;165:101–110. doi: 10.1016/s0021-9150(02)00203-4. [DOI] [PubMed] [Google Scholar]
- 16.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature. 1998;391:82–86. doi: 10.1038/34184. [DOI] [PubMed] [Google Scholar]
- 17.Babaev VR, Yancey PG, Ryzhov SV, Kon V, Breyer MD, Magnuson MA, Fazio S, Linton MF. Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:1647–1653. doi: 10.1161/01.ATV.0000173413.31789.1a. [DOI] [PubMed] [Google Scholar]
- 18.Moore KJ, Fitzgerald ML, Freeman MW. Peroxisome proliferator-activated receptors in macrophage biology: friend or foe? Curr Opin Lipidol. 2001;12:519–527. doi: 10.1097/00041433-200110000-00007. [DOI] [PubMed] [Google Scholar]
- 19.Thieringer R, Fenyk-Melody JE, Le Grand CB, Shelton BA, Detmers PA, Somers EP, Carbin L, Moller DE, Wright SD, Berger J. Activation of peroxisome proliferator-activated receptor gamma does not inhibit IL-6 or TNF-alpha responses of macrophages to lipopolysaccharide in vitro or in vivo. J Immunol. 2000;164:1046–1054. doi: 10.4049/jimmunol.164.2.1046. [DOI] [PubMed] [Google Scholar]
- 20.Guyton K, Bond R, Reilly C, Gilkeson G, Halushka P, Cook J. Differential effects of 15-deoxy-delta (12,14)-prostaglandin J2 and a peroxisome proliferator-activated receptor gamma agonist on macrophage activation. J Leukoc Biol. 2001;69:631–638. [PubMed] [Google Scholar]
- 21.Rossi A, Kapahi P, Natoli G, Takahashi T, Chen Y, Karin M, Santoro MG. Anti-inflammatory cyclopentenone prostaglandins are direct inhibitors of IkappaB kinase. Nature. 2000;403:103–108. doi: 10.1038/47520. [DOI] [PubMed] [Google Scholar]
- 22.Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7:48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- 23.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 24.Rosen CJ. The rosiglitazone story - lessons from an FDA advisory committee meeting. N Engl J Med. 2007;357:844–846. doi: 10.1056/NEJMp078167. [DOI] [PubMed] [Google Scholar]
- 25.Seo T, Qi K, Chang C, Liu Y, Worgall TS, Ramakrishnan R, Deckelbaum RJ. Saturated fat-rich diet enhances selective uptake of LDL cholesteryl esters in the arterial wall. J Clin Invest. 2005;115:2214–2222. doi: 10.1172/JCI24327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang CL, Seo T, Matsuzaki M, Worgall TS, Deckelbaum RJ. N-3 fatty acids reduce arterial LDL-cholesterol delivery and arterial lipoprotein lipase levels and lipase distribution. Arterioscler Thromb Vasc Biol. 2009;29:555–561. doi: 10.1161/ATVBAHA.108.182287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang CL, Seo T, Du CB, Accili D, Deckelbaum RJ. n-3 Fatty acids decrease arterial low-density lipoprotein cholesterol delivery and lipoprotein lipase levels in insulin-resistant mice. Arterioscler Thromb Vasc Biol. 2010;30:2510–2517. doi: 10.1161/ATVBAHA.110.215848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neve BP, Fruchart JC, Staels B. Role of peroxisome proliferators-activated receptors (PPAR) in atherosclerosis. Biochem Pharmacol. 2000;60:1245–1250. doi: 10.1016/s0006-2952(00)00430-5. [DOI] [PubMed] [Google Scholar]
- 29.Lopes-Virella MF. Interactions between bacterial lipopolysaccharides and serum lipoproteins and their possible role in coronary heart disease. Eur Heart J. 1993;14:118–124. [PubMed] [Google Scholar]
- 30.Thorp E, Kuriakose G, Shah YM, Gonzalez FJ, Tabas I. Pioglitazone increases macrophage apoptosis and plaque necrosis in advanced atherosclerotic lesions of nondiabetic low-density lipoprotein receptor-null mice. Circulation. 2007;116:2182–2190. doi: 10.1161/CIRCULATIONAHA.107.698852. [DOI] [PubMed] [Google Scholar]
- 31.Sakamoto J, Kimura H, Moriyama S, Odaka H, Momose Y, Sugiyama Y, Sawada H. Activation of human peroxisome proliferators-activated receptor (PPAR) subtypes by pioglitazone. Biochem Biophys Res Commun. 2000;278:704–711. doi: 10.1006/bbrc.2000.3868. [DOI] [PubMed] [Google Scholar]
- 32.Lee JY, Hwang DH. Docosahexaenoic acid suppresses the activity of peroxisome proliferator-activated receptors in a colon tumor cell line. Biochem Biophys Res Commun. 2002;298:667–674. doi: 10.1016/s0006-291x(02)02530-5. [DOI] [PubMed] [Google Scholar]
- 33.Edwards IJ, O’Flaherty JT. Omega-3 Fatty Acids and PPARgamma in Cancer. PPAR Res. 2008;2008:358052. doi: 10.1155/2008/358052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Khodadadi Iraj, Griffin Bruce, Thumser Alfred. Differential effects of long-chain fatty acids and clofibrate on gene expression profiles in cardiomyocytes. Arch Iranian Med. 2008;11:42–49. [PubMed] [Google Scholar]
- 35.Allman M, Gaskin L, Rivera CA. CCl4-induced hepatic injury in mice fed a Western diet is associated with blunted healing. J Gastroenterol Hepatol. 2010;25:635–643. doi: 10.1111/j.1440-1746.2009.06112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou J, Wilson KM, Medh JD. Genetic analysis of four novel peroxisome proliferator activated receptor-gamma splice variants in monkey macrophages. Biochem Biophys Res Commun. 2002;293:274–283. doi: 10.1016/S0006-291X(02)00138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sauma L, Stenkula KG, Kjolhede P, Stralfors P, Soderstrom M, Nystrom FH. PPAR-gamma response element activity in intact primary human adipocytes: effects of fatty acids. Nutrition. 2005;20:60–68. doi: 10.1016/j.nut.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 38.Schoonjans K, Peinado-Onsurbe J, Lefebvre AM, Heyman RA, Briggs M, Deeb S, Staels B, Auwerx J. PPARalpha and PPARgamma activators direct a distinct tissue-specific transcriptional response via a PPRE in the lipoprotein lipase gene. EMBO J. 1996;15:5336–5348. [PMC free article] [PubMed] [Google Scholar]
- 39.Wu X, Huang H, Tang F, Le K, Xu S, Liu P. Regulated expression of endothelial lipase in atherosclerosis. Mole Cell Endocrinol. 2010;315:233–238. doi: 10.1016/j.mce.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 40.Khanna AK. Enhanced susceptibility of cyclin kinase inhibitor p21 knockout mice to high fat diet induced atherosclerosis. J Biomed Sci. 2009;16:66. doi: 10.1186/1423-0127-16-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kambayashi T, Jacob CO, Strassmann G. IL-4 and IL-13 modulate IL-10 release in endotoxin-stimulated murine peritoneal mononuclear phagocytes. Cell Immunol. 1996;171:153–158. doi: 10.1006/cimm.1996.0186. [DOI] [PubMed] [Google Scholar]
- 42.Huber SA, Sakkinen P, Conze D, Hardin N, Tracy R. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 1999;19:2364–2367. doi: 10.1161/01.atv.19.10.2364. [DOI] [PubMed] [Google Scholar]
- 43.Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003;163:1117–1125. doi: 10.1016/S0002-9440(10)63471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pinderski Oslund LJ, Hedrick CC, Olvera T, Hagenbaugh A, Territo M, Berliner JA, Fyfe AI. Interleukin-10 blocks atherosclerotic events in vitro and in vivo. Arterioscler Thromb Vasc Biol. 1999;19:2847–2853. doi: 10.1161/01.atv.19.12.2847. [DOI] [PubMed] [Google Scholar]
- 45.Renier G, Skamene K, DeSanctis J, Radzioch D. Dietary n-3 Polyunsaturated Fatty Acids Prevent the Development of Atherosclerotic Lesions in Mice. Modulation of Macrophage Secretory Activities. Arterioscler Thromb Vasc Biol. 1993;13:1515–1524. doi: 10.1161/01.atv.13.10.1515. [DOI] [PubMed] [Google Scholar]
- 46.Qiu G, Ho AC, Yu W, Hill JS. Suppression of endothelial or lipoprotein lipase in THP-1 macrophages attenuates proinflammatory cytokine secretion. J Lipid Res. 2007;48:385–394. doi: 10.1194/jlr.M600304-JLR200. [DOI] [PubMed] [Google Scholar]
- 47.Jin W, Millar JS, Broedl U, Glick JM, Rader DJ. Inhibition of endothelial lipase causes increased HDL cholesterol levels in vivo. J Clin Invest. 2003;111:357–362. doi: 10.1172/JCI16146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee JY, Plakidas A, Lee WH. Differential modulation of Toll-like receptors by fatty acids: preferential inhibition by n-3 polyunsaturated fatty acids. J Lipid Res. 2003;44:479–486. doi: 10.1194/jlr.M200361-JLR200. [DOI] [PubMed] [Google Scholar]
- 49.Zhao Y, Joshi-Barve S, Barve S, Chen LH. Eicosapentaenoic acid prevents LPS-induced TNF-alpha expression by preventing NF-kappaB activation. J Am Coll Nutr. 2000;23:71–78. doi: 10.1080/07315724.2004.10719345. [DOI] [PubMed] [Google Scholar]
- 50.Ajuwon KM, Spurlock ME. Palmitate activates the NF-B transcription factor and induces IL-6 and TNF expression in 3T3-L1 adipocytes. J Nutr. 2005;135:1841–1846. doi: 10.1093/jn/135.8.1841. [DOI] [PubMed] [Google Scholar]
- 51.Harris WS. N-3 fatty acids and lipoproteins: comparison of results from human and animal studies. Lipids. 1996;31:243–252. doi: 10.1007/BF02529870. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.