

Abstract
Purpose
Nuclear receptor 2E1 (NR2E1) is a transcription factor with many roles during eye development and thus may be responsible for the occurrence of certain congenital eye disorders in humans. To test this hypothesis, we screened NR2E1 for candidate mutations in patients with aniridia and other congenital ocular malformations (anterior segment dysgenesis, congenital optic nerve malformation, and microphthalmia).
Methods
The NR2E1 coding region, 5′ and 3′ untranslated regions (UTRs), exon flanking regions including consensus splice sites, and six evolutionarily conserved non-coding candidate regulatory regions were analyzed by sequencing 58 probands with aniridia of whom 42 were negative for PAX6 mutations. Nineteen probands with anterior segment dysgenesis, one proband with optic nerve malformation, and two probands with microphthalmia were also sequenced. The control population comprised 376 healthy individuals. All sequences were analyzed against the GenBank sequence AL078596.8 for NR2E1. In addition, the coding region and flanking intronic sequences of FOXE3, FOXC1, PITX2, CYP1B1, PAX6, and B3GALTL were sequenced in one patient and his relatives.
Results
Sequencing analysis showed 17 NR2E1 variants including two novel rare non-coding variants (g.-1507G>A, g.14258C>T), and one novel rare coding variant (p.Arg274Gly). The latter was present in a male diagnosed with Peters’ anomaly who subsequently was found to have a known causative mutation for Peters’ plus syndrome in B3GALTL (c.660+1G>A). In addition, the NR2E1 novel rare variant Arg274Gly was present in the unaffected mother of the patient but absent in 746 control chromosomes.
Conclusions
We eliminated a major role for NR2E1 regulatory and coding mutations in aniridia and found a novel rare coding variant in NR2E1. In addition, we found no coding region variation in the control population for NR2E1, which further supports its previously reported high level of conservation and low genetic diversity. Future NR2E1 studies in ocular disease groups such as those involving retinal and optic nerve abnormalities should be undertaken to determine whether NR2E1 plays a role in these conditions.
Introduction
Congenital ocular malformations contribute to 17% of blindness cases in children worldwide [1]. Aniridia is a severe form of congenital ocular malformation characterized by iris hypoplasia or complete or partial absence of the iris, and is usually accompanied by a range of other ocular disorders such as macular and optic nerve hypoplasia, glaucoma, and cataract [2]. Aniridia can be found combined with interhemispheric brain abnormalities [3-7], obesity [6], and as part of the WAGR syndrome, which includes Wilms' tumor, genitourinary anomalies, and mental retardation [8]. Anterior segment dysgenesis (ASD) is a genetically diverse group of congenital ocular malformations that affect the cornea, iris, lens, and ciliary body. The clinical manifestations of ASD vary greatly between individuals. ASD can be classified as infantile glaucoma, Axenfeld-Rieger syndrome, and Peters’ anomaly (PA), among others [9]. The molecular mechanisms underlying congenital eye disorders involve mutations in genes that control the specification of the eye field, optic vesicle morphogenesis, growth patterning and closure of the optic cup, development of the retina and optic nerve, anterior segment morphogenesis, and lens development. Among those genes, PAX6 is the most prominent; it is the only known causative gene in classic aniridia and accounts for approximately 80% of these patients [10]. Other genes mutated in ASD include B3GALTL [11], CYP1B1 [12], FOXC1 [13,14], FOXE3 [15], and PITX2 [16,17]. Nevertheless, despite substantial efforts to identify causative mutations [10,18-23], the pathogenesis of many congenital ocular malformations remains unknown.
The nuclear receptor 2E1 (NR2E1, also known as TLX) is involved in controlling proliferation in neural stem cells during brain and eye development. The role of NR2E1 in human disease is starting to be recognized as the genomic variation has been associated with bipolar disorder [24], and overexpression in gliomas correlates with decreased survival of patients with brain tumors [25]. NR2E1 is expressed early during eye morphogenesis in the eye field together with Pax6 [26], as well as in the optic cup and optic stalk [27,28]. Mice lacking Nr2e1 display brain and eye defects resulting from abnormal neural stem cell proliferation and depletion of the neural stem cell pool [29,30]. At the molecular level, there are different ways in which NR2E1 affects pathways involved in eye development. In mice, Nr2e1 represses Pax2 expression [28], which is required for optic cup and optic nerve development [31]. Pax2 and Pax6 mutually inhibit each other to define the retina and optic stalk boundaries [32] and dysregulation of this process negatively affects optic nerve development [33]. Thus, Nr2e1 may indirectly influence the expression of Pax6, a master regulator of eye development that results in various eye developmental disorders when mutated [34]. Evidence for such an interaction comes from Xenopus studies where Nr2e1 positively affects Pax6 levels [26]. Interestingly, the genetic interaction between Nr2e1 and Pax6 regulates the establishment of the dorsal-ventral cortical boundary in the mouse telencephalon [35]. In addition, Nr2e1 is involved in the retinoic acid pathway by potentiating the retinoic acid–mediated induction of the retinoic acid receptor beta 2 (RARβ2) promoter [36]. Interestingly, the retinoic acid pathway is involved in retinal [37] and anterior chamber morphogenesis [38,39]. Nr2e1 also has a non-cell autonomous role in activating the wingless signaling pathway to promote neural stem cell proliferation and self-renewal [40]. This pathway has many roles during eye development, including patterning of the ocular surface ectoderm [41]. Finally, NR2E3, the closest relative to NR2E1 in the human genome, causes enhanced S-cone syndrome and retinitis pigmentosa in humans [42-44]. Thus, due to the important role that NR2E1 plays during eye development, we hypothesize that NR2E1 may be involved in human eye disorders impacting a wide range of eye structures whose development depend on NR2E1 genetic interactors such as PAX2 and PAX6.
Overall, the NR2E1 locus is unusually highly conserved, reminiscent of the HOX cluster, and displays low genetic diversity among humans [24,45]. We have previously screened for NR2E1 mutations in patients with brain malformations [46,47] and bipolar disorder [24] but did not find any amino acid variations. However, 14 non-synonymous variants have now been reported in public databases: the Single Nucleotide Polymorphism database (dbSNP), the 1000 Genomes Project, and the NHLBI Exome Sequencing Project (ESP). Among these variants, six are predicted to confer amino acid substitutions that would affect protein function by sorting intolerant from tolerant ( SIFT) and polymorphism phenotyping (PolyPhen) scores; two were found in cancerous tissues in the heterozygous state, three were found in European-descendent ESP cohorts (comprising heart, lung, and blood diseases) in the heterozygous state, and one of unknown zygosity was found in European-descendent cohorts with atherosclerotic heart disease from the ClinSeq project [48].
Surprisingly, no cohort group comprised of individuals with a specific eye disorder has been screened for variation in NR2E1. To initiate such studies, we focused on sequencing NR2E1 in patients with aniridia but included patients with ASD, microphthalmia, and optic nerve malformations known not to harbor PAX6 mutations. We chose aniridia since we hypothesized that NR2E1 could alter PAX6 expression or functioning and ultimately lead to a phenotype resembling PAX6 haploinsufficiency. In this study, we identified several NR2E1 polymorphisms as well as a new amino acid variant in a patient diagnosed with Peters’ anomaly (PA) who we subsequently found harbors a known causative mutation in B3GALTL. Sequencing of B3GALTL, CYP1B1, FOXC1, FOXE3, and PITX2 in the patient and his close relatives also revealed new variants in a subset of these genes. In conclusion, we did not find a causative mutation in NR2E1 that could explain aniridia.
Methods
Patients and control individuals
This study followed Canada’s Tri-Council Statement on Ethical Conduct for Research Involving Humans and was approved by the University of British Columbia (Certificate of Approval #C99–0524). Informed consent was obtained for all patients. Clinical and demographic data for all subjects are reported in Table 1. The study group consisted of 80 probands, 376 controls, and 22 unaffected relatives.
Table 1. Demographics of patients with ASD, microphthalmia, and optic nerve malformation.
| Pathology |
Gender |
Ethnicity |
PAX6 |
|||||
|---|---|---|---|---|---|---|---|---|
| Male | Female | Unknown | Caucasian | Unknown | Tested | Total | ||
| Aniridia |
17 |
28 |
22 |
8 |
59 |
44 |
67 |
|
| ASD | ||||||||
| Axenfeld-Rieger syndrome |
0 |
0 |
1 |
0 |
1 |
1 |
1 |
|
| Coloboma/congenital cataract |
0 |
1 |
0 |
1 |
0 |
1 |
1 |
|
| Peters' anomaly |
3 |
1 |
8 |
4 |
8 |
12 |
12 |
|
| Rieger syndrome |
1 |
1 |
3 |
2 |
3 |
5 |
5 |
|
| Other | ||||||||
| Microphthalmia |
2 |
0 |
0 |
2 |
0 |
2 |
2 |
|
| Optic nerve malformation |
1 |
0 |
0 |
1 |
0 |
1 |
1 |
|
| Control |
0 |
0 |
376 |
376 |
0 |
N/A |
376 |
|
| Unaffected relatives | 7 | 14 | 1 | 0 | 22 | N/A | 22 | |
The demographic features of 89 patients with congenital ocular malformations, 376 controls and 22 unaffected relatives are shown. Numbers indicate the number of patients participating in this study. The majority of patients were tested for PAX6 mutations and chosen to participate in this study after they were found negative. Thirty-one probands with aniridia were negative for PAX6 after sequencing and 11 probands with aniridia were negative for PAX6 after chromosomal analysis. N/A, not applicable.
Fifty-eight probands were diagnosed with aniridia, one proband had Axenfeld-Rieger syndrome, one proband had coloboma with congenital cataract, 12 probands had PA, five probands had Rieger syndrome, two probands had microphthalmia, and one proband had optic nerve malformation (Table 1). Slightly more than 70% of the samples collected had previously been examined for PAX6 pathogenic mutations and found to be negative using chromosomal analysis (11 aniridia samples [11 probands], one PA sample) and dideoxy fingerprinting or sequencing (33 aniridia samples [31 probands] and 21 samples of ASD, microphthalmia, and other disorders); see Table 1.
Thirty-six patients were contacted and DNA samples collected during the 2007 Aniridia International Medical Conference (Memphis, TN). Some DNA samples were obtained from collections belonging to the research groups of Dr. Brian Brooks (one sample; National Eye Institute, National Institutes of Health, Department of Health and Human Services, Bethesda, MD), Dr. Thomas Rosenberg and Dr. Karen Gronskov (17 samples; Kennedy Center, Glostrup, Denmark), Dr. Francesca Pasutto (14 samples; Institute of Human Genetics, Friedrich-Alexander University of Erlangen-Nuremberg, Erlangen, Germany), Dr. Michael Walter (seven samples; Department of Medical Genetics, University of Alberta, Edmonton, Canada), and Dr. Veronica Van Heyningen (14 samples; Medical Research Council, Human Genetics Unit, Edinburgh, Scotland).
The control group consisted of 282 individuals of Caucasian descent obtained from the Coriell Cell Repository, 188 of whom had been used in a previous study including 94 samples from individuals considered “neurologically normal” [24]. Ninety-four Caucasian patients diagnosed with Gilbert syndrome, a bilirubin disorder, also used in a previous study were included in this study as controls [24].
In addition, 22 unaffected relatives were included in the study to better assess the potential pathogenicity of the variants found. Eighteen were relatives of patients with aniridia, and four were relatives of patients with PA.
NR2E1 sequencing analysis
Oragene DNA self-collection kits (DNA Genotek, Gaithersburg, MD) were used to collect saliva from patients and relatives during the 2007 Aniridia International Medical Conference (Memphis, TN). Genomic DNA was extracted using MoleStrips DNA Blood Kit (Lysaker, Norway) according to the manufacturer’s instructions. Patient blood-purified DNA sent by collaborators was shipped and stored at 4 °C. Sequence analysis included bidirectional sequencing of the coding region, 5′ and 3′ untranslated regions (UTRs), exon flanking regions including consensus splice sites, and six evolutionarily conserved candidate regulatory non-coding regions using 20 polymerase chain reaction (PCR) amplicons as previously described (Kumar et al., 2007) [47]. Human genomic NR2E1 (GenBank AL078596.8) sequence was used as the reference sequence. The numbering of NR2E1 variants was based on Antonarakis and the Nomenclature Working Group [49]. Every human NR2E1 variant found was confirmed by repeating the PCR and sequencing process. DNA samples from a subset of patients displaying NR2E1 variants g.14121C>G and g.14258C>T with unknown PAX6 genotype were subjected to targeted array comparative genomic hybridization analysis with exon-level resolution to identify deletions or duplications of one or more exons of PAX6 [50] with GeneDx (Gaithersburg, MD). In addition, these samples were analyzed for mutations with bidirectional sequencing of exonic regions of PAX6 (exons 1–13, the alternatively spliced exon 5a, and splice junctions) by GeneDx. B3GALTL, CYP1B1, FOXC1, FOXE3, and PITX2 were analyzed in patient 21,000 and his family for sequence variations by bidirectional sequencing of exons and at least 10 bp of flanking intron sequence. Sanger sequencing was performed using BigDye terminator kit version 3.1 and capillary electrophoresis on an ABI3130XL (Applied Biosystems, Carlsbad, CA). Subsequent data analysis was performed using SeqScape (Life Technologies, Carlsbad, CA). Primers for B3GALTL were previously described [11], and novel primers are depicted in Table 2. Forward primers had 5’-ACC CAC TGC TTA CTG GCT TAT C-3’ and reverse primers 5’-GAG GGG CAA ACA ACA GAT GGC-3’ added for sequencing of the PCR product (bolded, Table 2).
Table 2. PCR primers designed for mutational analysis of CYP1B1, PITX2, FOXC1, FOXE3, and B3GALTL.
| Gene | Name | Sequence |
|---|---|---|
|
B3GALTL |
oEMS4859 |
GAATGAAATCAGAAAAAAGTCAGCG |
| |
oEMS4860 |
TATGTCCCATAAACATAGTATTTC |
|
CYP1B1 |
CYP1B1–2.1–2FH |
ACCCACTGCTTACTGGCTTATCTCCGACCTCTCCACCCAAC |
| |
CYP1B1–2.1–2RH |
GAGGGGCAAACAACAGATGGCCAGTGCTCCGAGTAGTGGCC |
| |
CYP1B1–2-FH2 |
ACCCACTGCTTACTGGCTTATCGCAGCTCCGGTCCGC |
| |
CYP1B1–2-2RH2 |
GAGGGGCAAACAACAGATGGCCAGCTCACGGAACTCGGG |
| |
CYP1B1–2.3–2FH |
ACCCACTGCTTACTGGCTTATCTTCCGTGTGGTGTCCGG |
| |
CYP1B1–2.3–2RH |
GAGGGGCAAACAACAGATGGCCGCCTTCTTTTCCGCAGAG |
| |
CYP1B1–2-4FH |
ACCCACTGCTTACTGGCTTATCACAACGAAGAGTTCGGGCG |
| |
CYP1B1–2-4RH |
GAGGGGCAAACAACAGATGGCGAAACCCCAAACCCGGG |
| |
CYP1B1–3-1FH |
ACCCACTGCTTACTGGCTTATCCTAGATAGCCTATTTAAGAAAAAGTGGAATTA |
| |
CYP1B1–3-1RH |
GAGGGGCAAACAACAGATGGCGTGAGCCAGGATGGAGATGAAG |
| |
CYP1B1–3-2FH |
ACCCACTGCTTACTGGCTTATCGTTTTTGTCAACCAGTGGTCTGTG |
| |
CYP1B1–3-2RH2 |
GAGGGGCAAACAACAGATGGCCTACTCATGAAGAACCGCTGGG |
|
FOXC1 |
FOXC1–1FH2 |
ACCCACTGCTTACTGGCTTATCCAGCGCAGCCGGACGCACAG |
| |
FOXC1–1RH2 |
GAGGGGCAAACAACAGATGGCGCCAGCCCTGCTTGTTGTCCCG |
| |
FOXC1–2FH |
ACCCACTGCTTACTGGCTTATCAGCTACATCGCGCTCATCACCA |
| |
FOXC1–2RH |
GAGGGGCAAACAACAGATGGCTGCTGTCGGGGCTCTCGATCTT |
| |
FOXC1–3FH |
ACCCACTGCTTACTGGCTTATCCCGTGCGCATCCAGGACATCAA |
| |
FOXC1–3RH |
GAGGGGCAAACAACAGATGGCATGGCTTGCAGGTTGCAGTGGT |
| |
FOXC1–4FH |
ACCCACTGCTTACTGGCTTATCCTACTCGCCCGGCCAGAGCTCC |
| |
FOXC1–4RH3 |
GAGGGGCAAACAACAGATGGCTTTCGATTTTGCCTTGATGG |
|
FOXE3 |
FOXE3–1FH |
ACCCACTGCTTACTGGCTTATCAGGAGGGGTGGAAAGGGAAGGGGA |
| |
FOXE3–1RH |
GAGGGGCAAACAACAGATGGCCGGTAGATGGCGGCCAGCGTGAG |
| |
FOXE3–2FH |
ACCCACTGCTTACTGGCTTATCCGAGCCAGGGCGGGAGCCAG |
| |
FOXE3–2RH |
GAGGGGCAAACAACAGATGGCAAGGCTGCGGCTGCGGCGTC |
| |
FOXE3–3FH |
ACCCACTGCTTACTGGCTTATCCGCCCGCGCGTCTGTTCAGC |
| |
FOXE3–3RH |
GAGGGGCAAACAACAGATGGCGAGTCCAGGAGGCCACGACGAGA |
|
PITX2 |
PITX2–2FH |
ACCCACTGCTTACTGGCTTATCAGTCTCATCTGAGCCCTGCTCAC |
| |
PITX2–2RH |
GAGGGGCAAACAACAGATGGCGCGATTTGGTTCTGATTTCCT |
| |
PITX2–3FH |
ACCCACTGCTTACTGGCTTATCGTCTTTGCTCTTTGTCCCTCTTTC |
| |
PITX2–3RH |
GAGGGGCAAACAACAGATGGCAATTTGGGGAAAGGAATTAACGTC |
| |
PITX2–4AFH |
ACCCACTGCTTACTGGCTTATCGCCCGCCTCTGGTTTTAAGATG |
| |
PITX2–4ARH |
GAGGGGCAAACAACAGATGGCTCCGGAAGGCTCAAGCGAAAAA |
| |
PITX2–4BFH |
ACCCACTGCTTACTGGCTTATCGGGAGGGAGAGAAGAAGGGGGT |
| |
PITX2–4BRH |
GAGGGGCAAACAACAGATGGCGAGCCAGGCGAACGACCACT |
| |
PITX2–5FH |
ACCCACTGCTTACTGGCTTATCCCAGCTCTTCCACGGCTTCTGC |
| |
PITX2–5RH |
GAGGGGCAAACAACAGATGGCTCGGAGAGGGAACTGTAATCTCGC |
| |
PITX2–6FH |
ACCCACTGCTTACTGGCTTATCTGAGTGCGCTAGCGTGTGTGTC |
| PITX2–6RH | GAGGGGCAAACAACAGATGGCTCCCTTTCTTTAGTGCCCACGACC |
Bolded, sequences used for sequencing primers.
Database search and in silico analysis of variants
Single Nucleotide Polymorphism Database was searched at dbSNP database (accessed July 2012). The 1000 Genomes database was searched at 1000 Genomes (accessed July 2012). The NHLBI Exome Sequencing Project (ESP) was searched at the Exome Variant Server, NHLBI Exome Sequencing Project (ESP), Seattle, WA (accessed July 2012). SIFT scores [51] were calculated using the online resource (accessed May 2011). PolyPhen scores [52] were calculated using the online resource (accessed July 2012).
Clinical assessment of patient 2,100
When patient 2,100 was an infant, a pediatric ophthalmological consultant performed bedside inspections, including assessment of visual acuity with large objects and preferential looking techniques. Examination of the exterior eye and eye movements was performed with a pencil light. Anterior segments were studied with a hand-held slit lamp, and visualization of the posterior segments by indirect ophthalmoscopy. Examinations under general anesthesia were performed with an operating microscope. Intraocular tension was assessed with applanation tonometry and a Schiötz tonometer. Retinal inspections were performed with a binocular indirect ophthalmoscope, and eye dimensions were measured with ultrasound.
Results
Studying a proband group made up primarily of aniridia (72.5% [58/80]) and ASD (23.75% [19/80]) and enriched for cases with no evidence of PAX6 mutation (82.5% [66/80]), we identified 17 NR2E1 variants (Table 3). Only one variant was located in the coding region. Among the non-coding region variants, five were in the 5′-UTR, seven within intronic regions, and four within upstream conserved candidate regulatory elements. To explore whether the variants found represented polymorphisms, rare variants, or candidate mutations in NR2E1, we sequenced a control group of 376 unaffected individuals (752 normal chromosomes). Not all the amplicons were successfully sequenced for every control, and thus, the exact number of control chromosomes is depicted in Table 3.
Table 3. Variation identified in NR2E1.
| Nucleotide changea | Amino-acid change | Locationb | Proband allele freq. | UFM | Control allele freq. | Total allele freq. | Previously reported | Flanking sequence |
|---|---|---|---|---|---|---|---|---|
| g.-2945A>G |
N/A |
CE11A (Upstream) |
2/160 |
1 |
0/324 |
2/484 (0.41)c |
[47] |
TCAGAACTGTATTGTGATTTA |
| g.-1507G>A |
N/A |
CE12A (Upstream) |
1/160 |
1 |
0/370 |
1/530 (0.19)c |
This study |
AATGGGGAGGGGGTAGGGGAT |
| g.-1492G>A |
N/A |
CE12A (Upstream) |
8/160 |
1 |
0/370 |
8/530 (1.51) |
[47] |
GGGGATGAGGGCCTCTCTTCA |
| g.-1453C>G |
N/A |
CE12A (Upstream) |
1/160 |
0 |
1/370 |
2/530 (0.38)c |
[47] |
AGCGGGAGCCCGCAACGCCCG |
| g.-555C>T |
N/A |
5′UTR |
1/160 |
0 |
2/370 |
3/530 (0.57)c |
[47] |
ATCTAGTTTTCCCACTCTGCG |
| g.-364C>A |
N/A |
5′UTR |
1/160 |
0 |
ND |
1/160 (0.63)c |
dbSNP |
CGTAGGAAGGCCATTTTCGTG |
| g.-200G>C |
N/A |
5′UTR |
8/160 |
1 |
ND |
8/160 (5.00) |
[47] |
AGAAACTTAAGGATGCTTAAA |
| g.-93A>G |
N/A |
5′UTR |
117/160 |
15 |
ND |
117/160 (73.13) |
[47] |
GCTGGAGGGCAGCTGGAGAGC |
| g.-34C>T |
N/A |
5′UTR |
7/160 |
1 |
ND |
7/160 (4.38) |
[47] |
ACTCGGGCAGCGCCCACCAAC |
| g.2040G>A |
N/A |
CE17B (Intron 1) |
74/160 |
11 |
ND |
74/160 (46.25) |
dbSNP |
CGCCTTGCCCGGCTTCTCGCG |
| g.3026C>G |
N/A |
CE19B (Intron 1) |
1/160 |
1 |
ND |
1/160 (0.63)c |
[47] |
GAGGGGGGCGCCGAGCCGGTG |
| g.3154C>T |
N/A |
CE19B (Intron 1) |
12/160 |
0 |
44/370 |
56/530 (10.57) |
dbSNP |
GTTGTAATTACCCGGCCGAGC |
| g. 4601–4602delTC |
N/A |
Intron 1 |
15/160 |
1 |
ND |
15/160 (9.38) |
[47] |
TTGCTTAGCATCTCTCTCTCC |
| g.10049–10050delTG |
N/A |
Intron 4 |
80/160 |
12 |
ND |
80/160 (50.00) |
dbSNP |
CTGAGCTGTGTGATTGGGGTC |
| g.14121C>G |
p.Arg274Gly |
Exon 7 |
1/160 |
0 |
0/746 |
1/906 (0.11)c |
This study |
GGTGGTGGCTCGATTTAGACA |
| g.14258C>T |
N/A |
Intron 7 |
1/160 |
0 |
0/746 |
1/906 (0.11)c |
This study |
TCAGCCACCTCGAAGTCTGAA |
| g.14672C>A | N/A | Intron 7 | 8/160 | 0 | ND | 8/160 (5.00) | dbSNP | AAGTGATCCGCCTGCCTCGGC |
Allele frequencies of sequence variations within NR2E1 in patients with congenital ocular malformations and controls. The number of unaffected family members (UFM) who have the same variation as their affected relatives is shown. aNumbering based on Antonarakis and the Nomenclature Working Group (Antonarakis SE, 1998) [49], where A of the initiator Met codon in exon 1 is denoted nucleotide +1 in human genomic NR2E1 sequence: GenBank AL078596.8. bCE, evolutionary conserved element within NR2E1 locus (Abrahams et al., 2002) [45]. cRare variants representing <1% of the population. N/A, not applicable; ND, not determined; UFM=Unaffected family members.
Although most of our patients with aniridia were negative for PAX6 mutations, a fraction of the probands (11/58) had only chromosomal aberrations at the PAX6 locus analyzed, so point mutations and small deletion/insertions were not detected. Similarly, some patients sequenced for PAX6 exons may have intronic or upstream deletions that were not detected by the method used. In this way, our aniridia PAX6 negative group might have been overall smaller than 42 probands, thus reducing the power of our study.
The rare variant g.-1507G>A was located in a conserved element and was not previously reported but was also found in an unaffected relative of the patient with aniridia and thus was not a strong candidate for a causative mutation. However, two rare variants had not been previously reported and not found in the control population (Table 3); variant g.14258C>T was located in intron 7 in a patient with aniridia, and variant g.14121C>G (Arg274Gly) was located in exon 7 in a patient diagnosed with PA. We further sequenced PAX6 in these patients and found a known causal mutation [53,54] cooccurring with the variant g.14258C>T, which suggested that there was no functional significance for this NR2E1 variant. However, we did not find a PAX6 mutation in the male patient 21,000 harboring the variant g.14121C>G (Arg274Gly). In summary, we did not find any candidate mutations in NR2E1 in patients with aniridia but found one candidate mutation in a patient diagnosed with PA whom we characterized further as described below.
Clinical characteristics of patient 21,000
The patient, a boy, was the third child of a 40-year-old woman after six pregnancies, three of which were terminated by spontaneous abortion. The child was delivered by spontaneous birth in gestational week 38 with a low birthweight (1,775 g [<first percentile]) and birth length (40 cm [<first percentile]). The placenta was small with one-third infarction. No neonatal asphyxia was noted. Immediately after birth, the patient’s large head circumference and corneal clouding were observed. Intracranial ultrasound showed intraventricular hemorrhage grade 3 with dilation of the ventricular system. A ventriculoperitoneal shunt was necessary to control his head circumference. Pediatric follow-up showed pronounced growth retardation. At three years of age, the bone age was retarded by two years. A laparoscopic examination established right testicular agenesis at six years of age. At 12 years, the beginning of puberty was noted, and his height had reached 126 cm (<third percentile). Puberty-suppressing treatment and growth hormone treatment were initiated despite normal hormone values to improve his final height. At 15 years, his height was 149 cm (<third percentile), and his weight 52.8 kg (20th to 50th percentile). He had normal proportions between the upper and lower trunk as well as extremities with normal hands and feet. He showed normal facial characteristics and had normal teeth and normal umbilicus. His fine and gross motor skills were appropriate for his age. His psychomotor development was described as normal by several examinations, the last one at the age of five. No renal failure was suspected, and no ultrasound exam of the kidneys was done. The patient had a normal karyotype, and no sign of inborn metabolic diseases in blood and urine.
When the patient was four days old, a corneal opacity on both eyes was noted during an ophthalmic examination, and a tentative diagnosis of Peters’ anomaly was made. He was visually alert and had no nystagmus. At the age of six weeks, examination under general anesthesia disclosed a corneal lenticular contact with thread-like structures from the pupillary margin to the posterior lens surface. When the patient was 14 months old, binocular visual acuity of 20/200 was assessed by preferential looking. Reexamination under general anesthesia showed corneal diameters (right/left) of 11/11 mm, axial lengths of 19.8/20.8 mm, and intraocular tension of 11/11 mmHg. Gonioscopy showed dysgenesis of the iridocorneal angle with a fine membrane covering the peripheral part of the iris root, and drag on the peripheral iris. The lenses were clear, and indirect ophthalmoscopy showed normal optic nerve heads, normal retinal vessels and pigmentation, and no sign of persistent hyaloid vessels. Both eyes had normal diameters, large central corneal opacities with central thinning, and clear peripheries. Therapy-resistant glaucoma developed in both eyes and was complicated by keratopathy, nearly collapsed anterior chambers, and dense cataracts. At age 13, he was virtually blind and used Logtext and Braille in school.
Genetic assessment of patient 21,000
To comprehensively study patient 21,000, we sequenced additional candidate genes. This patient was originally diagnosed with PA, but careful review of clinical information revealed short stature and developmental delay resembling Peters’ plus syndrome (PP). Thus, we screened the patient for additional genes known to be involved in the development of PA (CYP1B1, PITX2, FOXC1, and FOXE3) and PP (B3GALTL). During this work, we identified a known homozygous pathogenic variation c.660+1G>A in B3GALTL [11] indicative of PP (Table 4). In addition, we found three novel non-pathological variants and nine known variants in B3GALTL, FOXC1, and FOXE3 (Table 4). Subsequently, we sequenced B3GALTL in patient 21,000’s mother, father, and sister, and found the B3GALTL c.660+1G>A variation in the heterozygous state in all of them.
Table 4. Variants found in B3GALTL, CYP1B1, FOXC1, and FOXE3.
| Gene | Nucleotide changea | Amino acid change | Location | Previously reported |
|---|---|---|---|---|
|
B3GALTL |
c.597–23delA |
N/A |
Intron 7 |
dbSNP |
| |
c.781–34_31dup |
N/A |
Intron 9 |
This study |
| |
c.1065–142T>C |
N/A |
Intron 12 |
dbSNP |
| |
c.348T>C |
p.(=) |
Exon 6 |
dbSNP |
| |
c.660+1G>Ab |
N/A |
Intron 8 |
[11,59] |
|
CYP1B1 |
c.142C>G |
p.Arg48Gly |
Exon 2 |
dbSNP |
| |
c.1294G>C |
p.Val432Leu |
Exon 3 |
dbSNP |
| |
c.1347T>C |
p.(=) |
Exon 3 |
dbSNP |
| |
c.1358A>G |
p.Asn453Ser |
Exon 3 |
dbSNP |
|
FOXE3 |
c.587G>C |
p.Gly196Ala |
Exon 1 |
This study |
| |
c.510C>T |
p.(=) |
Exon 1 |
dbSNP |
| FOXC1 | c.1267G>T | p.Ala423Ser | Exon1 | This study |
Sequence variations within B3GALTL, CYP1B1, FOXE3, and FOXC1 found in patient 21,000 and/or his mother, father and sister. Human genomic sequences (GenBank): B3GALTL: NM_194318.3; CYP1B1: NM_000104.3; FOXC1: NM_001453.2; FOXE3: NM_012186.2. aNumbering based on Antonarakis and the Nomenclature Working Group (Antonarakis SE, 1998), where A of the initiator Met codon in exon 1 is denoted nucleotide +1 in the coding region. bPathological mutation found in patient 21,000. p.(=), no amino acid change.
We then explored the possibility that the phenotype of patient 21,000 might be the result of a combination of mutations in B3GALTL and NR2E1 by further characterizing the NR2E1 rare variant g.14121C>G (Arg274Gly). The sequence trace of this variant showed a double C/G peak, indicative of heterozygosity and thus the presence of a Wt arginine (Arg) and a variant glycine (Gly) in NR2E1 at amino acid 274 (Figure 1). To better understand the biochemical and possible biologic consequences of the amino acid change, we considered the SIFT score, which was 0.01, suggestive of no tolerance for this amino acid substitution. In addition, homology–Basic Alignment Search Tool analysis depicts a high (>90%) NR2E1-protein conservation among vertebrates and 100% identity at Arg274. NR2E3 protein is also highly (>70%) conserved and has 100% identity at Arg309, which aligns with NR2E1 Arg274. Furthermore, a database search for NR2E1 coding variants revealed the Arg274Gln variant (dbSNP, rs148906882), found in a melanoma sample and thus of potential biologic significance [55]. We also screened for the Arg274Gly variant in the relatives of patient 21,000, including the mother, father, and sister (Figure 1), and found that the mother was positive for variant Arg274Gly but presented with no phenotypic eye abnormalities even after detailed reexamination. These results suggest that variant Arg274Gly did not contribute to the phenotype in patient 21,000.
Figure 1.
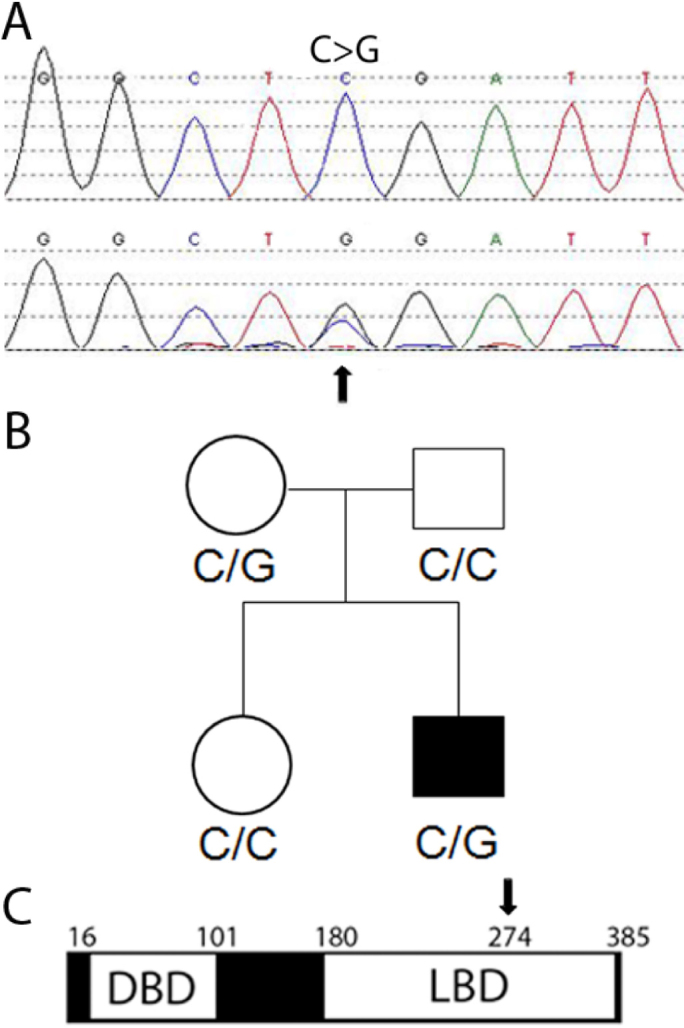
Patient 21,000 and his mother are heterozygous for a novel rare protein variant of NR2E1. A: The patient 21,000 chromatogram shows the base pair change C–>G and the normal allele. B: The pedigree of the family shows the affected boy 21,000. C: NR2E1 amino-acid change from Arg to Gly (arrow) is located in the ligand binding domain. DBD, DNA Binding Domain; LBD, Ligand Binding Domain; numbers represent amino-acids.
Discussion
NR2E1 is a candidate for human congenital ocular malformation based on its role in mouse eye development, and interaction with key eye developmental genes such as Pax2 and Pax6 as well as prominent signaling pathways that regulate eye morphogenesis such as wingless and retinoic acid. Based on these data, we undertook the first screening for NR2E1 mutations focused on human eye disorders. A patient population with congenital eye disorders enriched for lack of mutations in PAX6 was screened for sequence variation in functional regions of NR2E1 including candidate regulatory and coding regions. We extended the characterization of several known polymorphisms, and identified one novel rare variant in a conserved element (g.-1507G>A). In addition, we found one novel rare intronic variant (g.14258C>T) and one novel rare coding variant (g.14121C>G; p. Arg274Gly) not present in the control group. The latter represents one of the few amino acid changes found in NR2E1, all with unknown functional consequences, despite past substantial efforts to identify coding variants with sequencing-based mutation screening [24,46,47]; thus, we focused our further studies on this variant.
The novel rare NR2E1 coding variant was found heterozygous in a patient diagnosed with Peters’ anomaly; the variant results in a substitution from Arg to Gly in amino acid 274 (Arg274Gly). Substantial evidence suggests that this amino acid change alters NR2E1 protein functioning: 1) the high conservation of Arg274 not only in NR2E1 but also in NR2E3 (Arg309) and the association of the NR2E3 variant, Arg309Gly, with eye disease [56]; 2) the low SIFT score indicating the substitution would not be tolerated; and 3) the possible clinical relevance of the Arg274Gln variant found in melanoma tissue [55], which, interestingly, has a SIFT score of only 0.04. However, patient 21,000 also harbored a known causative homozygous mutation in B3GALTL, and his phenotypically normal mother was heterozygous for the NR2E1 Arg274Gly variant. Although the patient does not seem to have typical PP due to the lack of facial dysmorphic features [57], it is unlikely that the NR2E1 variant has a role in improving this condition. Thus, we conclude that the Arg274Gly variant does not cause disease in the heterozygous state, which is in accordance with studies in mice where homozygous loss-of-function of Nr2e1 is required for brain phenotypes [58]. However, the potential remains that this variant could be found in a future patient contributing to the phenotype in a homozygous or compound heterozygous state.
In conclusion, we have eliminated a major role for NR2E1 regulatory and coding mutations in aniridia. In addition, the lack of coding region variation we have found in the control population for NR2E1 further supports the high level of conservation and low genetic diversity known for this gene [24,45]. These genomic characteristics also argue that most changes in the coding region have important biologic consequences. Thus, future studies in other ocular disease groups are well justified, and we propose that diseases involving retinal defects or optic nerve malformations should constitute the next research focus.
Acknowledgments
The authors thank Dr. Veronica Van Heyningen (Human Genet. Unit, Med. Res. Council, Edinburgh, UK) for providing DNA samples, and Dr. Valerie Anne Wallace (Faculty of Medicine, University of Ottawa, ON, Canada) for providing training to AB. Importantly, the authors also thank the patients and their families for the kind donation of their time and DNA. This work was funded by a Sharon Stewart Aniridia Research Award to EMS. The authors indicate no financial conflict of interest.
References
- 1.Ferretti PCA, Tickle C, Moore G, eds. Embryos, Genes and Birth Defects. 2nd ed. Chichester: John Wiley & Sons Ltd; 2006. [Google Scholar]
- 2.Kokotas H, Petersen MB. Clinical and molecular aspects of aniridia. Clin Genet. 2010;77:409–20. doi: 10.1111/j.1399-0004.2010.01372.x. [DOI] [PubMed] [Google Scholar]
- 3.Abouzeid H, Youssef MA, ElShakankiri N, Hauser P, Munier FL, Schorderet DF. PAX6 aniridia and interhemispheric brain anomalies. Mol Vis. 2009;15:2074–83. [PMC free article] [PubMed] [Google Scholar]
- 4.Sisodiya SM, Free SL, Williamson KA, Mitchell TN, Willis C, Stevens JM, Kendall BE, Shorvon SD, Hanson IM, Moore AT, van Heyningen V. PAX6 haploinsufficiency causes cerebral malformation and olfactory dysfunction in humans. Nat Genet. 2001;28:214–6. doi: 10.1038/90042. [DOI] [PubMed] [Google Scholar]
- 5.Bamiou DE, Musiek FE, Sisodiya SM, Free SL, Davies RA, Moore A, van Heyningen V, Luxon LM. Deficient auditory interhemispheric transfer in patients with PAX6 mutations. Ann Neurol. 2004;56:503–9. doi: 10.1002/ana.20227. [DOI] [PubMed] [Google Scholar]
- 6.Netland PA, Scott ML. Boyle JWt, Lauderdale JD. Ocular and systemic findings in a survey of aniridia subjects. J AAPOS. 2011;15:562–6. doi: 10.1016/j.jaapos.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Thompson PJ, Mitchell TN, Free SL, Williamson KA, Hanson IM, van Heyningen V, Moore AT, Sisodiya SM. Cognitive functioning in humans with mutations of the PAX6 gene. Neurology. 2004;62:1216–8. doi: 10.1212/01.wnl.0000118298.81140.62. [DOI] [PubMed] [Google Scholar]
- 8.Fischbach BV, Trout KL, Lewis J, Luis CA, Sika M. WAGR syndrome: a clinical review of 54 cases. Pediatrics. 2005;116:984–8. doi: 10.1542/peds.2004-0467. [DOI] [PubMed] [Google Scholar]
- 9.Idrees F, Vaideanu D, Fraser SG, Sowden JC, Khaw PT. A review of anterior segment dysgeneses. Surv Ophthalmol. 2006;51:213–31. doi: 10.1016/j.survophthal.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 10.Traboulsi EI, Ellison J, Sears J, Maumenee IH, Avallone J, Mohney BG. Aniridia with preserved visual function: a report of four cases with no mutations in PAX6. Am J Ophthalmol. 2008;145:760–4. doi: 10.1016/j.ajo.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 11.Reis LM, Tyler RC, Abdul-Rahman O, Trapane P, Wallerstein R, Broome D, Hoffman J, Khan A, Paradiso C, Ron N, Bergner A, Semina EV. Mutation analysis of B3GALTL in Peters Plus syndrome. Am J Med Genet A. 2008;146A:2603–10. doi: 10.1002/ajmg.a.32498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vincent A, Billingsley G, Priston M, Williams-Lyn D, Sutherland J, Glaser T, Oliver E, Walter MA, Heathcote G, Levin A, Heon E. Phenotypic heterogeneity of CYP1B1: mutations in a patient with Peters' anomaly. J Med Genet. 2001;38:324–6. doi: 10.1136/jmg.38.5.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Honkanen RA, Nishimura DY, Swiderski RE, Bennett SR, Hong S, Kwon YH, Stone EM, Sheffield VC, Alward WL. A family with Axenfeld-Rieger syndrome and Peters Anomaly caused by a point mutation (Phe112Ser) in the FOXC1 gene. Am J Ophthalmol. 2003;135:368–75. doi: 10.1016/s0002-9394(02)02061-5. [DOI] [PubMed] [Google Scholar]
- 14.Kaur K, Ragge NK, Ragoussis J. Molecular analysis of FOXC1 in subjects presenting with severe developmental eye anomalies. Mol Vis. 2009;15:1366–73. [PMC free article] [PubMed] [Google Scholar]
- 15.Iseri SU, Osborne RJ, Farrall M, Wyatt AW, Mirza G, Nurnberg G, Kluck C, Herbert H, Martin A, Hussain MS, Collin JR, Lathrop M, Nurnberg P, Ragoussis J, Ragge NK. Seeing clearly: the dominant and recessive nature of FOXE3 in eye developmental anomalies. Hum Mutat. 2009;30:1378–86. doi: 10.1002/humu.21079. [DOI] [PubMed] [Google Scholar]
- 16.Arikawa A, Yoshida S, Yoshikawa H, Ishikawa K, Yamaji Y, Arita RI, Ueno A, Ishibashi T. Case of novel PITX2 gene mutation associated with Peters' anomaly and persistent hyperplastic primary vitreous. Eye (Lond) 2010;24:391–3. doi: 10.1038/eye.2009.114. [DOI] [PubMed] [Google Scholar]
- 17.Lines MA, Kozlowski K, Kulak SC, Allingham RR, Heon E, Ritch R, Levin AV, Shields MB, Damji KF, Newlin A, Walter MA. Characterization and prevalence of PITX2 microdeletions and mutations in Axenfeld-Rieger malformations. Invest Ophthalmol Vis Sci. 2004;45:828–33. doi: 10.1167/iovs.03-0309. [DOI] [PubMed] [Google Scholar]
- 18.Berker N, Alanay Y, Elgin U, Volkan-Salanci B, Simsek T, Akarsu N, Alikasifoglu M. A new autosomal dominant Peters' anomaly phenotype expanding the anterior segment dysgenesis spectrum. Acta Ophthalmol. 2009;87:52–7. doi: 10.1111/j.1600-0420.2007.01082.x. [DOI] [PubMed] [Google Scholar]
- 19.Chavarria-Soley G, Michels-Rautenstrauss K, Caliebe A, Kautza M, Mardin C, Rautenstrauss B. Novel CYP1B1 and known PAX6 mutations in anterior segment dysgenesis (ASD). J Glaucoma. 2006;15:499–504. doi: 10.1097/01.ijg.0000243467.28590.6a. [DOI] [PubMed] [Google Scholar]
- 20.Dansault A, David G, Schwartz C, Jaliffa C, Vieira V, de la Houssaye G, Bigot K, Catin F, Tattu L, Chopin C, Halimi P, Roche O, Van Regemorter N, Munier F, Schorderet D, Dufier JL, Marsac C, Ricquier D, Menasche M, Penfornis A, Abitbol M. Three new PAX6 mutations including one causing an unusual ophthalmic phenotype associated with neurodevelopmental abnormalities. Mol Vis. 2007;13:511–23. [PMC free article] [PubMed] [Google Scholar]
- 21.Edward D, Al Rajhi A, Lewis RA, Curry S, Wang Z, Bejjani B. Molecular basis of Peters anomaly in Saudi Arabia. Ophthalmic Genet. 2004;25:257–70. doi: 10.1080/13816810490902648. [DOI] [PubMed] [Google Scholar]
- 22.Vincent A, Billingsley G, Priston M, Glaser T, Oliver E, Walter M, Ritch R, Levin A, Heon E. Further support of the role of CYP1B1 in patients with Peters anomaly. Mol Vis. 2006;12:506–10. [PubMed] [Google Scholar]
- 23.Reis LM, Semina EV. Genetics of anterior segment dysgenesis disorders. Curr Opin Ophthalmol. 2011;22:314–24. doi: 10.1097/ICU.0b013e328349412b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar RA, McGhee KA, Leach S, Bonaguro R, Maclean A, Aguirre-Hernandez R, Abrahams BS, Coccaro EF, Hodgins S, Turecki G, Condon A, Muir WJ, Brooks-Wilson AR, Blackwood DH, Simpson EM. Initial association of NR2E1 with bipolar disorder and identification of candidate mutations in bipolar disorder, schizophrenia, and aggression through resequencing. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:880–9. doi: 10.1002/ajmg.b.30696. [DOI] [PubMed] [Google Scholar]
- 25.Park HJ, Kim JK, Jeon HM, Oh SY, Kim SH, Nam DH, Kim H. The neural stem cell fate determinant TLX promotes tumorigenesis and genesis of cells resembling glioma stem cells. Mol Cells. 2010;30:403–8. doi: 10.1007/s10059-010-0122-z. [DOI] [PubMed] [Google Scholar]
- 26.Zuber ME, Gestri G, Viczian AS, Barsacchi G, Harris WA. Specification of the vertebrate eye by a network of eye field transcription factors. Development. 2003;130:5155–67. doi: 10.1242/dev.00723. [DOI] [PubMed] [Google Scholar]
- 27.Monaghan AP, Grau E, Bock D, Schutz G. The mouse homolog of the orphan nuclear receptor tailless is expressed in the developing forebrain. Development. 1995;121:839–53. doi: 10.1242/dev.121.3.839. [DOI] [PubMed] [Google Scholar]
- 28.Yu RT, Chiang MY, Tanabe T, Kobayashi M, Yasuda K, Evans RM, Umesono K. The orphan nuclear receptor Tlx regulates Pax2 and is essential for vision. Proc Natl Acad Sci USA. 2000;97:2621–5. doi: 10.1073/pnas.050566897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu HK, Belz T, Bock D, Takacs A, Wu H, Lichter P, Chai M, Schutz G. The nuclear receptor tailless is required for neurogenesis in the adult subventricular zone. Genes Dev. 2008;22:2473–8. doi: 10.1101/gad.479308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang CL, Zou Y, Yu RT, Gage FH, Evans RM. Nuclear receptor TLX prevents retinal dystrophy and recruits the corepressor atrophin1. Genes Dev. 2006;20:1308–20. doi: 10.1101/gad.1413606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sehgal R, Karcavich R, Carlson S, Belecky-Adams TL. Ectopic Pax2 expression in chick ventral optic cup phenocopies loss of Pax2 expression. Dev Biol. 2008;319:23–33. doi: 10.1016/j.ydbio.2008.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwarz M, Cecconi F, Bernier G, Andrejewski N, Kammandel B, Wagner M, Gruss P. Spatial specification of mammalian eye territories by reciprocal transcriptional repression of Pax2 and Pax6. Development. 2000;127:4325–34. doi: 10.1242/dev.127.20.4325. [DOI] [PubMed] [Google Scholar]
- 33.Azuma N, Yamaguchi Y, Handa H, Tadokoro K, Asaka A, Kawase E, Yamada M. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am J Hum Genet. 2003;72:1565–70. doi: 10.1086/375555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hanson IM. PAX6 and congenital eye malformations. Pediatr Res. 2003;54:791–6. doi: 10.1203/01.PDR.0000096455.00657.98. [DOI] [PubMed] [Google Scholar]
- 35.Stenman J, Yu RT, Evans RM, Campbell K. Tlx and Pax6 co-operate genetically to establish the pallio-subpallial boundary in the embryonic mouse telencephalon. Development. 2003;130:1113–22. doi: 10.1242/dev.00328. [DOI] [PubMed] [Google Scholar]
- 36.Kobayashi M, Yu RT, Yasuda K, Umesono K. Cell-type-specific regulation of the retinoic acid receptor mediated by the orphan nuclear receptor TLX. Mol Cell Biol. 2000;20:8731–9. doi: 10.1128/mcb.20.23.8731-8739.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hyatt GA, Schmitt EA, Marsh-Armstrong N, McCaffery P, Drager UC, Dowling JE. Retinoic acid establishes ventral retinal characteristics. Development. 1996;122:195–204. doi: 10.1242/dev.122.1.195. [DOI] [PubMed] [Google Scholar]
- 38.McFadden SA, Howlett MH, Mertz JR, Wallman J. Acute effects of dietary retinoic acid on ocular components in the growing chick. Exp Eye Res. 2006;83:949–61. doi: 10.1016/j.exer.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 39.McFadden SA, Howlett MH, Mertz JR. Retinoic acid signals the direction of ocular elongation in the guinea pig eye. Vision Res. 2004;44:643–53. doi: 10.1016/j.visres.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 40.Qu Q, Sun G, Li W, Yang S, Ye P, Zhao C, Yu RT, Gage FH, Evans RM, Shi Y. Orphan nuclear receptor TLX activates Wnt/beta-catenin signalling to stimulate neural stem cell proliferation and self-renewal. Nat Cell Biol. 2010;12:31–40. doi: 10.1038/ncb2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller LA, Smith AN, Taketo MM, Lang RA. Optic cup and facial patterning defects in ocular ectoderm beta-catenin gain-of-function mice. BMC Dev Biol. 2006;6:14. doi: 10.1186/1471-213X-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Escher P, Gouras P, Roduit R, Tiab L, Bolay S, Delarive T, Chen S, Tsai CC, Hayashi M, Zernant J, Merriam JE, Mermod N, Allikmets R, Munier FL, Schorderet DF. Mutations in NR2E3 can cause dominant or recessive retinal degenerations in the same family. Hum Mutat. 2009;30:342–51. doi: 10.1002/humu.20858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayashi T, Gekka T, Goto-Omoto S, Takeuchi T, Kubo A, Kitahara K. Novel NR2E3 mutations (R104Q, R334G) associated with a mild form of enhanced S-cone syndrome demonstrate compound heterozygosity. Ophthalmology. 2005;112:2115. doi: 10.1016/j.ophtha.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 44.Schorderet DF, Escher P. NR2E3 mutations in enhanced S-cone sensitivity syndrome (ESCS), Goldmann-Favre syndrome (GFS), clumped pigmentary retinal degeneration (CPRD), and retinitis pigmentosa (RP). Hum Mutat. 2009;30:1475–85. doi: 10.1002/humu.21096. [DOI] [PubMed] [Google Scholar]
- 45.Abrahams BS, Mak GM, Berry ML, Palmquist DL, Saionz JR, Tay A, Tan YH, Brenner S, Simpson EM, Venkatesh B. Novel vertebrate genes and putative regulatory elements identified at kidney disease and NR2E1/fierce loci. Genomics. 2002;80:45–53. doi: 10.1006/geno.2002.6795. [DOI] [PubMed] [Google Scholar]
- 46.Kumar RA, Everman DB, Morgan CT, Slavotinek A, Schwartz CE, Simpson EM. Absence of mutations in NR2E1 and SNX3 in five patients with MMEP (microcephaly, microphthalmia, ectrodactyly, and prognathism) and related phenotypes. BMC Med Genet. 2007;8:48. doi: 10.1186/1471-2350-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar RA, Leach S, Bonaguro R, Chen J, Yokom DW, Abrahams BS, Seaver L, Schwartz CE, Dobyns W, Brooks-Wilson A, Simpson EM. Mutation and evolutionary analyses identify NR2E1-candidate-regulatory mutations in humans with severe cortical malformations. Genes Brain Behav. 2007;6:503–16. doi: 10.1111/j.1601-183X.2006.00277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Biesecker LG, Mullikin JC, Facio FM, Turner C, Cherukuri PF, Blakesley RW, Bouffard GG, Chines PS, Cruz P, Hansen NF, Teer JK, Maskeri B, Young AC, Manolio TA, Wilson AF, Finkel T, Hwang P, Arai A, Remaley AT, Sachdev V, Shamburek R, Cannon RO, Green ED. The ClinSeq Project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009;19:1665–74. doi: 10.1101/gr.092841.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Antonarakis SE. Recommendations for a nomenclature system for human gene mutations. Nomenclature Working Group. Hum Mutat. 1998;11:1–3. doi: 10.1002/(SICI)1098-1004(1998)11:1<1::AID-HUMU1>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 50.Redeker EJ, de Visser AS, Bergen AA, Mannens MM. Multiplex ligation-dependent probe amplification (MLPA) enhances the molecular diagnosis of aniridia and related disorders. Mol Vis. 2008;14:836–40. [PMC free article] [PubMed] [Google Scholar]
- 51.Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baum L, Pang CP, Fan DS, Poon PM, Leung YF, Chua JK, Lam DS. Run-on mutation and three novel nonsense mutations identified in the PAX6 gene in patients with aniridia. Hum Mutat. 1999;14:272–3. doi: 10.1002/(SICI)1098-1004(1999)14:3<272::AID-HUMU21>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 54.Singh S, Chao LY, Mishra R, Davies J, Saunders GF. Missense mutation at the C-terminus of PAX6 negatively modulates homeodomain function. Hum Mol Genet. 2001;10:911–8. doi: 10.1093/hmg/10.9.911. [DOI] [PubMed] [Google Scholar]
- 55.Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, Davis S, Stemke-Hale K, Davies MA, Gershenwald JE, Robinson W, Robinson S, Rosenberg SA, Samuels Y. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet. 2011;43:442–6. doi: 10.1038/ng.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haider NB, Jacobson SG, Cideciyan AV, Swiderski R, Streb LM, Searby C, Beck G, Hockey R, Hanna DB, Gorman S, Duhl D, Carmi R, Bennett J, Weleber RG, Fishman GA, Wright AF, Stone EM, Sheffield VC. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat Genet. 2000;24:127–31. doi: 10.1038/72777. [DOI] [PubMed] [Google Scholar]
- 57.Faletra F, Athanasakis E, Minen F, Fornasier F, Marchetti F, Gasparini P. Vertebral defects in patients with Peters plus syndrome and mutations in B3GALTL. Ophthalmic Genet. 2011;32:256–8. doi: 10.3109/13816810.2011.587082. [DOI] [PubMed] [Google Scholar]
- 58.Land PW, Monaghan AP. Expression of the transcription factor, tailless, is required for formation of superficial cortical layers. Cereb Cortex. 2003;13:921–31. doi: 10.1093/cercor/13.9.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lesnik Oberstein SA, Kriek M, White SJ, Kalf ME, Szuhai K, den Dunnen JT, Breuning MH, Hennekam RC. Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am J Hum Genet. 2006;79:562–6. doi: 10.1086/507567. [DOI] [PMC free article] [PubMed] [Google Scholar]