

Abstract
A direct intramolecular anti-Markovnikov hydroetherification reaction of alkenols is described. By employing catalytic quantities of commercially-available 9-mesityl-10-methylacridinium perchlorate and 2-phenylmalononitrile as a redox-cycling source of a hydrogen atom, we report the anti-Markovnikov hydroetherification of alkenes with complete regioselectivity. In addition, we present results demonstrating that this novel catalytic system can be applied to the anti-Markovnikov hydrolactonization of alkenoic acids.
The development of catalytic protocols for the direct addition of heteroatom nucleophiles to alkenes has been an area of intense study over the past two decades.1 The vast majority of these methods give rise to primarily Markovnikov-type addition products. Given the challenges associated with reversal of innate alkene polarization, there are comparatively few methods that allow for the direct anti-Markovnikov addition of nucleophiles to olefins.1,2 Although Hartwig3,4 and Grubbs5 have demonstrated transition metal catalyst systems for the anti-Markovnikov addition of amines and water, respectively, to alkenes, success has been limited to terminal styrenes. We were drawn to the possibility that single electron oxidation of olefins to their respective cation radicals could provide a basis to develop a general catalyst system for a range of heteroatom nucleophiles with unactivated alkenes.6–9 Herein, we report the direct intramolecular anti-Markovnikov addition of alcohols to alkenes via a unique two-component single electron photoredox system. This transformation provides a reactivity profile that complements traditional Markovnikov-based Brønsted acid-catalyzed reactions (Scheme 1).10,11
Scheme 1.

Divergent Regioselectivity in Alkene Addition reactions
Seminal work from Arnold12 and Gassman13,14 provided the first evidence for cation radical-mediated anti-Markovnikov reactivity. Arnold further characterized the initial nucleophile-cation radical adduct as the three-membered intermediate 8 by density functional theory calculations.15 It is likely that the observed anti-Markovnikov selectivity results from the rupture of the weaker of the two C–X bonds, giving rise to the more stable radical intermediate 9 (Scheme 2). Additionally, Gassman and Arnold have each demonstrated that single electron photooxidants can serve as effective single electron oxidants to access reactive olefin cation radicals (7). To date, however, this method remains significantly limited in scope and requires nearly stoichiometric quantities of the photooxidant that can often result in oxidant incorporation into the reaction products as well as undesired side reactivity.16 Truly catalytic photosensitized anti-Markovnikov alcohol additions are limited to 1,1-diarylethylenes.17–19
Scheme 2.

Anti-Markovnikov Reactivity of Cation Radicals
After analysis of this body of literature, we proposed that the critical step preventing the development of catalytic protocols is the fate of putative radical intermediate 9. We hypothesized that using an alternative class of photooxidants might enable a general approach to this problem. We recognized that good candidates for a single electron redox catalyst should: i) exhibit nearly complete redox reversibility, ii) be capable of oxidizing alkenes in the range of +1.0 V to +2.0 V and iii) be positively charged to minimize unproductive back electron transfer to 7 via minimization of Coulombic attraction in the reduced (neutral) form of the catalyst.
Reports of commercially-available 9-mesityl-10-methylacridinium perchlorate (2), first employed by Fukuzumi et al., drew our attention as a photooxidant for this application.20 Given the acridinium moiety’s strong absorption band in the visible region (λ = 430 nm), high excited state oxidation power (E1/2red* = +2.06 V vs. SCE)21 and utility in a number of reported transformations relying on single electron pathways,22,23 we predicted that cation radicals could be conveniently generated from an electronically diverse range of alkenes. Additionally, the reduced form of the acridinium catalyst (11) is a moderate single electron reductant (E1/2ox = −0.57 V vs. SCE)21 that we presumed would be capable of return electron transfer to radical intermediate 12.
As a starting point, we elected to focus on the development of a catalytic system for the direct intramolecular anti-Markovnikov hydroetherification of alkenols.2,24 To date, Mizuno has reported the only known direct anti-Markovnikov hydroetherification reaction of alkenols which is believed to proceed via exciplex formation, and is limited to diphenylethylene alkenes.18 To begin, we subjected alkenol 4 to 5 mol % of catalyst 2 in degassed 1,2-dichloroethane (DCE) under irradiation with 450 nm LEDs. The anti-Markovnikov adduct, tetrahydrofuran 6, was obtained, albeit in low yields (36% yield, Table 1, entry 1), though significantly higher than when employing the cyanoarene photooxidants (Entries 2 and 3) used by Mizuno and Gassman in their pioneering studies. No trace of the Markovnikov adduct (5) was observed and conversion of the starting alkenol was relatively high (83%), but yields were significantly diminished by extensive unidentifiable byproduct formation that likely arose from competing radical processes.
Table 1.
Catalyst Optimization Studiesa

| ||||
|---|---|---|---|---|
| Entry | Conditions | Conversionb | Yieldb | 6:5b |
| 1 | Standard Conditions | 83% | 36% | >20:1 |
| 2c,d | 0.2 equiv 9,10-dicyanoanthracene instead of 2 | 21% | 5% | >20:1 |
| 3c,e | 0.5 equiv 1-cyanonaphthalene instead of 2 | 47% | 15% | >20:1 |
| 4 | With 0.5 equiv N-hydroxyphthalimide | 48% | 41% | >20:1 |
| 5 | With 0.5 equiv 9-phenylfluorene | 78% | 51% | >20:1 |
| 6 | With 0.5 equiv PhCH(CN)2 (3) | 89% | 73% | >20:1 |
| 7f | No Photooxidant | < 5% | < 5% | – |
| 8f | No Light | < 5% | < 5% | – |
| 9f,g | Ru(bpy)3Cl2 instead of 2 | < 5% | < 5% | – |
Reactions irradiated with a 15W 450 nm LED flood lamp.
Determined by 1H NMR analysis.
Irradiated with 10 x 8W T5 fluorescent bulbs (output >290 nm).
Benzene as solvent.
MeCN as solvent with 0.5 equiv of biphenyl.
With 0.5 equiv of 3.
With 1.0 equiv of methyl viologen.
After evaluation of a number of known single electron photooxidants failed to afford synthetically useful yields of the desired adduct, we felt that a distinctly different approach to this problem was required. Speculating that the reduction of radical 9 was still limiting reactivity, we hypothesized that employing a hydrogen atom donor that could facilitate this process while simultaneously serving as a single electron redox mediator.
Potential hydrogen atom donors were selected on the basis of their respective homolytic bond dissociation energies (BDE). To ensure exothermic hydrogen atom transfer, we limited our survey of potential H-atom redox catalysts to moieties possessing R–H bonds with BDE <90 kcal/mol (Table 1, entries 3–5).25 Though 0.5 equivalents of either N-hydroxyphthalimide (BDE = 87 kcal/mol, entry 3) or 9-phenylfluorene (BDE = 74 kcal/mol, entry 4) gave modest increases in reaction efficiency, we were pleased to find that 2-phenylmalononitrile (3, BDE = 77 kcal/mol) furnished anti-Markovnikov adduct 6 in 73% yield (entry 5) with no trace of the undesired Markovnikov regioisomer. Further control experiments demonstrate that both the acridinium photocatalyst and light are necessary for reactivity (entries 6, 7).26 The utility of the acridinium catalyst as a single electron photooxidant is underscored when compared directly with the frequently employed Ru(bpy)32+,27 which failed to give any of the desired product (entry 8). This result demonstrates the advantage of acridinium catalysts as visible light single electron photooxidants and should allow for greater latitude in potential substrates with alkenes possessing oxidation potentials ranging up to +2.0 V.
Our mechanistic hypothesis outlined in Scheme 3 proposes that following H-atom transfer from 3, the resulting radical 13 could serve as an oxidant for radical 11, regenerating the ground state photooxidation catalyst (2). Following this redox event, proton transfer would regenerate the H-atom donor (3) and furnish the desired product.
Scheme 3.

Proposed Mechanism for the Anti-Markovnikov Hydroetherification of Alkenols
Having identified a viable catalyst system, we investigated the scope of the intramolecular anti-Markovnikov hydroalkoxylation of alkenols (Table 2). Electronically distinct styrenes (entries 1–3) ranging from electron rich (4-(MeO)C6H4, entry 1; 80% yield) to electron deficient (4-ClC6H4, entry 2; 60% yield) provided good yields of the desired 5-exo adducts. Additionally, Thorpe-Ingold assistance is not required in the backbone of the molecule, as the substrate in entry 3, which lacked the geminal dimethyl substituent, gave nearly identical levels of reaction efficiency (82% yield) as in entry 1 (80% yield). Furthermore, the mild reaction conditions are highlighted in entry 6, where a silyl-protected alcohol remains unperturbed by the cyclization conditions. A gram-scale reaction of the alkenol in entry 4 produced the expected tetrahydrofuran product in 77% isolated yield. Though long reaction times are required for most substrates, significantly shorter reaction times are possible by increasing the amount of 3 employed.28
Table 2.
Scope of the Intramolecular Anti-Markovnikov Hydroetherification Reaction of Alkenolsa

|
Yields of cyclic ether products averaged from two reactions after 36–196 h. In all cases, the anti-Markovnikov adduct was formed in >20:1 selectivity. All alkenol oxidation potentials were measured in MeCN with 0.1 M Bu4N+ClO4− and Ag/AgCl as the reference electrode.
With 2.0 equiv of PhCH(CN)2.
Determined by 1H NMR analysis of the crude reaction mixture.
In addition to the formal 5-exo cyclization mode (entries 1–6), other ring closure types were possible. The alkenol in entry 8 underwent 6-exo cyclization to furnish the anticipated disubstituted tetrahydropyran adduct in 68% yield and 2.5:1 diastereoselectivity. Treatment of β-citronellol to the catalyst conditions resulted in 7-exo cyclization in modest, but reproducible yields (46% yield, 1.2:1 d.r., entry 9). The reactions in entries 8 and 9 required 2.0 equivalents of PhCH(CN)2 to avoid longer reaction times. Given their high oxidation potentials, monosubstituted alkenes are inaccessible by this catalyst system; however, expansion of the substrate scope will be a focus of future efforts through catalyst development.
It is particularly noteworthy that all of the reactions in Table 2 furnished the anti-Markovnikov hydroalkoxylation adducts exclusively. To emphasize the unique regioselectivity of this process, a direct comparison of alkene reactivity with cation radicals or Brønsted acids is depicted in Eq 1&2. Alkenol 14 is known to undergo Brønsted acid-assisted Markovnikov hydroetherification to furnish tetrahydropyran 15, while tetrahydrofuran 16 is obtained exclusively using our catalytic protocol. Perhaps most intriguing was the tetra-hydropyran product obtained in Eq 4 from a formal 6-endo cyclization mode despite the availability of a more kinetically viable 5-exo pathway. A control experiment where 17 was subjected to triflic acid furnished Markovnikov adduct 18, further distinguishing this catalytic protocol from traditional Brønsted acid methods.
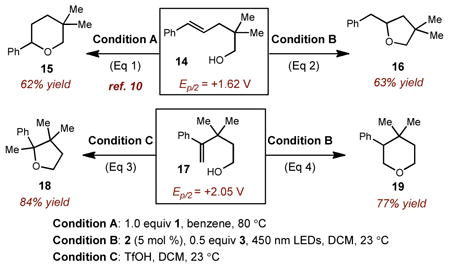 |
Finally, we have preliminary results pertaining to the use of alcohols and carboxylic acids as nucleophiles (Eq 5&6). Intermolecular addition of methanol to anethole (20) provided anti-Markovnikov adduct 21 exclusively in 81% isolated yield, further demonstrating the utility of this catalyst system (Eq 5). Finally, treatment of alkenoic acid 22 under the standard conditions in the presence of 2,6-lutidine resulted in exclusive anti-Markovnikov hydrolactonization with complete regioselectivity to afford 23 in 72% isolated yield. This reaction provides a potentially valuable disconnection to access a range of biologically-active γ-butyrolactones.14,29
 |
Eq 5 |
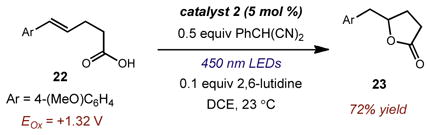 |
Eq 6 |
In summary, we have developed a direct anti-Markovnikov hydroetherification of alkenols employing a unique two-component organic photoredox catalyst system. We believe that this unique approach to managing open-shell pathways holds promise to develop additional anti-Markovnikov hydrofunctionalization reactions. Studies further investigating this transformation as well as other heteroatom nucleophiles in this context are currently underway.
Supplementary Material
Acknowledgments
The authors are grateful to J. Roberts for aid in obtaining the cyclic voltammograms of the alkenol substrates and C. Price for the preparation of some of these substrates. The project described was supported by The University of North Carolina at Chapel Hill, Award No. R01 GM098340 from the National Institute of General Medical Sciences and an Eli Lilly New Faculty Award.
Footnotes
ASSOCIATED CONTENT
Experimental procedures and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Sources
The authors declare no competing financial interests.
References
- 1.For comprehensive reviews on catalytic functionalization of olefins, see Beller M, Seavad J, Tillack A, Jiao H. Angew Chem Int Ed. 2004;43:3368. doi: 10.1002/anie.200300616.Hintermann L. Top Organomet Chem. 2010;31:123.Müller TE, Hultzsch KC, Yus M, Foubelo F, Tada M. Chem Rev. 2008;108:3795. doi: 10.1021/cr0306788.
- 2.Catalytic anti-Markovnikov additions of nucleophiles to olefins have been described as one of the “top 10 challenges for catalysis.” See: Haggin J. Chem Eng News. 1993;71:23.
- 3.Utsunomiya M, Kuwano R, Kawatsura M, Hartwig JF. J Am Chem Soc. 2003;125:5608. doi: 10.1021/ja0293608. [DOI] [PubMed] [Google Scholar]
- 4.Utsunomiya M, Kuwano R, Kawatsura M, Hartwig JF. J Am Chem Soc. 2004;126:2702. doi: 10.1021/ja031542u. [DOI] [PubMed] [Google Scholar]
- 5.Dong G, Teo P, Wickens ZK, Grubbs RH. Science. 2011;333:1609. doi: 10.1126/science.1208685. [DOI] [PubMed] [Google Scholar]
- 6.For a review on reactivity patterns of cation-radicals, see Schmittel M, Burghart A. Angew Chem Int Ed. 1997;36:2550.
- 7.For electrochemical intramolecular etherifications mediated by cation radicals, see Sutterer A, Moeller KD. J Am Chem Soc. 2000;122:5636.Moeller KD. Synlett. 2009;8:1208.
- 8.For hydroetherifications of alkene cation radicals generated by heterolysis of β-substituted radicals, see Crich D, Ranganathan K, Neelamkavil S, Huang X. J Am Chem Soc. 2003;125:7942. doi: 10.1021/ja035639s.Crich D, Ranganathan K. J Am Chem Soc. 2005;127:9924. doi: 10.1021/ja051657t.Crich D, Shirai M, Brebion F, Rumthao S. Tetrahedron. 2006;62:6501.Crich D, Brebion F, Suk DH. Top Curr Chem. 2006;263:1.
- 9.For a largely theoretical perspective on intramolecular nucleophilic capture of cation radicals by tethered alcohols, see: Roth HD, Herbertz T, Sauers RR, Wang H. Tetrahedron. 2006;62:6471.
- 10.Jeong Y, Kim D, Choi Y, Ryu J. Org Biomol Chem. 2011;9:374–378. doi: 10.1039/c0ob00701c. [DOI] [PubMed] [Google Scholar]
- 11.Smith MB, March J. March’s Advanced Organic Chemistry. John Wiley and Sons; New York: 2001. p. 993. [Google Scholar]
- 12.Neuteufel RA, Arnold DR. J Am Chem Soc. 1973;95:4080. [Google Scholar]
- 13.Gassman PG, Bottorff KJ. Tetrahedron Lett. 1987;28:5449. [Google Scholar]
- 14.Gassman PG, Bottorff KJ. J Am Chem Soc. 1987;109:7547. [Google Scholar]
- 15.Arnold DR, Chan MSW, McManus KA. Can J Chem. 1996;74:2143. [Google Scholar]
- 16.McManus KA, Arnold DA. Can J Chem. 1995;73:2158. [Google Scholar]
- 17.Mizuno K, Nakanishi I, Ichinose N, Otsuji Y. Chem Lett. 1989;41:1095. [Google Scholar]
- 18.Mizuno K, Tamai T, Nishiyama T, Tani K, Sawasaki M, Otsuji Y. Angew Chem Int Ed. 1994;33:2113. [Google Scholar]
- 19.Asaoka S, Kitazawa T, Wada T, Inoue Y. J Am Chem Soc. 1999;121:8486. [Google Scholar]
- 20.Fukuzumi S, Kotani H, Ohkubo K, Ogo S, Tkachenko NV, Lemmetyinen H. J Am Chem Soc. 2004;126:1600. doi: 10.1021/ja038656q. [DOI] [PubMed] [Google Scholar]
- 21.Ohkubo K, Mizushime K, Iwata R, Souma K, Suzuki S, Fukuzumi S. Chem Commun. 2010;46:601. doi: 10.1039/b920606j. [DOI] [PubMed] [Google Scholar]
- 22.Kotani H, Ohkubo K, Fukuzumi S. J Am Chem Soc. 2004;126:15999. doi: 10.1021/ja048353b. [DOI] [PubMed] [Google Scholar]
- 23.Ohkubo K, Nanjo T, Fukuzumi S. Org Lett. 2005;7:4265. doi: 10.1021/ol051696+. [DOI] [PubMed] [Google Scholar]
- 24.Sanford MS, Groves JT. Angew Chem Int Ed. 2004;43:588. doi: 10.1002/anie.200351941. [DOI] [PubMed] [Google Scholar]
- 25.Please see the Supporting Information for a more extensive list of the hydrogen atom donors that were evaluated.
- 26.The BF4− salt of catalyst 2 can be employed without significant variation in yields.
- 27.For reviews see: Yoon TP, Ischay MA, Du J. Nature Chem. 2010;2:527. doi: 10.1038/nchem.687.Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102. doi: 10.1039/b913880n.
- 28.Please see the Supporting Information for details.
- 29.Seitz M, Reiser O. Curr Opin Chem Biol. 2005;9:285. doi: 10.1016/j.cbpa.2005.03.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.