

Abstract
Glioblastoma multiforme (GBM) remains one of the most malignant primary central nervous system tumors. Personalized therapeutic approaches have not become standard of care for GBM, but science is fast approaching this goal. GBM’s heterogeneous genomic landscape and resistance to radiotherapy and chemotherapy make this tumor one of the most challenging to treat. Recent advances in genome-wide studies and genetic profiling show that there is unlikely to be a single genetic or cellular event that can be effectively targeted in all patients. Instead, future therapies will likely require personalization for each patient’s tumor genotype or proteomic profile. Over the past year, many investigations specifically focused simultaneously on strategies to target oncogenic pathways, angiogenesis, tumor immunology, epigenomic events, glioma stem cells (GSCs), and the highly migratory glioma cell population. Combination therapy targeting multiple pathways is becoming a fast growing area of research, and many studies put special attention on small molecule inhibitors. Because GBM is a highly vascular tumor, therapy that directs monoclonal antibodies or small molecule tyrosine kinase inhibitors toward angiogenic factors is also an area of focus for the development of new therapies. Passive, active, and adoptive immunotherapies have been explored by many studies recently, and epigenetic regulation of gene expression with microRNAs is also becoming an important area of study. GSCs can be useful targets to stop tumor recurrence and proliferation, and recent research has found key molecules that regulate GBM cell migration that can be targeted by therapy. Current standard of care for GBM remains nonspecific; however, pharmacogenomic studies are underway to pave the way for patient-specific therapies that are based on the unique aberrant pathways in individual patients. In conclusion, recent studies in GBM have found many diverse molecular targets possible for therapy. The next obstacle in treating this fatal tumor is ascertaining which molecules in each patient should be targeted and how best to target them, so that we can move our current nonspecific therapies toward the realm of personalized medicine.
Keywords: GBM, oncogenomics, genetics, signaling cascades, pharmacogenomics
Introduction
Glioblastoma multiforme (GBM) represents one of the greatest challenges in the management of cancer patients worldwide. Even with aggressive surgical resections using state-of-the-art neuroimaging, along with recent advances in radiotherapy and chemotherapy, the prognosis for GBM patients remains dismal (median survival after diagnosis is about 14 months).1 A search for better, more personalized, specific, and less toxic GBM therapies is being conducted worldwide at a remarkable pace, with most of the studies over the past year focused simultaneously on strategies to target oncogenic pathways, angiogenesis, tumor immunology, epigenomic events, glioma stem cells (GSCs), and the highly migratory glioma cell population.
The pace at which genome-wide studies (GWSs) are being conducted has increased dramatically, and has led to an interest in other cell-wide profiling studies such as single nucleotide polymorphism (SNP)-omics, epigenomics, transcriptomics, metabolomics, microRNA (miRNA)-omics, proteomics, and secretomics. Even though the data gained from these studies show that there are only a few key signaling pathways in GBM, combination therapies targeting multiple pathways may still be required. Small molecule inhibitors are of special interest because they can directly target these signaling cascades with specificity and reduced toxicity. GBM is also a highly vascular tumor, and factors such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), basic fibroblast growth factor (bFGF)/FGF2, and hepatocyte growth factor (HGF)/scatter factor are implicated in angiogenesis. Therapy that directs monoclonal antibodies (MAbs) or small molecule tyrosine kinase inhibitors (TKIs) toward angiogenic factors has been the focus of recent studies in this area. Glioblastoma cells also have a distinct immunologic impact and express tumor-associated antigens. Passive, active, and adoptive immunotherapies have been explored by many studies. Another approach to GBM therapy is targeting the epigenetic mechanisms to alter gene expression. Demethylation of hypermethylated promoters is one possible way to reactivate gene expression, but a major issue with this remains target specificity. The study of miRNAs is a growing new area of research, and presents more possible targets for epigenetic therapy. GSCs, the cells that are largely responsible for the recurrence of tumors, can also be targeted by specific therapy such as the inhibition of stem cell pathways or prodifferentiation. A major challenge to GBM treatment is the proliferation of migrating cells, and recent research has found key molecules that regulate GBM cell migration that can be targeted by therapy. Although current treatment for GBM is not individualized, the trend shown by studies of other tumors show that personalized medicine can become the standard of care in the near future.
Overview of glioblastoma
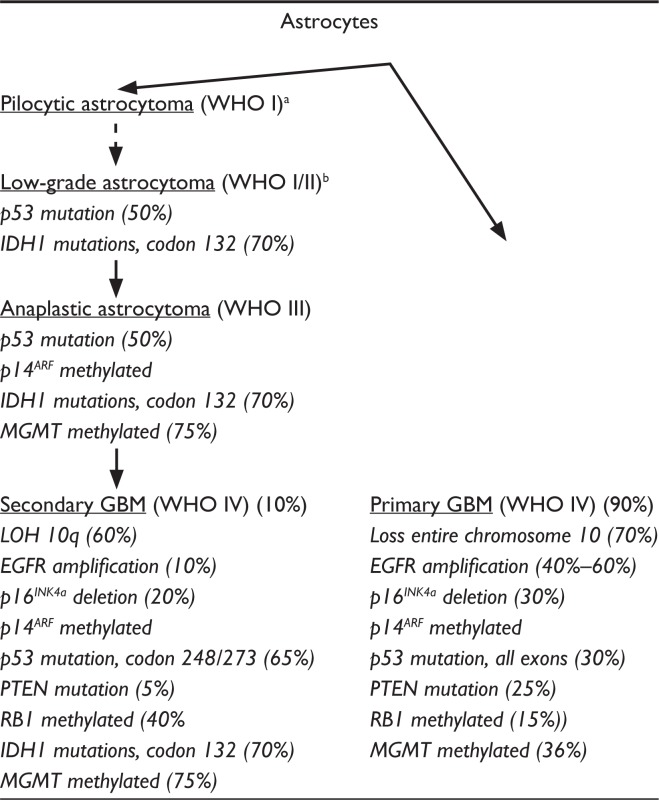
Primary central nervous system (CNS) gliomas account for about 1.4% of all cancers, but rank second in the causes of death from neurological disease.2 Glioblastoma is the most common primary CNS tumor, with about 3 in 100,000 people newly diagnosed each year, accounting for over 51% of all gliomas.3 Gliomas are categorized as World Health Organization (WHO) grades I through IV, based on histological characteristics, which carry prognostic and survival correlations. Glioblastoma is a WHO grade IV glioma, the most malignant grade. For decades, it has been known that some gliomas of lower WHO grades can recur, progress, or transform into GBM. These have been traditionally termed secondary GBMs, whereas de novo GBM tumors are termed primary GBMs. The genetic heterogeneity of GBMs underscores the existence of these two subtypes (Table 1). For example, recent GWSs have identified mutations in NADP(+)-dependent isocitrate dehydrogenase (IDH) genes that appear frequently in secondary GBM.4 The mean age of primary GBM patients is about 62, whereas the mean age of secondary GBM patients is about 45.5,6
Table 1.
Major genetic alterations in primary and secondary GBM initiation and progression
|
Some authors consider pilocytic astrocytoma as a distinct tumor that does not transform to higher grade astrocytomas (dotted arrow);
Some authors describe low-grade astrocytoma as WHO I or WHO II.
Abbreviations: EGFR, epidermal growth factor receptor; IDH, isocitrate dehydrogenase; LOH, loss of heterozygosity; MGMT, O6-methylguanine-DNA methyltransferase; PTEN, phosphatase and tensin homolog; RB1, retinoblastoma 1.
The etiology of GBM is unknown but is likely multifactorial, with several environmental and genetic agents implicated as risk factors.7 Although the incidence of gliomas has been reported by some to be on the rise,8,9 the predisposing factors are poorly understood.10–13 The only proven environmental risk factor for gliomas is exposure to ionizing radiation.14 Many other factors have been suggested, including chemical carcinogens in occupations such as rubber manufacturing, petroleum production, vinyl chloride, pesticides, forestry work, cleaning services, and passive smoking exposure.11 Interestingly, increasing socioeconomic status increases the risk for gliomas.15 Caucasians are more frequently affected than Asians or Africans.6,16–18 About 5% of gliomas represent familial aggregations, with some seen syndromes such as Cowden disease, Li-Fraumeni syndrome, and neurofibromatosis.10 Studies have noted some molecular correlations with longer survival, such as O6-methylguanine-DNA methyltransferase (MGMT) hypermethylation; however, no molecular event has become routinely accepted as a prognostic factor or routinely used for diagnostic or therapeutic purposes.19 Age and Karnofsky performance status provide the primary prognostic factors in commonly used recursive partitioning analyses seen in the Radiation Therapy Oncology Group and European Organization for Research and Treatment of Cancer prognostic classes.20,21
GBM patients may present with progressive focal neurologic deficits, headaches, and seizures. The aggressive growth of GBMs typically precludes incidental discovery. Diagnosis begins with suspicious findings on magnetic resonance imaging (MRI) that includes T1-weighted images with and without gadolinium and T2-weighted images. A contrast-enhanced computed tomography scan may be useful; however, this modality remains far inferior. Glioblastoma is hypointense to isointense with a ring-pattern of enhancement on gadoliniumenhanced T1 images and is hyperintense on both T2 and fluid attenuation inversion recovery images. It can be focal, multifocal, or diffuse (gliomatosis cerebri). The glioblastoma is mainly found in the frontal lobes, but they also occur in the cerebellum, brainstem, and spinal cord. The majority of neoplastic cells are found within the tumor bed and within 2 cm of the enhancing border; however, migrating cells can be found several centimeters away from the tumor and even in the contralateral hemisphere. Magnetic resonance spectroscopy, magnetic resonance perfusion, and positron emission tomography are more sophisticated imaging tools that can help delineate varied metabolic rates and therapeutic responses. They can be particularly useful for differentiating tumor recurrence from radiation necrosis.
GBM consists of poorly differentiated neoplastic astrocytes, cellular and nuclear atypia, brisk mitotic activity, diminished apoptosis, neoangiogenesis, vascular thrombosis, and pseudopallisading necrosis.22–24 Vascular hyperproliferation and necrosis are essential diagnostic features that set GBM apart from lower grade gliomas. Despite its highly invasive properties, GBM does not metastasize outside the CNS.
Surgery remains the cornerstone in the treatment of GBM despite a lack of phase 3 randomized clinical trials (RCTs). Resection of >98% doubles the survival over that following biopsy alone.25–28 GBM infiltrates adjacent brain parenchyma, so complete resection is not possible; however, modern neuroimaging allows tumor resection in cases where tumor does not involve eloquent structures (eg, Broca’s speech cortex). Only maximal cytoreduction of >98% significantly alters the survival and likely improves response to radiotherapy and chemotherapy.28,29 Surgery only helps in the short term. A recent meta-analysis shows that 2-year diseasefree survival is unchanged in patients who receive maximal cytoreductive surgery vs biopsy alone.30 Patients who undergo surgery at high-volume academic centers appear to have an advantage, as mortality at these centers is half (2.5%) of that seen at low-volume centers (4.9%).31
Radiation therapy (RT) has also been shown to add several months to survival and has become part of the standard of care. Postoperative fractionated external-beam radiotherapy is administered as 60 Gy in 30 fractions over a period of about 6 weeks to a target volume defined as a 2- to 3-cm ring of tissue beyond the contrast-enhancing rim of tumor seen on the preoperative MRI. All enhancing tumor is ideally resected. RT should not be started before 2 weeks after surgery to allow for adequate wound healing. Phase 3 RCTs have shown that RT can prolong survival after surgery to 9–10 months, doubling the 4–5 months of survival of patients with surgery alone.32,33
Meta-analyses looking at various heterogeneous chemotherapy regimens show an overall survival benefit of an additional 1–2 months.30 Therefore, in combination with surgery and RT, chemotherapy provides an overall median survival of about 14 months.30 However, this may include selection biases for patients involved in clinical trials.30,34 The most commonly tested agents that have shown some modest benefits are nitrosoureas. Only 2 agents, polifeprosan 20 with carmustine implant and temozolomide (TMZ), have made their way into standard of care treatments for newly diagnosed and recurrent GBM, both of which separately increase the survival about 2 months.1,35,36 Polifeprosan 20 with carmustine implant is a bis-chloronitrosourea (BCNU [carmustine])-impregnated polymer wafer placed intraoperatively along the resection cavity.35 TMZ is an imidazotetrazine derivative of the alkylating agent dacarbazine. It gets converted in the systemic circulation at physiologic pH to the active compound monomethyl triazeno imidazole carboxamide. It exhibits schedule-dependent antineoplastic activity by interfering with DNA replication. It acts nonspecifically on rapidly dividing cells, but has the benefit of readily crossing the blood–brain barrier with little toxicity. TMZ should be administered concomitantly with RT at 75 mg/m2 daily followed by 200 mg/m2 for 5 days every 4 weeks for a total of 6 months.1 Patients with methylation (and therefore silencing of gene expression) of MGMT that confers resistance to alkylating agents respond better to TMZ.37 The efficacy of the combination of BCNU and TMZ has not been established.
Pharmacogenomics of glioblastoma
The past decade has seen a tremendous growth in the exploration into the oncogenomics and molecular biology of GBM, revealing a heterogeneous and extensive genomic landscape.7 Current understanding of the molecular characteristics of this disease has demonstrated that there are unlikely to be single genetic or cellular events that can be effectively targeted in all patients. Future therapies will require personalization for each patient’s tumor genotype or proteomic profile. The birth of this century witnessed a new era of investigational drugs, transitioning from the traditional nonspecific chemotherapies of the past to target-specific, molecular-based drugs developed in response to our new understanding of the molecular biology of this deadly tumor. Even though many investigational agents have demonstrated preclinical success, none have yet to provide meaningful results at the clinical stage of testing. Our current standard of care chemotherapies remains nonspecific. Nevertheless, our rapidly expanding knowledge of GBM genomics has allowed us to enter an era of unprecedented pharmacogenomic investigation where highly specific and safer efficacious therapies are just around the corner. GWSs are continually discovering new molecular targets. Clearly, the investigation of GBM therapies is shifting over to one that is more patient-specific and personalized by targeting specific oncogenic pathways in each patient. Over the past year, most investigations have focused simultaneously on strategies to target oncogenic pathways, angiogenesis, tumor immunology, epigenomic events, GSCs, and the highly migratory glioma cell population.38–41 These areas dominated recent neuro-oncology conferences, including the Society of Neuro-oncology and the American Association of Neurological Surgeons’ Section on Tumors conferences.
Despite a common clinical presentation and histology, GBM is a genetically heterogeneous tumor.7 Recent comprehensive genetic screens of GBM have confirmed that genetic alterations are scattered across the entire genome; common regions of loss include areas on 1p, 6q, 9p, 10p, 10q, 13q, 14q, 15q, 17p, 18q, 19q, 22q, and Y.42–47 Many of these genetic losses represent loss of specific tumor suppressor genes with direct effects on gliomagenesis; however, some of these losses likely represent the inherent genomic instability that develops in tumor cells. Gains of gene expression due to genetic alterations at the genomic level have been demonstrated in GBM in the form of duplication of entire chromosomes, intrachromosomal amplification of specific alleles, extrachromosomal amplification (often in the form of double minutes), and activating mutations.47–49 Gene overexpression occurs less frequently than loss. Gliomagenesis also involves errors in DNA replication, DNA repair, chromosomal segregation, and alteration of numerous signaling cascades not directly attributed to genomic mutations. This collection of genetic and cellular alterations gives rise to the “mutator phenotype” in glioma cells.50 Central to this mutator phenotype are DNA repair mechanisms. There are at least 4 DNA repair pathways that may go awry in GBMs, including nucleotide excision repair, base excision repair, mismatch repair, and direct reversal of lesions in recombination.50,51 MGMT, one of the most studied DNA repair enzymes, has demonstrated increased levels in GBM.52 MGMT specifically removes promutagenic alkyl groups from the O6 position of guanine in DNA. Therefore, MGMT protects cells against carcinogenesis induced by alkylating agents. Repair of O6-alkylguanine adducts by tumor cells has been implicated in drug resistance since it reduces the cytotoxicity of alkylating chemotherapeutic agents.53 Loss of MGMT expression may be caused by methylation of promoter CpG islands, which has been observed in gliomas.54,55
Despite the development of a mutator phenotype and a plethora of genetic alterations, there are a discrete number of signaling pathways that appear to be most commonly affected and implicated in GBM.56,57 Aberrant epidermal growth factor receptor (EGFR) and other tyrosine kinase receptor autocrine signaling pathways lead to robust alterations in cellular development, proliferation, migration, and vacularization. Growth factor signaling activates an intricately complex network of intracellular cascades modulated by G-protein-coupled receptors and second messengers that converge at multiple sites, one of which is Ras. In GBM, specific mutations affecting Ras have not been detected; however, high levels of Ras guanosine triphosphate (GTP) have been documented in cell lines and primary tumors, suggesting that this signaling pathway is activated by upstream factors such as receptor tyrosine kinase activation.58 Another major way of activating this pathway is via the loss of neurofibromin function, the protein product of the large neurofibromatosis 1 (NF1) gene.59–61 Ras-GTP can also activate the Raf-MAPKK-ERK, PI3K-PKB, and PI3K-Rac-Rho pathways. These influence cell survival and migration.62 GBM also demonstrates inactivating mutations in the TP53 pathway and retinoblastoma (RB1) pathways.63–66 The TP53 transcription factor facilitates DNA repair by halting the cell cycle for repair enzymes to work, or if the damage is too great, it induces cell death. Loss of normal TP53 function can result from altered expression of either TP53 gene, MDM2, or p14ARF genes in GBM.67 RB1 controls the transition from G1 into S phase of the cell cycle by inhibiting the action of elongation factor E2F1, and its expression is commonly altered in GBM. The cyclin-dependent kinase 4 (CDK4)/cyclin D1 complex phosphorylates the RB1 protein, thereby increasing release of the E2F1 transcription factor that activates genes involved in the G1-to-S transition.68 Mutations of phosphatase tensin homolog (PTEN) on chromosome 10q23, also called MMAC1 and TEP1, occur frequently in GBM.69,70 PTEN contains a central catalytic phosphatase core domain that negatively regulates phosphatidylinositol-3 kinase (PI3K) by dephosphorylating phosphatidylinositol-3,4,5 triphosphate (PIP3) and phosphatidylinositol-3,4 diphosphate (PIP2).71 In the case of mutant PTEN, the elevated lipid second messenger PIP3 is used by PI3K to hyperphosphorylate protein kinase B (PKB)/Akt.72 This modulates the activity of proteins that play a critical role in cell survival, invasion, and proliferation.73 Isocitrate dehydrogenase (IDH1 and IDH2) mutations which reduce enzymatic activity are frequent in secondary GBMs.4
In addition to these known genetic events, recent GWS has provided additional insight on the genetic abnormalities of GBM. Over the past year, there has been an exponential increase in GWS, largely due to the marked decrease in their cost and time. Publically available databases such as The Cancer Genome Atlas (TCGA) and Rembrandt make the process even cheaper and quicker. Recent GWS analyses have identified signatures (metagene or multigene signatures74) to divide GBM tumors into genetically distinct subgroups, which may represent an improved classification system over histological classification.75–78 These studies can help to identify GBM subtypes that are sensitive to specific therapies which target the defective signaling networks existing in each of the subtypes and lead to more individualized therapy. Also, studies show that gene expression profiles of gliomas may be a better predictor of survival than histology.76,79 For example, Bredel et al80 show that 7 highly interactive genes are significantly associated with survival (POLD2, CYCS, MYC, AKR1C3, YME1L1, ANXA7, and PDCD4). Recent studies have added new TSGs such as p18INK4c, PTPRD, CASPR2, BNC2, PTPLAD2, and PTPRE to the ever growing list.77,81 Recently identified novel oncogenes include GBR10, MKLN1, PPARGC1A, HGF, NAV3, CNTN1, SYT1, and ADAMTSL3.81 By using an automated network-based approach with TCGA data, Cerami et al82 discovered a new driver candidate gene AGAP2/CENTG1, which can activate the PI3K pathway. They also found alterations in the genes DCTN2, TUBGCP2, TUBGCP6, and FGFR1OP, which are all located in the centrosome and could disrupt chromosomal segregation and/or the DNA damage response.82
The increased ease of GWS has allowed more in-depth and genome-wide investigation of SNPs in GBM. Most SNPs are nonfunctional; however, some do appear to create genetic expression alterations that favor tumorigenesis or provide prognostic information. SNPs recently identified by GWS that are associated with GBM include HLA-A, ALOX5, IRAK3, ITGB2, NCF2, NFKB1, SELP, SOD1, STAT1, PARP1, PRKDC, ERCC2, ERCC1, and GLTSCR1.83–85 Therapies that target some of these oncogenic events are being developed, but hurdles still exist. Gene delivery systems such as viral vectors (herpes simplex, adenovirus, and poliovirus), nanoparticle constructs, expression plasmids, and liposomal preparations are under study.
The ease with which we now do GWS has lead to a thrust in performing similar comprehensive profiles of virtually all cellular processes affecting tumorigenesis, ranging from genomic and SNP-omic to epigenomic (methylomic), transcriptomic, metabolomic, miRNA-omic, proteomic, and secretomic (Figure 1).75,78,86–88 Comprehensive epigenomic profiles of DNA methylation are providing many areas of glioma cell regulation ready for exploitation by demethylating agents. Fouse et al recently used unique methylation profiles to easily distinguish between GBM cells.77 Brennan et al89 used the TCGA as part of their study to analyze the GBM proteome for signal transduction pathway activation and mutation in pathway member genes, classifying GBM into 3 subclasses that are associated with EFGR activation, PDGFR activation, or the loss of NF1, a RAS regulator. This type of classification is useful for the development of targeted therapies. The metabolomics of GBM cells gained a renewed interest recently with the interesting discovery that IDH1 and IDH2 mutations are frequent in gliomas and are associated with younger age and a better prognosis.5,90,91 Other recent discoveries in the study of metabolomics include the identification of ATP citrate lyase as a positive regulator of glycolic function in GBM.92 Studies using miRNA-omic profiles have found many miRNAs that are involved in the GBM, and studies using the transcriptome have defined subtypes of glioma.75,93 All of these new comprehensive profiles are providing detailed knowledge about the molecular and cellular biology of GBM development to a degree never before appreciated, identifying potential targets at a rapid pace.
Figure 1.

The major profiles of glioma cell processes under investigation.
Targeted combination therapy
The data gained from GWS show that there appears to be a few key redundant signaling pathways in GBM. Therefore, the upstream location of targets (eg, growth factor receptors) makes the drugs that target them susceptible to downstream resistance due to other factors driving the pathway of interest. Recent studies highlight that combination therapies may be required to significantly target a specific pathway or a combination of important pathways (Figure 2).94 For example, multiple mechanisms involving polo-like kinase 1 (Plk-1), protein kinase B (Akt-1), and/or p53 pathways are implicated in the glioma cell response to DNA damage caused by chemotherapy and radiotherapy.95 The small molecule inhibitor of protein phosphatase 2A, LB-1.2, activates Plk-1 and Akt-1 and paradoxically enhances cytotoxic chemotherapy efficacy.95 When LB-1.2 is used in conjunction with TMZ, there is a complete regression of GBM in vivo, whereas treatment with either drug alone only induces short-term inhibition.95 When LB-1.2 is combined with another drug, doxorubicin (which targets the topoisomerase II complex), the same effect is observed.95 Therapies targeting both EGFR and c-Met also have shown better efficacy. Cotreatment with panitumumab (an anti-EFGR antibody) and AMG 102 (a neutralizing antibody to HGF, a ligand for c-Met) induces tumor inhibition in vitro, whereas each drug used alone fails to completely inhibit tumor growth.96 Another possible target for therapy is FOXO1, which is at the convergence of several growth factor receptor tyrosine kinase pathways. Targeting FOXO1 can induce glioma cell death and inhibit tumor growth in vitro.97
Figure 2.

Cellular targets for current investigational agents in clinical trials for GBM.
Understandably, older studies focused on proteins overexpressed on the surface of GBM cells as promising targets. Antibodies directed at EGFR and its mutated variants have been studied for many years, but they have not quickly translated into effective therapies. More work is being done.98 The more recent elucidation of the aberrant signaling pathways and their redundant, crosstalking factors (eg, mTOR; Figure 2) has now moved a significant amount of focus toward studying small molecules that can directly target these downstream factors intracellularly (Table 2). Many signaling factors are now being targeted by investigational small molecular inhibitors in clinical trials (Figure 2). Small molecular inhibitors that target unique proteins have gained considerable popularity. Factors targeted by small molecular inhibitors include Akt (perifosine), EGFR (erlotinib), farnesyltransferase (lonafarnib), histone deacetylase (vorinostat), heat shock proteins (AT13387), Met (XL184), mTOR (sirolimus), PI3K (BEZ235), PKCβ (enzastaurin), PDGFR (imatinib), proteasomes (bortezomib), Raf (sorafenib), Src (dasatinib), and transforming growth factor (TGFβ, AP12009; Figure 2). Many of these are in trials of combination therapies. This approach offers the advantage of remarkable specificity, reduced toxicity, and applicability to other cancers with similar aberrant signaling cascades. For example, a phase 2 trial of an antiangiogenic antibody, bevacizumab, combined with a topoisomerase I small molecule inhibitor, irinotecan, shows efficacy in recurrent GBM.99 Once efficacious GBM therapies are established (still a great hurdle), we anticipate a personalized approach with targeted combination therapies. Each patient will be tested for his or her tumor’s unique genomic, SNP-omic, epigenomic, transcriptomic, metabolomic, miRNA-omic, proteomic, and secretomic profiles. Thus, a plethora of treatment options specifically targeting unique molecules in the patient will be available.
Table 2.
Small molecule targets for investigational agents
| Target | Agent |
|---|---|
| AKT inhibitor | Perifosine |
| EGF RTK inhibitor | Erlotinib, gefitinib, lapatinib, BIBW2992, nimotuzumab, cetuximab, AEE788 |
| Growth factor inhibitor | Leflunomide, suramin |
| FTI inhibitor | Tipifarnib, lonafarnib |
| HDAC inhibitor | Vorinostat, depsipeptide, panobinostat, romidepsin |
| HSP90 inhibitor | AT13387 |
| LDL receptor peptide | ANG1005 (taxane derivative targets LDL receptors) |
| Met inhibitor | XL184 |
| mTOR inhibitor | Everolimus, sirolimus, temsirolimus, deforolimus, rapamycin |
| PDGF RTK inhibitor | Dasatinib, imatinib, tandutinib, pazopanib |
| PI3K inhibitor | BEZ235, XL765 |
| PKCβ STK inhibitor | Enzastaurin |
| Proteosome inhibitor | Bortezomib |
| Raf inhibitor | Sorafenib |
| Ras inhibitor | TLN-4601 |
| Sp1 inhibitor | Terameprocol |
| Src TK inhibitor | Dasatinib |
| TGFβ inhibitor | AP12009 (antisense) |
| Topoisomerase inhibitors | RTA744, etoposide, topotecan, irinotecan, AQ4N, edotecarin, rubitecan, pyrazoloacridine, karenitecin, gimatecan |
| VEGF RTK inhibitor | PTK787, semaxanib |
| Others | 131I-TM601 (scorpion venom peptide), CC-8490 |
Abbreviations: EGF, epidermal growth factor; FTI, farnesyltransferase inhibitor; HDAC, histone deacetylase; LDL, low-density lipoprotein; mTOR, mammalian target of rapamycin; PDGF, platelet-derived growth factor; PI3K, phosphoinositide 3-kinase; PKC, protein kinase C; RTK, receptor tyrosine kinase; STK, Src tyrosine kinase; TGF, transforming growth factor; TK, tyrosine kinase; VEGF, vascular endothelial growth factor.
Importantly, comprehensive profiles may provide prognostic data for drug response but not necessarily the drug target. SNP-omic and other types of profiles have identified genetic events that identify patients who are susceptible to specific drugs. For example, Halatsch et al100 found 2 genes (DUSP4 and STAT1) that are significantly associated with sensitivity to erlotinib (which targets EGFR and inhibits autophosphorylation), and 10 genes (CACNG4, FGFR4, HSPA1B, HSPB1, NFATC1, NTRK1, RAC1, SMO, TCF7L1, and TGFB3) that are associated with resistance to erlotinib. An MDR1 SNP was recently identified as a novel-independent predictive factor for patient response to TMZ.101 The characterization of the features of each patient’s tumor is already slowly entering some clinics but has not become uniformly accepted. A test for the methylation status of MGMT promoter exists, and in the near future, should be available everywhere to predict response to TMZ.102
Antiangiogenesis therapy
Glioblastoma is a highly vascular tumor that shows great potential as a target for antiangiogenic therapy.103 There are several angiogenic factors that are expressed in glioma cells, and these factors are involved in signaling pathways that promote neovascularization and tumor growth.104 VEGF, PDGF, basic FGF (bFGF)/FGF2, and HGF/scatter factor are the most common factors to be implicated in the angiogenesis of GBM.103 Other markers of abnormal vasculature include endocan (endothelial cell-specific molecule-1), which is expressed in high-grade gliomas.105 Current antiangiogenic targeted molecular therapy directs MAbs or small molecule TKIs toward angiogenic factors such as VEGF.104 VEGF promotes the proliferation and migration of endothelial cells, and thus is one of the more potent growth factors.104 Bevacizumab, an MAb that targets VEGF, was US Food and Drug Administration (FDA) approved in May 2009 as a single agent in GBM patients with progressive disease following prior therapy (TMZ was FDA approved as upfront GBM treatment in 2005). The 2 clinical trials that lead to the FDA approval were AVF3708g and NCI 06-C-0064E. The dose for both trials was 10 mg/kg IV.106 In AVF3708g, responses were observed in 25.9% of the patients, and the median response duration was 4.2 months.106 In NCI 06-C-0064E, an objective response rate was 19.6% and median response duration of 3.9 months was seen.106 Bevacizumab works by preventing VEGF from activating its receptors, thus abrogating subsequent effects of the signaling pathway.103 Studies of the efficacy of therapy combining bevacizumab with irinotecan are already underway, and phase 3 trials looking at the effects of adding bevacizumab to RT and TMZ for newly diagnosed GBM will soon begin.103,104 Other therapeutic agents targeting VEGF include aflibercept (VEGF-Trap), which works by depleting the circulating VEGF because aflibercept has greater affinity for VEGF than bevacizumab. A recent phase 2 study shows similar response rates between the two agents.103 Inhibitors of VEGFRs have produced promising results too, such as the pan-VEGFR inhibitor cediranib (AZD2171).103 Antiangiogenic agents targeting other pathways are less successful than VEGF/VEGFR; for example, imatinib mesylate, which inhibits PDGFR, is ineffective in part due to poor penetration across the blood–brain barrier, and cilengitide, which inhibits integrins, only shows modest activity in GBM.103 Studies of many more therapeutic agents are in progress, targeting other factors such as HGF (AMG-102) and EGFR (cetuximab; Table 3).104 One challenge to overcome in clinical studies with MAbs is the difficulty in delivering the agents across the blood–brain barrier.104 Other concerns of these therapies are the possible increased risk of intracranial hemorrhagic complications, venous thromboembolism, impaired wound healing, proteinuria, and hypertension.103 Because of the heterogeneous nature of the tumor, a future goal of antiangiogenic therapies should be to individualize treatment for each patient according to the tumor’s angiogenic phenotype and molecular pathways.104
Table 3.
Molecularly targeted agents in antiangiogenic therapy
| Agent | Target(s) |
|---|---|
| Bevacizumab | VEGF-A |
| Aflibercept (VEGF-Trap) | VEGF |
| Vandetanib | VEGFR, EGFR |
| Pazopanib | VEGFR, PDGFR, c-kit |
| Cediranib | VEGFR, PDGFR, c-kit |
| Vatalanib | VEGFR, PDGFR, c-kit |
| CT322 | VEGFR |
| Brivanib | VEGFR |
| DC101 | VEGFR |
| XL184 | c-met, VEGFR |
| XL880 | c-met, VEGFR, PDGFR, Tie-2 |
| Gefitinib | HER1/EGFR, Ras |
| Erlotinib | HER1/EGFR, Ras |
| Imatinib | PDGFRa, c-kit, BCR-ABL |
| MLN518 | PDGFR, Fit-3, c-kit |
| AEE788 | EGFR, VEGFR |
| Cetuximab | EGFR |
| Sorafenib | VEGFR, PDGFR, c-kit, Raf |
| Sunitinib | VEGFR, PDGFR, c-kit, FLT-3 |
| AMG-102 | HGF/SF |
| Cilengitide | αvβ3 and αvβ5 |
| ATN-161 | α5β1 |
Abbreviations: EGFR, epidermal growth factor receptor; HER, human epidermal receptor; HGF, hepatocyte growth factor; PDGFR, platelet-derived growth factor receptor; SF, scatter factor; VEGF, vascular endothelial growth factor.
Immunotherapy
Glioblastoma also alters the immunologic profile of the normal CNS environment, providing additional opportunities for exploring immune-based therapies.72 Glioma cells express numerous tumor-associated antigens107,108 that mostly exhibit immunosuppressive activities, such as hindering cell-mediated immunity. Other signs of an altered immune system seen in GBM include cutaneous anergy, lymphopenia, impaired antibody production, reduced lymphocyte protein synthesis, and diminished lymphocyte responsiveness.109–120 Many complex interactions between glioma cells and immune cells are thought to be mediated by glioma-derived cytokines such as interleukin (IL)-6, IL-8, TGF-β2, and VEGF. In addition, glioma cells are thought to induce expression of immunosuppressive factors from other cells within the environment, such as IL-10 and prostaglandins from monocytes.121 Some of these factors that are overexpressed in GBM have multiple effects. Vascular endothelial growth factor, which plays a key role in tumor neoangiogenesis, also inhibits the maturation of dendritic cells from progenitor cells originating from the bone marrow and promotes GBM tumor cell proliferation. Exploiting the immune system for investigational agents may go a long way to enhance the patient’s own immunotherapy against malignant gliomas and directly kill glioma cells.
Many exciting immunotherapies for GBM are now being pursued, including passive, active, and adoptive immunotherapeutic approaches. Passive immunotherapy involves administering antibodies or toxins to patients without specifically inducing or expanding a host antitumor response. Examples of antibodies currently under study include iodine-131-labeled antitenascin antibody (also known as 81C6), astatine-211-labeled antitenascin antibody, and the bispecific anti-EGFR and anti-CD64 antibody. Immunotoxin therapies include transferrin-CRM107 (transferrin conjugated with diphtheria toxin) and TP38 (TGF conjugated with Pseudomonas exotoxin). These passive immunotherapies all target proteins overexpressed on the surface of glioma cells. TP38 represents an elegant example of molecular engineering. This immunotoxin is a recombinant chimeric protein composed of the EGFR-binding ligand TGF and a genetically engineered Pseudomonas exotoxin. Once this protein binds cells expressing EGFR, a domain of the exotoxin undergoes proteolytic cleavage and mediates translocation of the carboxyl terminal toxin into the cytosol. Another domain of the exotoxin contains an adenosine 5′-diphosphate ribosylating activity that inactivates elongation factor 2, resulting in the death of the cell. In active immunotherapy, the tumor-bearing host is immunized with a “vaccine” to expand an antitumor immune response in vivo, whereas adoptive immunotherapy employs the ex vivo expansion of effector cells and return of these effectors to the tumor-bearing host. Examples of active immunotherapies include autologous tumor cell vaccines, vitespan (heat shock protein gp96 vaccine), and PEP-3-KLH (EGFRvIII peptide conjugated to an immunostimulatory agent). Exciting adoptive immunotherapy strategies include various dendritic cell vaccines. In these complex treatments, autologous dendritic cells are isolated from patients, pulsed with tumor-specific molecules (eg, tumor-specific peptides [EGFRvIII], tumor cell lysates, and tumor stem cell mRNA), expanded ex vivo, and then reintroduced to the patient.122 This field of investigation has slowly moved from nonspecific immunostimulatory approaches toward efforts eliciting very specific immune responses against tumor antigens, either by use of active immunization (cancer vaccines) or adoptive transfer of tumor-specific effector cells or antibodies (adoptive immunotherapy).59
Epigenetic therapy
In addition to targeting the genes themselves, targeting the epigenetic mechanisms to alter gene expression can also offer new approaches to GBM therapy. Epigenetic modifications do not cause changes in the primary DNA sequence, so theoretically the process is reversible and can be exploited for drug targeting.123 A well-studied epigenetic gene regulation mechanism is DNA methylation, which results from the addition of methyl groups at 5′-CpG-3′ dinucleotides by DNA methyltransferases.123 DNA methylation does not only occur aberrantly in tumors, but also participates in controlling gene expression in normal cells through processes such as silencing genes on the inactive X chromosome.123 Hypermethylation of promoter regions downregulates gene expression and allows for cancer development by silencing genes involved in cell cycle (p16INK4a and p15INK4b), tumor suppression (RB1, VHL, EMP3, RASSF1A, GATA6, and BLU), tumor invasion (AJAP1), and apoptosis (DAPK, TIMP3, CDH1, PCDH-γ-A11, and TMS1/ASC).87,124,125 Promoter methylation can alter the chemotherapy and radiotherapy sensitivity too, as is the case with the well-known example of MGMT promoter methylation. MGMT hypermethylation is associated with better response to alkylating agents and longer survival in GBM patients.126 Demethylation of hypermethylated promoters can reactivate gene expression and function, but the major issue for therapy based on this method is target specificity.123,124 Drugs that reactivate silent genes may globally demethylate, bringing the risk of activating undesired genes.123 Other mechanisms of epigenetic gene regulation include the posttranslation modification of histone proteins by acetylation, methylation, phosphorylation, and other modifications.123 Histone deacetylase inhibitors (HDACis) work by promoting a more open chromatin conformation to permit better access of other therapeutic agents and by reversing aberrant epigenetic gene silencing.123 Suberoylanilide hydroxamic acid, an HDACi, is shown to slow the growth of GBM, and other HDACi can inhibit the growth of GBM stemlike cells.124,127
Another rapidly growing area of study in epigenetic mechanisms is gene regulation by miRNAs (Figure 3). MiRNAs are endogenous, small (19–24 nucleotides) nonprotein encoding, single-stranded RNA molecules that participate in the posttranscription regulation of gene expression.128,129 MiRNAs act by imperfect complementary base pairing to the 3′ untranslated regions of their target mRNAs, leading to translational repression and/or mRNA degradation.129 Studies have estimated that at least one-third of all protein-coding genes in the human genome may be regulated in part by miRNAs, so this is clearly an important area of investigation.130 MiRNAs can potentially target many molecules and pathways at one time, and each mRNA can be regulated by multiple miRNAs.129 Large-scale miRNA expression profiling studies have found a distinguishing miRNA expression pattern in GBM,131 and functional miRNA studies have identified multiple upregulated miR-NAs (miR-21, miR-221/222, miR-339, and miR10b) that can suppress apoptosis, enhance cell proliferation and migration, and increase drug resistance.129,130,132–135 Multiple downregulated miRNAs have also been found (miR-128-1, miR-7, miR-124/137, miR-34a, miR-153, and miR-181) that inhibit glioma cell self-renewal, proliferation, invasion, and growth, and promote cell cycle arrest/induce apoptosis (Table 4).129–132 Gaire et al136 used TCGA to identify the relationships between miRNA expression and changes in genome copy numbers, and found coordinating pairs such as miR-29b and PTEN, miR-222 and EGFR, and miR-34a and SNX13. Kim et al79 also analyzed data from TCGA and found that miR-26a is part of an oncomir/oncogene cluster along with CDK4 and CENTG1. Antisense miRNA oligonucleotides (AMOs or antagomirs) can target the oncogenic miRNAs (oncomirs), whereas the downregulated miRNAs can be replaced with miRNA mimetics.129 Current and future miRNA-wide studies may be able to find more targets, and more knowledge about the mechanisms and targets of miRNAs will assist in developing novel GBM pharmacogenomic therapies.
Figure 3.

Biosynthesis and processing of microRNA.
Table 4.
MicroRNAs implicated in GBM and their role in tumor development
| MicroRNA | Expression | Function | Target(s) |
|---|---|---|---|
| miR-21 | High | Suppress apoptosis, promote invasion, resist chemotherapy | HNRPK, Tap63, PDCD4, TIMP3, RECK, LRRFIP1 |
| miR-7 | Low | Suppress growth, reduce invasion | EGFR, IRS-2 |
| miR-124/miR-137 | Low | Promote cell cycle arrest, induce differentiation of glioblastoma-derived stem cells, inhibit proliferation | CDK6, PTBP1 |
| miR-128–1 | Low | Impair self-renewal in glioma stem cell, inhibit proliferation | Bmi-1, E2F3a |
| miR-221/miR-222 | High | Enhance proliferation | CDKN1B, p27 |
| miR-26a | High | Promote aggressive tumor growth | PTEN, RB1, MAP3K2/MEKK2 |
| miR-10b | High | Promote invasion/migration | HOXD10 |
| miR-34a | Low | Suppress growth | c-Met, Notch |
| miR-153 | Low | Induce apoptosis | Bcl-2, Mcl-1 |
| miR-181a/181b | Low | Inhibit proliferation | unknown |
| miR-296 | High | Promote angiogenesis | HGS |
Abbreviations: EGFR, epidermal growth factor receptor; HGS, hepatocyte growth factor-regulated tyrosine kinase substrate.
GSC therapy
There exists a small population of GSCs in GBM that seem to be resistant to radiotherapy and chemotherapy and can stay dormant for sometime after treatment. However, these cells eventually reenter the cell cycle and cause a local recurrence of tumors. GSCs are undifferentiated cells that can self-renew, differentiate, and initiate tumors that mimic the parent tumor, representing a unique tumor cell subpopulation that should be targeted.137 There is currently no single set of markers to identify all GSCs, but the most commonly used is CD133.137 CD133+ cells show characteristics of stem cells such as multilineage differentiation. However, up to 40% of freshly isolated GBM tumors do not express CD133 but may still exhibit stem cell-like properties such as self-renewal and multilineage differentiation.137,138 Other GSC markers under investigation are A2B5 and SSEA-1.137,138 One possible way to directly target the GSC population and enhance therapy is by the inhibition of stem cell pathways such as Notch and Hedgehog.137 The Notch signaling pathway can be inhibited via small molecule γ-secretase inhibitors, and studies have shown this to suppress glioma cell growth and increase differentiation.137 Hedgehog-GLI signaling regulates the expression of stemness genes, and studies have suggested that inhibiting the pathway increases GBM therapy effi-cacy.137,139 Other potential molecular targets for GSC therapy include STAT3, REST, HIF2a, EZH2, and FABP7.77,139 MiR-124, miR-7, and miR-128 play important roles in stem cell renewal, differentiation, and invasion, and may serve as future targets.131 MiR-128 is significantly downregulated in GBM, and it targets Bmi-1 and E2F3a, which promotes GSC self-renewal.131 Therapeutic agents based on other epigenetic mechanisms such as HDACi can induce specific gene products such as DNER to inhibit the growth of GBM-derived neurospheres, showing promise as a novel GSC-targeting therapy.127 Other potential GSC therapeutic agents are oncolytic viruses, such as the oncolytic adenovirus δ-24-RGD or the oncolytic herpes simplex virus oHSV-G47Δ, which can kill neoplastic cells specifically while sparing normal cells.139 Another specific anti-GSC technique that is worth exploring is the prodifferentiation approach, which uses agents such as BMP4 to enforce a more differentiated phenotype in GSC.139 Pharmacogenomic therapies that target GSCs will clearly be a very exciting area of investigational drug design in the future.
Glioma cell migration/invasion therapy
Glioblastoma universally recurs due to proliferation of cells that have migrated away from the tumor focus. Current therapies have significant difficulty targeting these cells. Surgical resection and local chemotherapies (carmustine wafers placed in the resection cavities) are unlikely to target migrated cells. Investigators are using agents such as d-aminolevulinic acid to fluorescently label tumor cells to help assist surgeons during resection;140 however, strategies such as this are also unlikely to allow surgeons to remove all tumor cells that have migrated away. Recent research has identified several key molecules that regulate GBM cell migration and invasion, and may serve as attractive targets. Burgoyne et al141 found that when the receptor protein tyrosine phosphatase μ (PTPμ) is proteolytically cleaved, its fragments can promote GBM cell migration. They also showed that a peptide inhibitor of PTPμ can block the cell migration induced by the fragments. Another pathway implicated in tumor cell migration is the bidirectional receptor/ligand tyrosine kinase system EphB/ephrin-B.142 Other possible targets recently studied that affect migratory potential include tGLI1 (a novel splice variant of GLI1), SDF-1, Pin1, OPN, MGP, ADAM19, VAT-1, Sema3A-neuropilin-1, and AJAP1.143–150 Even miRNAs such as miR-10b have been implicated in glioma cell migration.128,134 Curative therapies will have to be able to cross the highly selective blood–brain barrier, infiltrate throughout the entire brain, and specifically attack migrating cells without harming surrounding normal glia. Our current knowledge of GBM suggests a progressive accumulation of genomic and proteomic changes. This is clearly evident in the multitude of differences between primary and secondary GBM. However, it is not unreasonable that the progressive nature of genomic instability of these tumors never stops. Successful therapies will have to work quickly and thoroughly; recurrent tumors may require reevaluation for different therapies due to a new genomic and proteomic profile that has created highly migratory cells.
Personalized medicine
Personalized medicine uses information specific to each patient to optimize therapeutic care. The goal is to use specific genomic, proteomic, transcriptomic, epigenomic, and other comprehensive profiles of the patients to select for the most appropriate molecular target for the most efficacious medication that would meet the individual’s needs. In oncology patients, molecular diagnostics can test for specific mutations and aberrant molecular pathways in cancer cells, which can guide physicians toward the best therapy for each patient. This type of personalized therapy has had some success in cancers such as breast cancer, non–small cell lung cancer (NSCLC), and colon cancer. In breast cancer, the overexpression of human epidermal growth factor receptor 2 (HER2)/neu is considered to be a predictive marker for response to the drug trastuzumab.151 Breast cancer patients are getting routine genetic screening for the presence of HER2 to identify the ones who can be appropriately treated by trastuzumab.152 Our institution has also started clinical trials to evaluate the performance of chemotherapy assigned by the genomic profiles of patients, specifically the lack of HER2, in early stage breast cancer. For NSCLC, EGFR mutations are also predictors for response to the drug gefitinib, and Mok et al153 found that patients who screened positive for EGFR mutations responded significantly better to treatment with gefitinib. Phase 2 prospective trials are ongoing at our site to evaluate the efficacy of using genomic expression profiles to guide chemotherapy in early stage NSCLC. Personalized medicine is also being used in colon cancer treatment, such as the genomic testing for a specific glucuronosyltransferase 1A1 (UGT1A1) polymorphism that affects UGT1A1 enzyme function.154 This enzyme is responsible for the breakdown of irinotecan, a drug used to treat colon cancer, and patients with the polymorphism will lose the enzyme function, leading to increased toxicity.155 UGT1A1 testing in the clinic helps physicians to choose the right dosage and type of drug to prevent severe irinotecan toxicity.
Clearly, personalized medicine is starting to become an important part of clinical care because of the need for better treatments with less toxicity. However, this type of personalized care does not yet exist for GBM. Although tests for MGMT promoter methylation status have been in use for clinical trials, it is not yet the standard of care and physicians do not routinely conduct genomic profile screenings as part of their clinical decision-making.156 One issue with the application of personalized medicine is with the lack of reliable standardized companion diagnostics that could be easily conducted in clinics to test for biomarkers. More innovations in the field of personalized medicine are needed for it to become part of the standard of care so that physicians can provide better treatments for individual patients.
Conclusion
Glioblastoma remains one of the most common and most malignant primary CNS tumors of mankind. Unfortunately, over half a century of investigation has not drastically altered survival. GBM has a long history of clinical investigation that has shaped our current understanding of this fatal disease. Even though the number of studies of this disease has grown almost exponentially, we are still awaiting breakthroughs that can extend survival beyond the typical 14 months. There are currently no common, easily identifiable etiologies, or risk factors; however, a complete unraveling of the GBM genome may provide important clues in the near future. Histopathologic descriptions have remained unchanged for years, but will likely be complimented by relevant genetic alterations (MGMT methylation status) as we gain a better understanding of the oncogenomic events and signaling pathways. During the past 5 years, we have witnessed only a single addition to the standard of care (TMZ) in upfront chemotherapy, and with only modest benefit. This will be improved upon with the current surge of investigational drugs. Therapy for newly diagnosed GBM patients is cytoreductive surgery (>98%), followed by concurrent TMZ and radiotherapy for 2–6 weeks after surgery and then TMZ alone for 6 months. These therapies are woefully inadequate. Surgery is never successful in removing all tumor cells, catching highly migratory cells early enough, or preventing recurrence. Radiotherapy helps only slightly, significantly hampered by radioresistent GSCs and the lack of any effect radiosensitizing pharmacogenomic therapy. Standard GBM chemotherapy lacks specificity and significant benefit.
However, there is hope. With the explosion in gene expression profiling, signaling pathway characterization, GSC identification, and immunomodulation strategies in GBM that has occurred over the last decade, there is now a tremendous surge in clinical studies evaluating new agents. In the next 5–10 years, these will be the major areas of investigational drug studies for GBM: targeted combination therapies, antiangiogenic therapies, immunotherapies, epigenetic therapies, cancer stem cell therapies, and cell migration and invasion therapies. The results from phase 3 RCTs of these exciting new areas of investigation will soon begin appearing and will help to ascertain which one of these therapeutic strategies will hold the most promise. The oncogenomic heterogeneity in oncology has supported the original belief that future therapies will need to be very individualistic to be successful. Although personalized approaches are not yet routinely conducted in all clinics, examples from the therapy of other tumors have shown that they have tremendous potential to become the new standard of care for GBM. Innovations in screening techniques will help propel personalized medicine into doctors’ offices and help the patients to receive better, more individualized care.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 2.Sehati N, Liau L. Adjuvant treatment for gliomas. Contemp Neurosurg. 2003;25(15):1–10. [Google Scholar]
- 3.Central Brain Tumor Registry of the United States . Statistical report: primary brain tumors in the United States, 2000–2004. Chicago: Central Brain Tumor Registry of the United States; 2008. [Google Scholar]
- 4.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohgaki H, Dessen P, Jourde B, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004;64(19):6892–6899. doi: 10.1158/0008-5472.CAN-04-1337. [DOI] [PubMed] [Google Scholar]
- 6.Ohgaki H. Genetic pathways to glioblastomas. Neuropathology. 2005;25(1):1–7. doi: 10.1111/j.1440-1789.2004.00600.x. [DOI] [PubMed] [Google Scholar]
- 7.Kanu OO, Hughes B, Di C, et al. Glioblastoma multiforme oncogenomics and signaling pathways. Clin Med Oncol. 2009;3:39–52. doi: 10.4137/cmo.s1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hess KR, Broglio KR, Bondy ML. Adult glioma incidence trends in the United States, 1977–2000. Cancer. 2004;101(10):2293–2299. doi: 10.1002/cncr.20621. [DOI] [PubMed] [Google Scholar]
- 9.Christensen HC, Kosteljanetz M, Johansen C. Incidences of gliomas and meningiomas in Denmark, 1943 to 1997. Neurosurgery. 2003;52(6):1327–1333. doi: 10.1227/01.neu.0000064802.46759.53. discussion 1333–1324. [DOI] [PubMed] [Google Scholar]
- 10.Wrensch M, Lee M, Miike R, et al. Familial and personal medical history of cancer and nervous system conditions among adults with glioma and controls. Am J Epidemiol. 1997;145(7):581–593. doi: 10.1093/oxfordjournals.aje.a009154. [DOI] [PubMed] [Google Scholar]
- 11.Wrensch M, Minn Y, Chew T, Bondy M, Berger MS. Epidemiology of primary brain tumors: current concepts and review of the literature. Neuro Oncol. 2002;4(4):278–299. doi: 10.1093/neuonc/4.4.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hodges LC, Smith JL, Garrett A, Tate S. Prevalence of glioblastoma multiforme in subjects with prior therapeutic radiation. J Neurosci Nurs. 1992;24(2):79–83. doi: 10.1097/01376517-199204000-00005. [DOI] [PubMed] [Google Scholar]
- 13.Nichols KE, Malkin D, Garber JE, Fraumeni JF, Jr, Li FP. Germ-line p53 mutations predispose to a wide spectrum of early-onset cancers. Cancer Epidemiol Biomarkers Prev. 2001;10(2):83–87. [PubMed] [Google Scholar]
- 14.Fisher JL, Schwartzbaum JA, Wrensch M, Wiemels JL. Epidemiology of brain tumors. Neurol Clin. 2007;25(4):867–890. vii. doi: 10.1016/j.ncl.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 15.Demers PA, Vaughan TL, Schommer RR. Occupation, socioeconomic status, and brain tumor mortality: a death certificate-based case-control study. J Occup Med. 1991;33(9):1001–1006. [PubMed] [Google Scholar]
- 16.Shinojima N, Tada K, Shiraishi S, et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003;63(20):6962–6970. [PubMed] [Google Scholar]
- 17.McLendon RE, Robinson JS, Jr, Chambers DB, Grufferman S, Burger PC. The glioblastoma multiforme in Georgia, 1977–1981. Cancer. 1985;56(4):894–897. doi: 10.1002/1097-0142(19850815)56:4<894::aid-cncr2820560432>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 18.Parkin DM, Muir CS. Cancer incidence in five continents Comparability and quality of data. IARC Sci Publ. 1992;(120):45–173. [PubMed] [Google Scholar]
- 19.Krex D, Klink B, Hartmann C, et al. Long-term survival with glioblastoma multiforme. Brain. 2007;130(Pt 10):2596–2606. doi: 10.1093/brain/awm204. [DOI] [PubMed] [Google Scholar]
- 20.Curran WJ, Jr, Scott CB, Horton J, et al. Recursive partitioning analysis of prognostic factors in three Radiation Therapy Oncology Group malignant glioma trials. J Natl Cancer Inst. 1993;85(9):704–710. doi: 10.1093/jnci/85.9.704. [DOI] [PubMed] [Google Scholar]
- 21.Mirimanoff RO, Gorlia T, Mason W, et al. Radiotherapy and temozolomide for newly diagnosed glioblastoma: recursive partitioning analysis of the EORTC 26981/22981-NCIC CE3 phase III randomized trial. J Clin Oncol. 2006;24(16):2563–2569. doi: 10.1200/JCO.2005.04.5963. [DOI] [PubMed] [Google Scholar]
- 22.Kleihues P, Cavenee WK. WHO Classification of Tumors Pathology and Genetics of Tumors of the Nervous System. Lyon: IARCpress; 2000. [Google Scholar]
- 23.Reardon DA, Rich JN, Friedman HS, Bigner DD. Recent advances in the treatment of malignant astrocytoma. J Clin Oncol. 2006;24(8):1253–1265. doi: 10.1200/JCO.2005.04.5302. [DOI] [PubMed] [Google Scholar]
- 24.Burger PC, Vogel FS, Green SB, Strike TA. Glioblastoma multiforme and anaplastic astrocytoma. Pathologic criteria and prognostic implications. Cancer. 1985;56(5):1106–1111. doi: 10.1002/1097-0142(19850901)56:5<1106::aid-cncr2820560525>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 25.Chang CH, Horton J, Schoenfeld D, et al. Comparison of postoperative radiotherapy and combined postoperative radiotherapy and chemotherapy in the multidisciplinary management of malignant gliomas. A joint Radiation Therapy Oncology Group and Eastern Cooperative Oncology Group study. Cancer. 1983;52(6):997–1007. doi: 10.1002/1097-0142(19830915)52:6<997::aid-cncr2820520612>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 26.Simpson JR, Horton J, Scott C, et al. Influence of location and extent of surgical resection on survival of patients with glioblastoma multiforme: results of three consecutive Radiation Therapy Oncology Group (RTOG) clinical trials. Int J Radiat Oncol Biol Phys. 1993;26(2):239–244. doi: 10.1016/0360-3016(93)90203-8. [DOI] [PubMed] [Google Scholar]
- 27.Wood JR, Green SB, Shapiro WR. The prognostic importance of tumor size in malignant gliomas: a computed tomographic scan study by the Brain Tumor Cooperative Group. J Clin Oncol. 1988;6(2):338–343. doi: 10.1200/JCO.1988.6.2.338. [DOI] [PubMed] [Google Scholar]
- 28.Lacroix M, Abi-Said D, Fourney DR, et al. A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J Neurosurg. 2001;95(2):190–198. doi: 10.3171/jns.2001.95.2.0190. [DOI] [PubMed] [Google Scholar]
- 29.Stummer W, Reulen HJ, Meinel T, et al. Extent of resection and survival in glioblastoma multiforme: identification of and adjustment for bias. Neurosurgery. 2008;62(3):564–576. doi: 10.1227/01.neu.0000317304.31579.17. discussion 564–576. [DOI] [PubMed] [Google Scholar]
- 30.Stewart LA. Chemotherapy in adult high-grade glioma: a systematic review and meta-analysis of individual patient data from 12 randomised trials. Lancet. 2002;359(9311):1011–1018. doi: 10.1016/s0140-6736(02)08091-1. [DOI] [PubMed] [Google Scholar]
- 31.Long DM, Gordon T, Bowman H, et al. Outcome and cost of craniotomy performed to treat tumors in regional academic referral centers. Neurosurgery. 2003;52(5):1056–1063. discussion 1063–1055. [PubMed] [Google Scholar]
- 32.Kristiansen K, Hagen S, Kollevold T, et al. Combined modality therapy of operated astrocytomas grade III and IV. Confirmation of the value of postoperative irradiation and lack of potentiation of bleomycin on survival time: a prospective multicenter trial of the Scandinavian Glioblastoma Study Group. Cancer. 1981;47(4):649–652. doi: 10.1002/1097-0142(19810215)47:4<649::aid-cncr2820470405>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 33.Walker MD, Alexander E, Jr, Hunt WE, et al. Evaluation of BCNU and/or radiotherapy in the treatment of anaplastic gliomas. A cooperative clinical trial. J Neurosurg. 1978;49(3):333–343. doi: 10.3171/jns.1978.49.3.0333. [DOI] [PubMed] [Google Scholar]
- 34.Fine HA, Dear KB, Loeffler JS, Black PM, Canellos GP. Meta-analysis of radiation therapy with and without adjuvant chemotherapy for malignant gliomas in adults. Cancer. 1993;71(8):2585–2597. doi: 10.1002/1097-0142(19930415)71:8<2585::aid-cncr2820710825>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 35.Brem H, Piantadosi S, Burger PC, et al. Placebo-controlled trial of safety and efficacy of intraoperative controlled delivery by biodegradable polymers of chemotherapy for recurrent gliomas. The Polymer-brain Tumor Treatment Group. Lancet. 1995;345(8956):1008–1012. doi: 10.1016/s0140-6736(95)90755-6. [DOI] [PubMed] [Google Scholar]
- 36.Westphal M, Hilt DC, Bortey E, et al. A phase 3 trial of local chemotherapy with biodegradable carmustine (BCNU) wafers (Gliadel wafers) in patients with primary malignant glioma. Neuro-oncol. 2003;5(2):79–88. doi: 10.1215/S1522-8517-02-00023-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 38.Sathornsumetee S, Rich JN, Reardon DA. Diagnosis and treatment of high-grade astrocytoma. Neurol Clin. 2007;25(4):1111–1139. x. doi: 10.1016/j.ncl.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 39.Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma:genetics, biology, and paths to treatment. Genes Dev. 2007;21(21):2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 40.Chi AS, Wen PY. Inhibiting kinases in malignant gliomas. Expert Opin Ther Targets. 2007;11(4):473–496. doi: 10.1517/14728222.11.4.473. [DOI] [PubMed] [Google Scholar]
- 41.Sathornsumetee S, Reardon DA, Desjardins A, Quinn JA, Vredenburgh JJ, Rich JN. Molecularly targeted therapy for malignant glioma. Cancer. 2007;110(1):13–24. doi: 10.1002/cncr.22741. [DOI] [PubMed] [Google Scholar]
- 42.Kim DH, Mohapatra G, Bollen A, Waldman FM, Feuerstein BG. Chromosomal abnormalities in glioblastoma multiforme tumors and glioma cell lines detected by comparative genomic hybridization. Int J Cancer. 1995;60(6):812–819. doi: 10.1002/ijc.2910600615. [DOI] [PubMed] [Google Scholar]
- 43.Mohapatra G, Bollen AW, Kim DH, et al. Genetic analysis of glioblastoma multiforme provides evidence for subgroups within the grade. Genes Chromosomes Cancer. 1998;21(3):195–206. [PubMed] [Google Scholar]
- 44.Nishizaki T, Ozaki S, Harada K, et al. Investigation of genetic alterations associated with the grade of astrocytic tumor by comparative genomic hybridization. Genes Chromosomes Cancer. 1998;21(4):340–346. doi: 10.1002/(sici)1098-2264(199804)21:4<340::aid-gcc8>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 45.von Deimling A, Fimmers R, Schmidt MC, et al. Comprehensive allelotype and genetic anaysis of 466 human nervous system tumors. J Neuropathol Exp Neurol. 2000;59(6):544–558. doi: 10.1093/jnen/59.6.544. [DOI] [PubMed] [Google Scholar]
- 46.Collins VP. Brain tumours: classification and genes. J Neurol Neurosurg Psychiatry. 2004;75(Suppl 2):2–11. ii. doi: 10.1136/jnnp.2004.040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vranova V, Necesalova E, Kuglik P, et al. Screening of genomic imbalances in glioblastoma multiforme using high-resolution comparative genomic hybridization. Oncol Rep. 2007;17(2):457–464. [PubMed] [Google Scholar]
- 48.Hui AB, Lo KW, Yin XL, Poon WS, Ng HK. Detection of multiple gene amplifications in glioblastoma multiforme using array-based comparative genomic hybridization. Lab Invest. 2001;81(5):717–723. doi: 10.1038/labinvest.3780280. [DOI] [PubMed] [Google Scholar]
- 49.Ruano Y, Mollejo M, Ribalta T, et al. Identification of novel candidate target genes in amplicons of Glioblastoma multiforme tumors detected by expression and CGH microarray profiling. Mol Cancer. 2006;5:39. doi: 10.1186/1476-4598-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991;51(12):3075–3079. [PubMed] [Google Scholar]
- 51.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 52.Silber JR, Bobola MS, Ghatan S, Blank A, Kolstoe DD, Berger MS. O6-methylguanine-DNA methyltransferase activity in adult gliomas: relation to patient and tumor characteristics. Cancer Res. 1998;58(5):1068–1073. [PubMed] [Google Scholar]
- 53.Belanich M, Pastor M, Randall T, et al. Retrospective study of the correlation between the DNA repair protein alkyltransferase and survival of brain tumor patients treated with carmustine. Cancer Res. 1996;56(4):783–788. [PubMed] [Google Scholar]
- 54.Watts GS, Pieper RO, Costello JF, Peng YM, Dalton WS, Futscher BW. Methylation of discrete regions of the O6-methylguanine DNA methyltransferase (MGMT) CpG island is associated with heterochromatinization of the MGMT transcription start site and silencing of the gene. Mol Cell Biol. 1997;17(9):5612–5619. doi: 10.1128/mcb.17.9.5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nakamura M, Watanabe T, Yonekawa Y, Kleihues P, Ohgaki H. Promoter methylation of the DNA repair gene MGMT in astrocytomas is frequently associated with G:C → A:T mutations of the TP53 tumor suppressor gene. Carcinogenesis. 2001;22(10):1715–1719. doi: 10.1093/carcin/22.10.1715. [DOI] [PubMed] [Google Scholar]
- 56.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kanu OO, Mehta A, Di C, et al. Glioblastoma multiforme: a review of therapeutic targets. Expert Opin Ther Targets. 2009;13(6):701–718. doi: 10.1517/14728220902942348. [DOI] [PubMed] [Google Scholar]
- 58.Guha A, Feldkamp MM, Lau N, Boss G, Pawson A. Proliferation of human malignant astrocytomas is dependent on Ras activation. Oncogene. 1997;15(23):2755–2765. doi: 10.1038/sj.onc.1201455. [DOI] [PubMed] [Google Scholar]
- 59.Wallace MR, Marchuk DA, Andersen LB, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249(4965):181–186. doi: 10.1126/science.2134734. [DOI] [PubMed] [Google Scholar]
- 60.Viskochil D, Buchberg AM, Xu G, et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell. 1990;62(1):187–192. doi: 10.1016/0092-8674(90)90252-a. [DOI] [PubMed] [Google Scholar]
- 61.Cawthon RM, Weiss R, Xu GF, et al. A major segment of the neurofibromatosis type 1 gene: cDNA sequence, genomic structure, and point mutations. Cell. 1990;62(1):193–201. doi: 10.1016/0092-8674(90)90253-b. [DOI] [PubMed] [Google Scholar]
- 62.Shaw RJ, Paez JG, Curto M, et al. The Nf2 tumor suppressor, merlin, functions in Racdependent signaling. Dev Cell. 2001;1(1):63–72. doi: 10.1016/s1534-5807(01)00009-0. [DOI] [PubMed] [Google Scholar]
- 63.Fulci G, Labuhn M, Maier D, et al. p53 gene mutation and ink4a-arf deletion appear to be two mutually exclusive events in human glioblastoma. Oncogene. 2000;19(33):3816–3822. doi: 10.1038/sj.onc.1203700. [DOI] [PubMed] [Google Scholar]
- 64.He J, Olson JJ, James CD. Lack of p16INK4 or retinoblastoma protein (pRb), or amplification-associated overexpression of cdk4 is observed in distinct subsets of malignant glial tumors and cell lines. Cancer Res. 1995;55(21):4833–4836. [PubMed] [Google Scholar]
- 65.Ishii N, Maier D, Merlo A, et al. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999;9(3):469–479. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Labuhn M, Jones G, Speel EJ, et al. Quantitative real-time PCR does not show selective targeting of p14(ARF) but concomitant inactivation of both p16(INK4A) and p14(ARF) in 105 human primary gliomas. Oncogene. 2001;20(9):1103–1109. doi: 10.1038/sj.onc.1204197. [DOI] [PubMed] [Google Scholar]
- 67.Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. Am J Pathol. 2007;170(5):1445–1453. doi: 10.2353/ajpath.2007.070011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13(12):1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- 69.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275(5308):1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 70.Steck PA, Pershouse MA, Jasser SA, et al. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15(4):356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 71.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273(22):13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 72.Maehama T, Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 1999;9(4):125–128. doi: 10.1016/s0962-8924(99)01519-6. [DOI] [PubMed] [Google Scholar]
- 73.Maier D, Jones G, Li X, et al. The PTEN lipid phosphatase domain is not required to inhibit invasion of glioma cells. Cancer Res. 1999;59(21):5479–5482. [PubMed] [Google Scholar]
- 74.Colman H, Zhang L, Sulman EP, et al. A multigene predictor of outcome in glioblastoma. Neuro Oncol. 2010;12(1):49–57. doi: 10.1093/neuonc/nop007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li A, Walling J, Ahn S, et al. Unsupervised analysis of transcriptomic profiles reveals six glioma subtypes. Cancer Res. 2009;69(5):2091–2099. doi: 10.1158/0008-5472.CAN-08-2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gravendeel L, Kouwenhoven M, Gevaert O, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69(23):9065–9072. doi: 10.1158/0008-5472.CAN-09-2307. [DOI] [PubMed] [Google Scholar]
- 77.Abstracts from the 2009 Joint Meeting of the Society for Neuro-Oncology (SNO) and the American Association of Neurological Surgeons/Congress of Neurological Surgeons (AANS/CNS) Section on Tumors. Neuro Oncol. 2009;11(5):563–699. [Google Scholar]
- 78.Verhaak RGW, Hoadley KA, Purdom E, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17(1):98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim H, Huang W, Jiang X, Pennicooke B, Park PJ, Johnson MD. Integrative genome analysis reveals an oncomir/oncogene cluster regulating glioblastoma survivorship. Proc Natl Acad Sci. 2010;107(5):2183–2188. doi: 10.1073/pnas.0909896107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bredel M, Scholtens DM, Harsh GR, et al. A network model of a cooperative genetic landscape in brain tumors. JAMA. 2009;302(3):261–275. doi: 10.1001/jama.2009.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nord H, Hartmann C, Andersson R, et al. Characterization of novel and complex genomic aberrations in glioblastoma using a 32K BAC array. Neuro Oncol. 2009;11(6):803–818. doi: 10.1215/15228517-2009-013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cerami E, Demir E, Schultz N, Taylor BS, Sander C. Automated network analysis identifies core pathways in glioblastoma. PLoS One. 2010;5(2):e8918. doi: 10.1371/journal.pone.0008918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Song W, Ruder AM, Hu L, et al. Genetic epidemiology of glioblastoma multiforme: confirmatory and new findings from analyses of human leukocyte antigen alleles and motifs. PLoS One. 2009;4(9):e7157. doi: 10.1371/journal.pone.0007157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rajaraman P, Brenner AV, Butler MA, et al. Common variation in genes related to innate immunity and risk of adult glioma. Cancer Epidemiol Biomarkers Prev. 2009;18(5):1651–1658. doi: 10.1158/1055-9965.EPI-08-1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McKean-Cowdin R, Barnholtz-Sloan J, Inskip PD, et al. Associations between polymorphisms in DNA repair genes and glioblastoma. Cancer Epidemiol Biomarkers Prev. 2009;18(4):1118–1126. doi: 10.1158/1055-9965.EPI-08-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yin D, Ogawa S, Kawamata N, et al. High-resolution genomic copy number profiling of glioblastoma multiforme by single nucleotide polymorphism DNA microarray. Mol Cancer Res. 2009;7(5):665–676. doi: 10.1158/1541-7786.MCR-08-0270. [DOI] [PubMed] [Google Scholar]
- 87.Martinez R, Martin-Subero JI, Rohde V, et al. A microarray-based DNA methylation study of glioblastoma multiforme. Epigenetics. 2009;4(4):255–265. doi: 10.4161/epi.9130. [DOI] [PubMed] [Google Scholar]
- 88.Zscharnack K, Kessler R, Bleichert F, Warnke JP, Eschrich K. The PFKFB3 splice variant UBI2K4 is downregulated in high-grade astrocytomas and impedes the growth of U87 glioblastoma cells. Neuropathol Appl Neurobiol. 2009;35(6):566–578. doi: 10.1111/j.1365-2990.2009.01027.x. [DOI] [PubMed] [Google Scholar]
- 89.Brennan C, Momota H, Hambardzumyan D, et al. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One. 2009;4(11):e7752. doi: 10.1371/journal.pone.0007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nobusawa S, Watanabe T, Kleihues P, Ohgaki H. IDH1 mutations as molecular signature and predictive factor of secondary glioblastomas. Clin Cancer Res. 2009;15(19):6002–6007. doi: 10.1158/1078-0432.CCR-09-0715. [DOI] [PubMed] [Google Scholar]
- 91.Zhao S, Lin Y, Xu W, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1{alpha} Science. 2009;324(5924):261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Beckner ME, Fellows-Mayle W, Zhang Z, et al. Identification of ATP citrate lyase as a positive regulator of glycolytic function in glioblastomas. Int J Cancer. 2010;126(10):2282–2295. doi: 10.1002/ijc.24918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Malzkorn B, Wolter M, Liesenberg F, et al. Identification and functional characterization of microRNAs involved in the malignant progression of gliomas. Brain Pathol. doi: 10.1111/j.1750-3639.2009.00328.x. Epub 2009 Aug 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stommel JM, Kimmelman AC, Ying H, et al. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science. 2007;318(5848):287–290. doi: 10.1126/science.1142946. [DOI] [PubMed] [Google Scholar]
- 95.Lu J, Kovach JS, Johnson F, et al. Inhibition of serine/threonine phosphatase PP2A enhances cancer chemotherapy by blocking DNA damage induced defense mechanisms. Proc Natl Acad Sci U S A. 2009;106(28):11697–11702. doi: 10.1073/pnas.0905930106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pillay V, Allaf L, Wilding AL, et al. The plasticity of oncogene addiction: implications for targeted therapies directed to receptor tyrosine kinases. Neoplasia. 2009;11(5):448–458. doi: 10.1593/neo.09230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lau CJ, Koty Z, Nalbantoglu J. Differential response of glioma cells to FOXO1-directed therapy. Cancer Res. 2009;69(13):5433–5440. doi: 10.1158/0008-5472.CAN-08-4540. [DOI] [PubMed] [Google Scholar]
- 98.Sampson JH, Akabani G, Archer GE, et al. Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors. Neuro Oncol. 2008;10(3):320–329. doi: 10.1215/15228517-2008-012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Vredenburgh JJ, Desjardins A, Herndon JE, II, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13(4):1253–1259. doi: 10.1158/1078-0432.CCR-06-2309. [DOI] [PubMed] [Google Scholar]
- 100.Halatsch ME, Löw S, Mursch K, et al. Candidate genes for sensitivity and resistance of human glioblastoma multiforme cell lines to erlotinib. J Neurosurg. 2009;111(2):211–218. doi: 10.3171/2008.9.JNS08551. [DOI] [PubMed] [Google Scholar]
- 101.Schaich M, Kestel L, Pfirrmann M, et al. A MDR1 (ABCB1) gene single nucleotide polymorphism predicts outcome of temozolomide treatment in glioblastoma patients. Ann Oncol. 2009;20(1):175–181. doi: 10.1093/annonc/mdn548. [DOI] [PubMed] [Google Scholar]
- 102.Purow B, Schiff D. Advances in the genetics of glioblastoma: are we reaching critical mass? Nat Rev Neurol. 2009;5(8):419–426. doi: 10.1038/nrneurol.2009.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wen PY. New developments in targeted molecular therapies for glioblastoma. Expert Rev Anticancer Ther. 2009;9(1):7–10. doi: 10.1586/14737140.9.1.7. [DOI] [PubMed] [Google Scholar]
- 104.Argyriou AA, Giannopoulou E, Kalofonos HP. Angiogenesis and anti-angiogenic molecularly targeted therapies in malignant gliomas. Oncology. 2009;77(1):1–11. doi: 10.1159/000218165. [DOI] [PubMed] [Google Scholar]
- 105.Maurage CA, Adam E, Mineo JF, et al. Endocan expression and localization in human glioblastomas. J Neuropathol Exp Neurol. 2009;68(6):633–641. doi: 10.1097/NEN.0b013e3181a52a7f. [DOI] [PubMed] [Google Scholar]
- 106.FDA Approval for Bevacizumad. National Cancer Institute.
- 107.Kurpad SN, Zhao XG, Wikstrand CJ, Batra SK, McLendon RE, Bigner DD. Tumor antigens in astrocytic gliomas. Glia. 1995;15(3):244–256. doi: 10.1002/glia.440150306. [DOI] [PubMed] [Google Scholar]
- 108.Chi DD, Merchant RE, Rand R, et al. Molecular detection of tumor-associated antigens shared by human cutaneous melanomas and gliomas. Am J Pathol. 1997;150(6):2143–2152. [PMC free article] [PubMed] [Google Scholar]
- 109.Brooks WH, Netsky MG, Normansell DE, Horwitz DA. Depressed cell-mediated immunity in patients with primary intracranial tumors. Characterization of a humoral immunosuppressive factor. J Exp Med. 1972;136(6):1631–1647. doi: 10.1084/jem.136.6.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Brooks WH, Caldwell HD, Mortara RH. Immune responses in patients with gliomas. Surg Neurol. 1974;2(6):419–423. [PubMed] [Google Scholar]
- 111.Brem H, Mahaley MS, Jr, Vick NA, et al. Interstitial chemotherapy with drug polymer implants for the treatment of recurrent gliomas. J Neurosurg. 1991;74(3):441–446. doi: 10.3171/jns.1991.74.3.0441. [DOI] [PubMed] [Google Scholar]
- 112.Menzies CB, Gunar M, Thomas DG, Behan PO. Impaired thymus-derived lymphocyte function in patients with malignant brain tumour. Clin Neurol Neurosurg. 1980;82(3):157–168. doi: 10.1016/0303-8467(80)90033-5. [DOI] [PubMed] [Google Scholar]
- 113.Brooks WH, Roszman TL, Mahaley MS, Woosley RE. Immunobiology of primary intracranial tumours. II. Analysis of lymphocyte subpopulations in patients with primary brain tumours. Clin Exp Immunol. 1977;29(1):61–66. [PMC free article] [PubMed] [Google Scholar]
- 114.Brooks WH, Roszman TL, Rogers AS. Impairment of rosette-forming T lymphocytes in patients with primary intracranial tumors. Cancer. 1976;37(4):1869–1873. doi: 10.1002/1097-0142(197604)37:4<1869::aid-cncr2820370435>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 115.Braun DP, Penn RD, Flannery AM, Harris JE. Immunoregulatory cell function in peripheral blood leukocytes of patients with intracranial gliomas. Neurosurgery. 1982;10(2):203–209. [PubMed] [Google Scholar]
- 116.Elliott LH, Brooks WH, Roszman TL. Inability of mitogen-activated lymphocytes obtained from patients with malignant primary intracranial tumors to express high affinity interleukin 2 receptors. J Clin Invest. 1990;86(1):80–86. doi: 10.1172/JCI114719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Miescher S, Whiteside TL, Carrel S, von Fliedner V. Functional properties of tumor-infiltrating and blood lymphocytes in patients with solid tumors: effects of tumor cells and their supernatants on proliferative responses of lymphocytes. J Immunol. 1986;136(5):1899–1907. [PubMed] [Google Scholar]
- 118.Roszman TL, Brooks WH, Elliott LH. Immunobiology of primary intracranial tumors. VI. Suppressor cell function and lectin-binding lymphocyte subpopulations in patients with cerebral tumors. Cancer. 1982;50(7):1273–1279. doi: 10.1002/1097-0142(19821001)50:7<1273::aid-cncr2820500709>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 119.Roszman TL, Elliott LH, Brooks WH. Proliferative potential of T-cell lymphocytes from gliomas. J Neurosurg. 1992;77(5):820–821. doi: 10.3171/jns.1992.77.5.0820. [DOI] [PubMed] [Google Scholar]
- 120.Roszman TL, Brooks WH. Immunobiology of primary intracranial tumours. III. Demonstration of a qualitative lymphocyte abnormality in patients with primary brain tumours. Clin Exp Immunol. 1980;39(2):395–402. [PMC free article] [PubMed] [Google Scholar]
- 121.Zou JP, Morford LA, Chougnet C, et al. Human glioma-induced immunosuppression involves soluble factor(s) that alters monocyte cytokine profile and surface markers. J Immunol. 1999;162(8):4882–4892. [PubMed] [Google Scholar]
- 122.Parney IF, Hao C, Petruk KC. Glioma immunology and immunotherapy. Neurosurgery. 2000;46(4):778–791. doi: 10.1097/00006123-200004000-00002. discussion 791–772. [DOI] [PubMed] [Google Scholar]
- 123.Nagarajan RP, Costello JF. Epigenetic mechanisms in glioblastoma multiforme. Semin Cancer Biol. 2009;19(3):188–197. doi: 10.1016/j.semcancer.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 124.Mueller WC, von Deimling A. Gene regulation by methylation. Recent Results Cancer Res. 2009;171:217–239. doi: 10.1007/978-3-540-31206-2_13. [DOI] [PubMed] [Google Scholar]
- 125.Wemmert S, Bettscheider M, Alt S, et al. p15 promoter methylation – a novel prognostic marker in glioblastoma patients. Int J Oncol. 2009;34(6):1743–1748. doi: 10.3892/ijo_00000305. [DOI] [PubMed] [Google Scholar]
- 126.Sonoda Y, Kumabe T, Watanabe M, et al. Long-term survivors of glioblastoma: clinical features and molecular analysis. Acta Neurochir (Wien) 2009;151(11):1349–1358. doi: 10.1007/s00701-009-0387-1. [DOI] [PubMed] [Google Scholar]
- 127.Sun P, Xia S, Lal B, et al. DNER, an epigenetically modulated gene, regulates glioblastoma-derived neurosphere cell differentiation and tumor propagation. Stem Cells. 2009;27(7):1473–1486. doi: 10.1002/stem.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xia H, Qi Y, Ng SS, et al. microRNA-146b inhibits glioma cell migration and invasion by targeting MMPs. Brain Res. 2009;1269:158–165. doi: 10.1016/j.brainres.2009.02.037. [DOI] [PubMed] [Google Scholar]
- 129.Pang JC, Kwok WK, Chen Z, Ng HK. Oncogenic role of microRNAs in brain tumors. Acta Neuropathol. 2009;117(6):599–611. doi: 10.1007/s00401-009-0525-0. [DOI] [PubMed] [Google Scholar]
- 130.Lawler S, Chiocca EA. Emerging functions of microRNAs in glioblastoma. J Neurooncol. 2009;92(3):297–306. doi: 10.1007/s11060-009-9843-2. [DOI] [PubMed] [Google Scholar]
- 131.Godlewski J, Newton HB, Chiocca EA, Lawler SE. MicroRNAs and glioblastoma; the stem cell connection. Cell Death Differ. 2009;17(2):221–228. doi: 10.1038/cdd.2009.71. [DOI] [PubMed] [Google Scholar]
- 132.Novakova J, Slaby O, Vyzula R, Michalek J. MicroRNA involvement in glioblastoma pathogenesis. Biochem Biophys Res Commun. 2009;386(1):1–5. doi: 10.1016/j.bbrc.2009.06.034. [DOI] [PubMed] [Google Scholar]
- 133.Li Y, Li W, Yang Y, et al. MicroRNA-21 targets LRRFIP1 and contributes to VM-26 resistance in glioblastoma multiforme. Brain Res. 2009;1286:13–18. doi: 10.1016/j.brainres.2009.06.053. [DOI] [PubMed] [Google Scholar]
- 134.Sasayama T, Nishihara M, Kondoh T, Hosoda K, Kohmura E. MicroRNA-10b is overexpressed in malignant glioma and associated with tumor invasive factors, uPAR and RhoC. Int J Cancer. 2009;125(6):1407–1413. doi: 10.1002/ijc.24522. [DOI] [PubMed] [Google Scholar]
- 135.Ueda R, Kohanbash G, Sasaki K, et al. Dicer-regulated microRNAs 222 and 339 promote resistance of cancer cells to cytotoxic T-lymphocytes by down-regulation of ICAM-1. Proc Natl Acad Sci U S A. 2009;106(26):10746–10751. doi: 10.1073/pnas.0811817106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gaire RK, Bailey J, Bearfoot J, Campbell IG, Stuckey PJ, Haviv I. MIRAGAA – a methodology for finding coordinated effects of microRNA expression changes and genome aberrations in cancer. Bioinformatics. 2010;26(2):161–167. doi: 10.1093/bioinformatics/btp654. [DOI] [PubMed] [Google Scholar]
- 137.Gilbert CA, Ross AH. Cancer stem cells: cell culture, markers, and targets for new therapies. J Cell Biochemistry. 2009;108(5):1031–1038. doi: 10.1002/jcb.22350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Son MJ, Woolard K, Nam DH, Lee J, Fine HA. SSEA-1 is an enrichment marker for tumor-initiating cells in human glioblastoma. Cell Stem Cell. 2009;4(5):440–452. doi: 10.1016/j.stem.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Piccirillo SM, Binda E, Fiocco R, Vescovi AL, Shah K. Brain cancer stem cells. J Mol Med. 2009;87(11):1087–1095. doi: 10.1007/s00109-009-0535-3. [DOI] [PubMed] [Google Scholar]
- 140.Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen HJ. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol. 2006;7(5):392–401. doi: 10.1016/S1470-2045(06)70665-9. [DOI] [PubMed] [Google Scholar]
- 141.Burgoyne AM, Phillips-Mason PJ, Burden-Gulley SM, et al. Proteolytic cleavage of protein tyrosine phosphatase mu regulates glioblastoma cell migration. Cancer Res. 2009;69(17):6960–6968. doi: 10.1158/0008-5472.CAN-09-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nakada M, Anderson EM, Demuth T, et al. The phosphorylation of ephrin-B2 ligand promotes glioma cell migration and invasion. Int J Cancer. 2010;126(5):1155–1165. doi: 10.1002/ijc.24849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mertsch S, Schurgers L, Weber K, Paulus W, Senner V. Matrix gla protein (MGP): an overexpressed and migration-promoting mesenchymal component in glioblastoma. BMC Cancer. 2009;9(1):302. doi: 10.1186/1471-2407-9-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lo HW, Zhu H, Cao X, Aldrich A, All-Osman F. A novel splice variant of GLI1 that promotes glioblastoma cell migration and invasion. Cancer Res. 2009;69(17):6790–6798. doi: 10.1158/0008-5472.CAN-09-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kenig S, Alonso MB, Mueller MM, Lah TT. Glioblastoma and endothelial cells cross-talk, mediated by SDF-1, enhances tumour invasion and endothelial proliferation by increasing expression of cathepsins B, S, and MMP-9. Cancer Lett. 2010;289(1):53–61. doi: 10.1016/j.canlet.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 146.Bagci T, Wu JK, Pfannl R, Ilag LL, Jay DG. Autocrine semaphorin 3A signaling promotes glioblastoma dispersal. Oncogene. 2009;28(40):3537–3550. doi: 10.1038/onc.2009.204. [DOI] [PubMed] [Google Scholar]
- 147.Atkinson GP, Nozell SE, Harrison DK, Stonecypher MS, Chen D, Benveniste EN. The prolyl isomerase Pin1 regulates the NF-[kappa] B signaling pathway and interleukin-8 expression in glioblastoma. Oncogene. 2009;28(42):3735–3745. doi: 10.1038/onc.2009.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lamour V, Mercier ML, Lefranc F, et al. Selective osteopontin knockdown exerts anti-tumoral activity in a human glioblastoma model. Int J Cancer. 2010;126(8):1797–1805. doi: 10.1002/ijc.24751. [DOI] [PubMed] [Google Scholar]
- 149.Qi B, Newcomer RG, Sang QX. ADAM19/adamalysin 19 structure, function, and role as a putative target in tumors and inflammatory diseases. Curr Pharm Des. 2009;15(20):2336–2348. doi: 10.2174/138161209788682352. [DOI] [PubMed] [Google Scholar]
- 150.Mertsch S, Becker M, Lichota A, Paulus W, Senner V. Vesicle amine transport protein-1 (VAT-1) is upregulated in glioblastomas and promotes migration. Neuropathol Appl Neurobiol. 2009;35(4):342–352. doi: 10.1111/j.1365-2990.2009.00993.x. [DOI] [PubMed] [Google Scholar]
- 151.Ferretti G, Felici A, Papaldo P, Fabi A, Cognetti F. HER2/neu role in breast cancer: from a prognostic foe to a predictive friend. Curr Opin Obstet Gynecol. 2007;19(1):56–62. doi: 10.1097/GCO.0b013e328012980a. [DOI] [PubMed] [Google Scholar]
- 152.Gown AM. Current issues in ER and HER2 testing by IHC in breast cancer. Mod Pathol. 2008;21(Suppl 2):S8–S15. doi: 10.1038/modpathol.2008.34. [DOI] [PubMed] [Google Scholar]
- 153.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 154.Innocenti F, Ratain MJ. Pharmacogenetics of irinotecan: clinical perspectives on the utility of genotyping. Pharmacogenomics. 2006;7(8):1211–1221. doi: 10.2217/14622416.7.8.1211. [DOI] [PubMed] [Google Scholar]
- 155.Patel KK, Babyatsky MW. Medical Education: a key partner in realizing personalized medicine in gastroenterology. Gastroenterology. 2008;134(3):656–661. doi: 10.1053/j.gastro.2008.01.064. [DOI] [PubMed] [Google Scholar]
- 156.Weller M, Stupp R, Reifenberger G, et al. MGMT promoter methylation in malignant gliomas: ready for personalized medicine? Nat Rev Neurol. 2010;6(1):39–51. doi: 10.1038/nrneurol.2009.197. [DOI] [PubMed] [Google Scholar]