

Abstract
Agents that chemically modify DNA form a backbone of many cancer treatments. A key problem for DNA modifying agents is lack of specificity. To address this issue, we designed novel molecular scaffolds, termed An-Hq and An-Hq2, which are activated by a hallmark of some cancers: elevated concentrations of reactive oxygen species. Elevated reactive oxygen species are linked to oncogenesis and is found to increase in several aggressive cancers. The agents are quinones that, upon oxidation, form highly electrophilic species. In vitro studies identified the mode of addition to DNA. The aniline portion of An-Hq serves to enhance nucleophilic addition to the ethyl phenyl ether instead of forming common Michael additions. Structural characterization showed the agents add to 2′-deoxyguanosine at the N2,N3-positions. The product formed is a bulky hydroxy-N2,3-benzetheno-2′-deoxyguanosine adduct. In addition, the oxidatively activated agents added to 2′-deoxyadenosine and 2′-deoxycytidine, but not thymidine or 2′-deoxyinosine. These findings are confirmed by primer extension analysis of a 392 base pair DNA. The full-length primer extension product was reduced by 69.0 ± 0.6% upon oxidative activation of An-Hq2 compared to controls. Little sequence dependence was observed with 76% of guanine, adenine, and cytosine residues showing an increase in extension stops between two and four fold above controls. Benzetheno-nucleobase addition to double stranded DNA was confirmed by LC/MS of a self-complementary oligonucletide. Experiments were carried out to confirm in vivo DNA damage. Because of the lesion identified in vitro, we reasoned that nucleotide excision repair should be involved in reversing the effects of these oxidatively activated agents and enhance toxicity in Drosophila melanogaster. Using an RNAi based approach, Ercc1 was silenced and survival monitored after injection of an agent. As expected, bulky cross-linking DNA modifying agents, cisplatin and chlorambucil, showed statistically significant enhanced toxicity in Drosophila with silenced Ercc1. In addition, 5-fluorouracil, which does not produce bulky lesions, showed no selective toxicity. An-Hq and An-Hq2 showed statistically significant toxicity in Drosophila with silenced Ercc1. Examination of cytotoxicity shows renal carcinoma cell lines as a target of these agents with a median IC50 of 1.8 μM. Taken together, this data shows that the designed oxidatively-activated agents form distinct, bulky DNA modifications that prove difficult for cancer cells possessing an elevated reactive oxygen species phenotype to overcome. The modification produced is relatively unique among anticancer agents.
Keywords: Reactive oxygen species, ROS, DNA-modifying agent, 2′-deoxyguanosine, anti-cancer agent, Ercc1, Renal carcinoma
INTRODUCTION
The harsh side effects of current chemotherapy treatments are one of the motivating factors for increasing selectivity in novel anticancer agents.1 Agents that chemically modify DNA form the backbone of many cancer treatments. A typical DNA-modifying agent is cisplatin, which is used to treat fifty percent of cancer patients.2 These agents represent a large portion of current and phased-out anticancer agents. The design of DNA-modifying agents has slowed drastically due to the perception that these agents will have high levels of side effects due to the modification of DNA, and subsequent cytotoxicity, in non-target cells. This lack of specificity is termed off-target reactivity, which limits tolerated doses and, therefore, decreases efficacy.3 Instead, these agents have been attached to transport active scaffolds that enhance uptake into target cancer cells.4,5 Side effects, however, are still problematic, and improving selectivity is a major goal in the development of novel anticancer agents. We are developing novel agents to address these problems.
A potential path forward in cancer drug development is to take advantage of a hallmark of many cancer cells, elevated reactive oxygen species (ROS).6,7 Persistent elevation of ROS has been found in numerous cancers, including renal cell carcinoma, melanoma, and leukemia.8–10 ROS occurs in four major endogenous forms within the cell: superoxide, hydrogen peroxide, singlet oxygen, and hydroxyl radical.11–13 Mitochondrial dysfunction, common to cancer, is known to increase ROS.14 Increased ROS damages DNA, leading to mutation.15 Consequently, it is no surprise that levels of ROS-induced DNA damage correlates with cancer prognosis.16 In turn, these mutations cause enhanced activation of oncogenes.17 A critical role for ROS in oncogene transformed cancer cells has been shown when investigating c-Myc induced damage.18 Furthermore, once the Ras oncogene is locked into an active state the concentration of cellular antioxidant enzymes are lowered causing elevated ROS.19,20 As a result, several chemical research teams are actively searching for means to utilize elevated ROS in order to generate selective anticancer agents.21–23 Our lab has designed oxidatively activated DNA modifying agents which selectivity target the elevated ROS phenotype in cancer cells.24
Our new oxidatively activated agents possess attractive anticancer properties.24 These agents, such as An-Hq and An-Hq2, utilize a novel mechanism of activation and are highly selective (Figure 1A). The agents are unreactive and stable in water, but they are strongly activated by all the ROS forms, excluding hydrogen peroxide. Addition of oxidative equivalents leads to a rate enhancement of greater than 1700-fold compared to when no oxidant is present. When reactivity was examined, we found that rather than a nitrogen mustard, our agents were actually oxidatively activated quinones. More specifically, we have demonstrated that these agents are capable of adding to a guanine. The product was a hydroxy-benzetheno-2′-deoxyguanosine adduct, where the guanine possessed an added phenol.
Figure 1. Identification of Determinants for Selective Activation of an Oxidatively-Activated Agent.

(A) Structure of the oxidatively activated agents An-Hq and An-Hq2. When ROS is present, An-Hq is activated, leading to the addition of phenol to guanine. We seek to determine structure and dependence of damage. (B) Genes responsible for producing proteins involved in DNA repair can be targeted using RNAi-mediated silencing. Delivery of known DNA modifying agents will then lead to selective death in the D. melanogaster model.
The possible formation of a benzethenoguanine left several important questions to be addressed. Specifically, we questioned if these new agents were actually DNA modifying agents. We set out to structurally characterize the lesion produced on nucleosides and on double stranded DNA, as well as examine the reaction mechanism in vitro (Figure 1A). To determine in vivo DNA adduct formation, studies using a powerful genetic model system,25 Drosophila melanogaster, were accomplished (Figure 1B). D. melanogaster has been shown to be an effective, whole-animal model in cancer research and in drug discovery.26–29 Many key features of DNA repair and cell death are conserved between Drosophila and mammals.30 Approximately 75% of human disease related genes have a functional ortholog in flies.31,32 Moreover, studies on Drosophila have been instrumental in elucidating Ras signaling, with major components conserved in mammals.33
Here, we first investigated the in vitro DNA lesions produced by our novel oxidatively activated agents by NMR. We found that these agents required a nucleoside base amine to initiate reactivity. A primer extension assay was used to explore the lesion produced on a 392-nucleotide PCR product. We discovered that the three nucleosides possessing amines are indeed modified in double stranded DNA, producing bulky lesions. These agents product the same adduct on a small double stranded DNA. Such lesions led us to predict that nucleotide excision repair was an important mechanism in repairing such adducts.34 Hence, we used a targeted RNAi knockdown approach based on the GAL4/UAS system (Figure 1B) to evaluate if bulky DNA modification was occurring in vivo.35 We silenced the Ercc1 gene, whose protein forms a complex with XPF as a key step in nucleotide excision and double strand break repair pathway.36 We found that Drosophila lacking Ercc1 expression showed elevated toxicity towards known DNA modifying agents, cisplatin and chlorambucil. The elevated Ercc1-dependent sensitivity did not occur when Drosophila were treated with 5-fluorouracil, which does not produce a bulky lesion. These results confirmed D. melanogaster as an effective model organism for ascertaining the genetic mechanisms of DNA modifying agents. We then examined our novel agents, An-Hq and An-Hq2. We demonstrate that flies in which Ercc1 expression was knocked down also show enhanced sensitivity toward the novel agents, particularly An-Hq, supporting a role for these oxidatively activated agents in modifying DNA in vivo.
MATERIAL AND METHODS
Synthesis of An-Hq and An-Hq2
The synthesis of An-Hq and An-Hq2 were accomplished.24 NMR (1H and 13C) spectra were recorded on Bruker AMX 400MHz spectrometer. Chemical resonances are reported in δ (ppm) units using 13C and residual 1H signals from deuterated solvents as references. High-resolution mass spectra (ESI) were recorded on a Micromass Q-TOF 2 (Waters). Analytical thin layer chromatography (TLC) was performed on silica gel 60 GF254 (Merck). Compounds were greater than 99% pure according to HPLC.
Reaction of An-Hq with Nucleoside Analogs
For the nucleoside reactivity studies, An-Hq (22 μl dissolved in DMSO, 1mmol) was added to phosphate buffer (2 mL, 25mM NaH2PO4 in 5% acetonitrile and 95% H2O, pH 8.0) which contained 0.4 mmol of the listed nucleoside. Na2Ir2Cl6 (22 μl dissolved in water, 1 mmol) was added, and the reaction was mixed. Product was monitored using a Beckman Coulter System Gold HPLC equipped with a diode array detector (260 nm detection) for lesion formation twice a day for 7 days. HPLC conditions were as follows: a Cosmosil 5C18-PAQ Waters column was used (4.6ID, 150mm in length). The gradient (solvent A=95% water, 5% acetonitrile and solvent B=5% water, 95% acetonitrile) was linear: 0% B for 5 min, 100% B over 20 min, 100% B for 4 min, 0% B over 2 min and held for 4 min. For mass spectrometry (MS), peaks were collected, dried to remove solvent (if stable, if not they were directly injected into MS), and then analyzed by MS. All yields were between 0.5 and 3%, depending on the nucleoside adduct.
The MS was performed as follows: isolated products were resuspended in 100 μL of 0.25% acetic acid and 15% acetonitrile. Infusion into the instrument occurred at a rate of 5 μL/min. The mass spectrometry was performed on a Thermo Fisher Scientific LTQ-FT, a hybrid instrument consisting of a linear ion trap and a Fourier transform ion cyclotron resonance mass spectrometer. The entire elutant was introduced into the LTQ-FT, using the standard electrospray ionization source for the instrument with a spray voltage of 5 kV and a capillary temperature of 275°C. Autogain control was used set to 500,000 with a maximum injection time of 1250 ms for FT-ICR full scans. Collision induced dissociation, MS/MS, was executed in the linear trap with an AGC setting of 10,000 and a maximum injection time of 500 ms. FT-ICR full scans were acquired in the positive ion mode at 100,000 resolving power at m/z 400.
2′-Deoxyguanosine-An-Hq Adduct NMR Analysis
All 1H NMR experiments were carried out on a Bruker DGX-501, 500 MHz instrument. Chemical resonances are reported in δ (ppm) units, using residual 1H signals from deuterated solvents as references. The reaction listed was scaled to 100-fold (total volume 200 mL). The reaction was incubated in the dark at 25°C for four days. Solvent was evaporated and the adduct of interest was purified in two stages. First, a Biotage SP1 Flash system, outfitted with a 5.5g RediSep Rf Gold C18 column, was used to purify the reaction mixture, using a 97% water and 3% methanol buffer A and methanol for buffer B. The gradient was 0% methanol to 10% over 10 column volumes, 10% to 100% over 3 column volumes and held there for 5 column volumes. The sample (~75% pure by HPLC) was collected after 13 column volumes and was lyophilized to remove solvent. The second stage employed HPLC purification, using a Grace Alltima HP C8 semi-preparative column (3μm, 7 × 53mm) at 2.5ml/min. Solvent A was 95% water and 5% acetonitrile and solvent B was 95% acetonitrile and 5% water. The gradient was 0% B for 5 min, 30% B over 8 min, 100% B over 3 min and then held there for 4 min. Absorbance was monitored at 260nM. The collected sample eluted around 8 min. After HPLC purification, the product was dried and dissolved into 500 μL d6-DMSO and evaluated by NMR for 15 hrs to determine the 1H-NMR.
Solvolysis of An-Hq in Methanol Using Na2IrCl6 as the Oxidant
An-Hq (10 mg, 0.039 mmol) dissolved in methanol (2 mL). Na2IrCl6·6H2O (43 mg, 0.078 mmol) and N,N-diisopropylethylamine (4.5 mg, 0.039 mmol) was added to the mixture. The mixture was reacted at rt for 0.5 hr. The reaction was filtered through silica gel and washed with acetonitrile. The filtrate was concentrated and the residue was purified by flash chromatography on silica gel to provide the product (9 mg, 0.017 mmol, 90% yield) as a yellow oil. 1H NMR (CDCl3, 400 MHz) δ 7.23 (t, J=8.2 Hz, 2 H), 6.80 (d, J=10.4 Hz, 2 H), 6.69 (m, 3 H), 6.26 (d, J=10.0 Hz, 2 H), 3.76 (t, J=6.2 Hz, 2 H), 3.54 (t, J=6.2 Hz, 2 H), 3.41 (q, J=7.1 Hz, 2 H), 3.36 (s, 3 H), 0.87 (t, J=7.1 Hz, 3 H); HRMS (ESI, positive) m/z calcd. for C17H22NO3 [M+H]+: 288.1600, found: 288.1614.
DNA Damage Visualization on a 392-Nucleotide PCR Product
Primer extension experiments were performed using a 392-nucleotide dsDNA, synthesized from pUC19 plasmid vector (New England Biolabs). A 30 cycle PCR amplification (55°C for 30 s, 75°C for 45 s, and 95°C for 30 s) was performed, using a 19-nucleotide forward primer, GGCCTCTTCGCTATTACGC, starting at the nucleotide at position 287 of the vector, and a 19-nucleotide reverse primer, ATACGCAAACCGCCTCTC, starting at position 672.37 The oligonucleotide was purified using a Cycle Pure Kit (Omega BioTek), yielding a final concentration of 40 μg/μL. DNA was reacted with the agent, An-Hq2, and Na2IrCl6 for 5 hrs at 37°C (720 μg DNA, 0.2 mM sodium phosphate, pH=8, 0.5 mM An-Hq2, and 2 mM iridium). For the pre-quenched control, DNA was added 24 hrs after the agent and the oxidant were mixed. The end products are benzoquinone and N-phenyldiethanolamine. Each sample of reacted DNA was then added to the primer extension mix (1X vent buffer, 100 μM dNTP, 200 nM primer, 0.05 U/μL vent(exo-) DNA polymerase). The primer was fluorescently-labeled with IRDye700. Twelve cycles were accomplished (55°C for 15 s, 72°C for 1 min, and 95°C for 30 s). Denaturing load dye was then added to the samples, and a 12% denaturing PAGE gel was performed. The gel was visualized using the Odyssey Infrared Imaging System (LiCor) with 169 μm resolution and the 700-channel. Sequencing was performed by standard methods, using manual sequencing of the 392-nucleotide PCR product and acyclo-terminators, except the fluorescently labeled M13 primer was substituted. Experiments were performed in triplicate, and standard errors were calculated.
LC/MS Product Analysis of Reaction of An-Hq2 with Oligonucleotide
High purity oligonucleotides were purchased from Eurofins MWG operon. Electrophoresis showed the oligonucletide to be greater than 95% pure. The DNA sequence was 5′-GCGCAATTGCGC-3′. The DNA was annealed in 25 mM sodium phosphate. Prior to reaction, the DNA was desalted and placed in 5mM Ammonium Acetate buffer, pH 8.0. The 100 μL reaction contained 2 mM DNA, 10 mM An-Hq2, and 10 mM of a (diacetoxyiodo)benzene oxidant. The reaction was left at room temperature in the dark for three days. Analysis by mass spectroscopy utilized a Thermo Scientific LTQ-FT, a hybrid instrument consisting of a linear ion trap and a Fourier transform ion cyclotron resonance mass spectrometer. The injection volume was 10 μL. Liquid chromatography was accomplished using a Waters Symmetry C18 5 μm, 2.1 × 150 m column, Finnigan Surveyor MS pump, and Finnigan Micro AS autosampler. The flow rate was 200 μL/min and the gradient ranged from 2% (v/v) acetonitrile in 5mM ammonium formate to 15% over 35 min. Autogain control was used and set at 500,000 with a maximum injection time of 1250 ms for FT-ICR full scans. FT-ICR full scans were acquired in the negative ion mode at 100,000 resolving power at m/z 400. Mass accuracy errors were below 500 ppb for full scan.
Drosophila Stocks and Husbandry
Drosophila were maintained on standard cornmeal, agar, and molasses media at 25°C under a 12:12 hr light:dark cycle. To induce targeted gene silencing, the following RNAi line was obtained from the Vienna Drosophila RNAi Center:35 UAS-Ercc1RNAi (v12622VDRC). This line was crossed to the daughterless-GAL4 (da-GAL4) driver line in order to ubiquitously inactivate gene expression. The isogenic host strain, w1118 (60100VDRC), was crossed to da-GAL4 as a control for genetic background effects. The da-GAL4 line was kindly provided by Dr. Mike Grotewiel (Virginia Commonwealth University.38
RNAi Mediated Suppression of Targeted Genes
To confirm knockdown efficiency of the transgenic RNAi approach, RT-PCR assays were conducted. For each sample, fifteen adult male Drosophila were homogenized, and total RNA was isolated using Trizol (Invitrogen, Carlsbad, CA). Total RNA was then DNase treated using DNA-free (Ambion, Austin, TX), according to manufacturer’s instructions. Total RNA (0.5 μg) was reverse transcribed using the Accuscript High Fidelity First Strand cDNA synthesis kit (Agilent Technologies, Santa Clara, CA), and the resulting cDNA was used in RT-PCR assays. Primers were as follows: Ercc1-F 5′-CGTGCTGTACCTCTCGC-3′ and Ercc1-R 5′-CTGAGGAACGGTTCCTG-3′. QuantumRNA β-actin Internal Standards (Ambion, Austin, TX) were used to amplify β-actin (control) according to manufacturer’s instructions, following optimization of the β-actin:competimer ratio. PCR products were separated electrophoretically on a 2% agarose gel and visualized with ethidium bromide. RT-PCR assays on two independent RNA isolations per genotype were performed.
Microinjection of Drosophila
Microinjections were performed on individual adult male Drosophila between three and seven days old. Individuals were held in position for microinjection using a gentle vacuum. A thin pulled glass micropipette attached to a Picospritzer III (Parker Hannifin, Cleveland, OH) was used to deliver anticancer agents into the fly abdomen. A 0.5 μL volume of agent was injected at ~40 psi of compressed air. Agents were dissolved in 100% DMSO, and solutions were made fresh daily. Blue food dye (0.125 mg/mL) was added to the solutions to confirm the delivery of the agent. Agents tested included An-Hq, An-Hq2, chlorambucil, cisplatin, and 5-fluorouracil. Microinjections of the vehicle alone (control) were also performed. For each agent, ten individuals of each genotype were injected at a 10 mM concentration. A concentration of 10 mM was used because preliminary analysis showed this to be effective at eliciting a response. Post injection, flies were placed on fresh standard Drosophila media at 25°C and on a 12:12 hr light:dark cycle. Individuals were scored for survival after one day and followed for a total of 7 days to examine longer-term effects of the agents. To analyze the significance of the data, chi-squared was calculated.
IC50 Determination of An-Hq2
An-Hq2 was submitted to the NCI Developmental Therapeutics Program for screening. The method uses total protein content and was developed by Monks et. al.39 Briefly, cells are treated with An-Hq2 and then fixed to the plate surface. The number of cells is proportional to the relative amount of protein as measured by sulforhodamine B.
RESULTS
In vitro Reactions with Nucleosides and Oligonucleotides
Our data established that An-Hq and An-Hq2 are highly selective cytotoxic agents against certain cancers. As is the new convention,40 the numbering for 2′-deoxyguanosine (dG) is retained throughout this manuscript. The numbering for the An-Hq phenol derivatives is denoted by a double prime, even when discussing the phenol without addition to dG. We designed these agents to release the hydroquinone moiety upon oxidation and leave an activated nitrogen mustard. Instead, we found that the hydroquinone was the active portion of the molecule. Initially, An-Hq was reacted with dG, using a Na2IrCl6 oxidant to determine if addition occurred. We found that the mass of the product corresponded to formation of a benzetheno-2′-deoxyguanosine. Previous literature demonstrates that benzoquinone can add to 2′-deoxyguanosine and produce a 3″-hydroxy-1,N2-benzetheno-2′-deoxyguanosine.41 The first step in the reaction mechanism is a Michael addition. Thus, we probed the ability of An-Hq to react and form the same benzetheno-2′-deoxyguanosine adduct and found it to be different.
In order to determine if these agents modify DNA as benzoquinone, An-Hq was oxidized in the presence of methanol and the product determined. Na2IrCl6 was used to induce oxidation in methanol (Figure 2A). Solvolysis by methanol serves as a simple nucleophile and as a means to trap the site of nucleophilic addition. Upon oxidation, An-Hq was 90% oxidized to a new compound after 15 min. 1H-NMR was used to analyze the isolated product; the region between 3 and 8 ppm is shown in Figure 2A. There are eighteen protons in this region, indicating a product with an additional methoxy group. The singlet peak at 3.4 ppm with an integration of 2.9 corresponds to the added methoxy group. The aromatic region is symmetric, indicating that addition did not occur at C3″ or C2″ of the phenol through Michael addition. We used mass spectrometry (MS) to ensure a product consistent with a single methanol addition. Therefore, An-Hq does not behave like benzoquinone. Instead, oxidative activation leads to a potent electrophile, via addition at C4″. This infers that the DNA lesion produced may be different than benzoquinone, and these agents are not simply releasing benzoquinone as their mechanism of action.
Figure 2. An-Hq and its Derivatives React with Guanine Differently than Benzoquinone.

(A) Quinones can react via two paths: C4″ addition, black, and C2″-Michael addition, grey. The product for each path is shown. To determine the reaction path, the product from oxidation in methanol was isolated. and the NMR and MS are shown. Oxidative activation of An-Hq in methanol proceeds via C4″ addition based on the grey NMR assignments and integrals. (B) Top. NMR of the reaction between An-Hq, 2′-deoxyguanosine, and an iridium oxidant in phosphate buffer. On the left is the reaction with the assigned NMR resonances superimposed on the product. On the right is the H1-NMR between 4–10 ppm. The assignments are in grey. An N1-proton signal is observed. Bottom. The reaction between 2′-deoxyguanosine, base, and benzoquinone according to literature. On the left is the reaction with assignments. On the right is the H1-NMR. Importantly, the N1 peak at around 10ppm is missing from the benzoquinone reaction product NMR. The different reaction path of An-Hq leads to a different guanine lesion.
As there was a difference in reactivity due to oxidative activation, we investigated the lesion produced through NMR analysis (Figure 2B). For consistency, the NMR are labeled starting from A for both products. The DNA lesion formed upon incubation of An-Hq was compared to that formed with benzoquinone. The product of An-Hq, Na2IrCl6, and dG in water-phosphate buffer was used to isolate the dG-adduct formed. The oxidant used was Na2IrCl6 because it possesses the correct potential to oxidize the agents but not dG.42 The reaction of benzoquinone and dG has been established by Jowa et al. as well as by Chenna and Singer.41,43 We followed the procedure, which entails incubation of dG in dimethylformamide (DMF) and potassium carbonate. This non-aqueous system formed the benzetheno-adduct in high yield. After reaction, we purified the products to greater than 95% purity levels (see Figure S1). Analysis by positive ion MS showed products with an m/z value of 358.1146 and an elemental composition of C16H16N5O5+ (Figure 3). MS/MS shows deglycosylation, indicating that the ribose portion of the nucleoside was not modified (see Figure S2).
Figure 3. An-Hq Reacts with Nucleoside Base Amines.

(A) Analogs that react. Reactions with An-Hq (left), HPLC analysis (center), and mass spectrometry (right). 2′-deoxyguanosine, 2′-deoxyadenosine, 2′ deoxycytidine, and 7-deaza-2′-deoxyguanosine all form products with similar spectra (inset). For HPLC analyses, the grey chromatograms are control reactions. The grey traces include no An-Hq, no oxidant, and a minus An-Hq and oxidant in the reaction mixture. The reaction trace (black) contains An-Hq, oxidant, and the nucleoside analog. Purification and mass spectrometry of the products indicates each has an added phenol. 2′-deoxyadenosine had two phenols added. (B) Analogs that do not react. 2′-deoxyinosine, which lacks an N2-amine, does not react. Thymidine does not form identifiable adducts. Importantly, this provides evidence that the N2-amine is necessary for addition on the nucleoside.
Reaction of benzoquinone revealed addition at N1,N2 as had been previously observed (Figure 2B, bottom). Ten resonances between the 4–10 ppm are shown. The B shift was a singlet at 9.4 ppm and was ascribed to the terminal phenol hydrogen at C1″. Two strong singlets, C and D, were observed at 8.0. These singlets were the C8-hydrogen and the C2″-hydrogens, with the C2″ being more deshielded. Importantly, the C8-hydrogen indicated that the five-membered ring of guanine was not modified. Accordingly, N7 was not the addition site. Resonances E and F were the remaining C5″ and C6″ singlet phenol hydrogens. Taken together, the three phenol resonances indicate two dG positions have added to the phenol. The triplet at 6.2 ppm, G, is the C3′-hydrogen. The resonances H, I, and J were all ribose protons, both hydroxyl and ribose. Finally, shift K is the hydrogen at N2. We found this hydrogen to be easily exchangeable and, hence, its integration was low. We then added a drop of D2O to differentiate exchangeable and non-exchangeable protons (Figure S3). We found resonances B, H, I, and K to be exchangeable further confirming our NMR assignments.
Analysis of the An-Hq-dG adduct was accomplished in a similar manner (Figure 2B, top). Again, ten resonances are observed with some key differences. Several resonances were identical to the 3″-hydroxy-1,N2-benzetheno-2′-deoxyguanosine adduct. Shift D, the C8-hydrogen, confirmed that An-Hq does not modify N7 on the five-membered ring. Likewise, resonances B, G, H, I, and J directly overlap (compare Figure 2B top and bottom) indicating modification was not at the ribose. The NMR identified several distinct hydrogens. Two new hydrogen peaks were seen. First, the A shift at 9.9 ppm was a strong singlet. Such a strongly deshielded hydrogen can only come from the amido-hydrogen at N1. In addition, the K-shift at the N2 hydrogen was observed at 4.3 ppm. Importantly, shift A (bolded in Figure 2B) indicated that the N1 position was no longer part of the phenol-dG adduct. This was interesting because, if taken with the appearance of the C8-hydrogen, the NMR reveals that the phenol added to N2 and N3. To further support our data that An-Hq is producing a different hydroxy-benzetheno-2′-deoxyguanosine adduct, we determined stability in acid. It has been observed that N2,3-benzetheno-2′-deoxyguanosine adducts are prone to degradation by incubation in acid since a weaker glycosidic bond is present.44 Incubation at pH 1 and elevated temperatures led to degradation of the adduct when compared to the 3″-hydroxy-1,N2-benzetheno adducts (see Figure S4). It should be noted that several tautomers can be drawn that assign correctly to the NMR data (see Figure S5). In addition a proposed mechanism to form the guanine adduct is listed in Figure S5. Importantly, this data showed that An-Hq generates a distinct lesion from benzoquinone. The distinct lesion stemmed from the oxidative activation mechanism of An-Hq.
We performed a series of HPLC analyses to determine which DNA bases are modified by An-Hq (Figure 3). Nucleosides were incubated with An-Hq and Na2IrCl6. After the incubation period, reactions were analyzed by HPLC with UV detection set to 260 nm. For each nucleoside, a reaction chromatogram (black) and control chromatograms (grey) are shown. Each of the controls excludes a single reaction component: An-Hq, dG, and Na2IrCl6. The reaction of dG with An-Hq is shown on the top left of Figure 3. A clearly observable product is seen at 13 min when comparing the controls and reaction traces. The product has a characteristic three band absorbance spectrum (inset). MS analysis showed a predominate ion at m/z of 358, and the elemental composition is C16H16N5O5+ with less than 300 ppb error. It should be noted that there is a small product at 18 min which possessed an m/z value equal to the deglycosylation product (data not shown). To the top right in Figure 3, we examined 7-deaza-dG for reactivity. The NMR revealed that N7 was not involved in the reaction. Based on this data, substitution of dG with 7-deaza-dG should not interfere with formation of reaction product. By performing the same reaction with 7-deaza-dG, a new series of products are observed at 15 min. Again, these products have similar UV absorbance. The MS spectra had a mass change of 1 amu or C17H17N4O5+, which is correct for a 7-deaza-dG adduct with an added phenol. This data proved that N7 is not involved in the reaction. To further validate the NMR results, we reacted 2′-deoxyinosine with An-Hq (Figure 3B, bottom left). 2′-deoxyinosine lacks N2 and, according to the NMR results, should not react. The reaction was prepared identically to the reaction with dG. We saw no product formation, further confirming the NMR results that N2 is required for product formation.
We examined An-Hq adduct formation with each nucleoside. Analysis of the reaction between 2′-deoxyadenosine and An-Hq revealed a new product at 15 min (Figure 3, middle left). The absorbance was similar with the dG adduct formed. The product was formed in low yield. The yield was ~0.4%. The MS analysis for this adduct gave a mass indicative that two phenols had added (addition of 188 amu). Addition of two phenols has been observed in the literature involving quinone addition to nucleosides.43 The reaction with 2′-deoxycytidine (Figure 3, middle right) gave even less product, which limited characterization to HPLC. Finally, we did not observe any adduct formation upon incubation with thymidine (Figure 3, bottom right). The key difference is that thymidine lacks a nucleoside aryl amine. Altogether, this data demonstrates that the oxidative activation of An-Hq and its derivatives generates an electrophile that can add to exocyclic amino groups of adenine, guanine, and cytosine. The yield of these reactions on double stranded DNA will differ from nucleosides as the aryl amines of adenine and cytosine are accessible from the major groove, while N2 of guanine is accessible from the minor groove.
With the ability to modify three out of the four nucleosides through different parts of the helix, we set out next to investigate our agents’ reactivity with double stranded DNA (dsDNA), a biologically relevant substrates. Reaction of An-Hq2 with Na2IrCl6 and a large DNA strand would allow us to identify specific DNA base sequences susceptible to damage by the agents (Figure 4). A 392-nucleotide section of the pUC19 plasmid, positions 287–678, was amplified by PCR. The fluorescently labeled primer used in our studies overlaps at positions 370–389. Vent(exo-) polymerase was used for both extension and manual sequencing. The nucleotides listed in Figure 4 sequence lanes correspond to the template strand. For example, bands in the G lane are equivalent to the G nucleotides in the 392-nucleotide template. Agent and oxidant are required to stop primer extension. When the unmodified DNA was extended, a full-length PCR product of 301 bp was observed. Comparison of the unreacted primer and full-length product allowed calculation of percent yield of the primer extension (Figure 4A). Several controls were utilized to identify unique damage produced by the activated An-Hq2. The controls included a minus Na2IrCl6 control (−OX), the end products produced after An-Hq2 oxidation (EP), a pre-quenched reaction control (PQ), and a negative control containing only DMSO (NR). Each control yielded 3.1–3.2% full-length product and demonstrated that oxidative activation is required. The end products gave by An-Hq2 oxidation produced a 3.1 ± 0.4% extension yield, similar to the minus oxidant control. This control established that the end products do not cause extension stops in high yield. When the 392-nucleotide DNA was incubated with already oxidized An-Hq2, a similar yield of 3.2 ± 0.1% full-length extension product was observed. The no reagent control yielded a 3.1 ± 0.1% extension yield. As a positive control, we incubated the 392-nucleotide DNA with cisplatin. Cisplatin is a known DNA modifying agent and, therefore, will induce replication stops at DNA damage sites. As expected, we observed no full-length extension product in the cisplatin positive control. Next, An-Hq2 was tested to further investigate its DNA modifying capabilities. Treatment of the 392-nucleotide DNA with An-Hq2 followed by oxidative activation led to a reduction in the full-length product from 3.2% to 1.0 ± 0.2%, or a decrease of 69 ± 0.6%. The decline in product formation shows that An-Hq2 and its derivatives modify dsDNA and stop the progression of the DNA polymerase in vitro.
Figure 4. An-Hq2 Modifies dsDNA In Vitro.
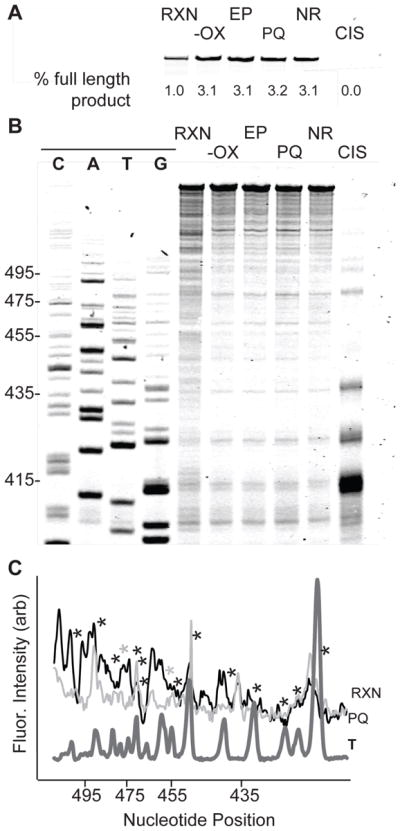
(A) Primer extension from a 392-nucleotide dsDNA resulted in a full-length extension product. Controls lanes include: lacking oxidant (−OX), incubation with the end products (EP), a pre-quenched reaction (PQ), and a negative control containing DMSO (NR). Control lanes did not alter the amount of full-length product produced. Reaction with either cisplatin (CIS) or An-Hq2 (RXN) in the presence of an oxidant led to a loss of full-length extension. (B) Gel showing sequence dependent damage. Sequence lanes on left. Unmodified DNA and controls gave similar patterns of extension stops. DNA incubated with cisplatin led to extension stops at polyguanine sequences. In contrast, An-Hq2 and oxidant caused damage at most sequences. (C) Quantification of An-Hq2 damage. Extension stops in the reaction (black) and in the pre-quenched control (light grey) were compared to a thymine sequence lane (T, dark grey). A decrease in extension stops was observed at thymine sequences.
The primer extension assay provides further evidence that damage induced by An-Hq2 and its derivatives is not only guanine specific (Figure 4B). The positive control, cisplatin, reacts at guanine repeats. This can be seen in the CIS lane of the gel, where the largest replication stop was at position 413. The corresponding sequence is 5′-GGGG. The negative controls (−OX, EP, PQ, and NR) had limited replication stops. We attributed these stops to the polymerase’s inability to pass certain structural features on the hard to replicate cloning region of pUC19. In contrast, activated An-Hq2 in the RXN lane was capable of modifying bases at most positions. When looking at positions 410–470, most nucleotides showed a greater than two-fold enhancement in the early terminations. In particular, guanine at positions 412–414, 422–423, and 430–436 all exhibited replication stops. Stops at adenine also occur. For example, replication stops can be seen at 410 and 430. Similar extension stops at cytosine positions 415–419 and 440–441 were observed. There were few predominate stops in the extension, with 76% of all three of these base types showing an increase in extension stops between two and four fold. In contrast, we observed little modification at thymine. The thymine positions on the 392-nucleotide sequence are denoted by asterisks in Figure 4C. The sequence 5′-TT at position 421 and 422 displayed limited extension stops when compared to the pre-quench control. Several other thymine residues showed no change in extension stops. In fact, only two thymine nucleotides out of nineteen had any difference between the PQ and RXN (Figure 4C, grey asterisks). This data illustrates that An-Hq2 and its derivatives can modify DNA in vitro, and that modification occurs at several types of DNA bases. This data is in-line with our nucleoside reactivity studies in Figure 3.
Reactivity with DNA was further explored in vitro using a 12-nucleotide oligonucleotide. The sequence chosen was a self-complementary sequence of 5′-GCGCAATTGCGC-3′, which has a molecular weight of 3646 g/mol with an elemental composition of C116H147N46O70P11. Reaction with An-Hq2 will produce addition of one benzetheno-group to the oligonucleotide that would increase the molecular weight to 3736 g/mol with an elemental composition of C122H149N46O71P11. This difference is quite large and is easily detected via FT-MS. The oligonucleotide was annealed to form dsDNA, reacted with An-Hq2 and an oxidant, and incubated for three days. The reaction mixture was then analyzed by LC/MS to determine whether An-Hq2 formed the benzetheno-adduct on a double stranded DNA in vitro (Figure 5). The total ion chromatogram can be seen at the top of Figure 5A, with a predominate ion peak at 12.0 min and another ion peak at 13.5 min. The MS spectrum of the 12.0 min peak is shown in Figure 5B. The 12.0 min spectrum has a large ion current with an m/z of 1214, which is the triply negatively charged unmodified oligonucleotide (Figure 5B). Also observed is the doubly negatively charged ion of the unmodified oligonucleotide with an m/z value of 1821. The MS spectrum of the 13.5 min peak is shown in Figure 5C. The major ion observed on the total ion current at 13.5 min corresponds to a m/z value of 1244, which is the expected m/z for the triply negatively charged benzetheno-oligonucleotide. Also observed is the doubly negatively charged modified oligonucleotide with an m/z of 1866. The elemental composition was determined to be C122H149N46O71P11 with an error or 447 ppb (Figure S6). The only other major ion observed in the 13.5 min FT-MS is an ion pair of the triply negatively charged oligonucleotide with trifluoroacetic acid (TFA) which has an m/z value of 1282; the TFA ion pair with the unmodified oligonucleotide is also seen in the 12.0 min FT-MS at m/z 1252. We should note that the modification can be at many of the nucleotide positions on the dsDNA, since modification at any location would give the same mass. This result is significant as it proves that An-Hq2 can add to dsDNA and form a lesion of the same mass shown in the nucleoside studies.
Figure 5. LC/MS Identification of the Product Formed Between An-Hq2 and a Self-Complementary Oligonucleotide.

(A) Reaction of An-Hq2 with an oligonucleotide, 5′-GCGCAATTGCGC, in the presence of an oxidant was monitored by LC/MS. The top chromatogram shows the total ion current for the reaction. The middle, grey, shows the ion chromatogram for the unmodified DNA. The unmodified DNA is observed at 12.0 min and predominates the total ion current. The bottom, black, is the ion chromatogram for 1244 m/z. This ion mass corresponds to the DNA with a single benzetheno-modification in the triply negatively charged state. The modified DNA was found to elute at 13.5 min and is also observed in total ion current chromatogram. (B) The top shows the sequence of the DNA. FT-MS spectra at 12.0 min shows unmodified DNA with a mass of 1214.208 for the triply charged and 1821.8176 for the doubly charged unmodified oligonucleotide. (C) FT-MS spectra 13.5 min shows a modified DNA with masses of 1244.2129 for the triply charged and 1866.821 for the doubly charged state. These masses correspond to a DNA with a single benzetheno-modification. The top structure shows the DNA sequence with a modified guanine, though the actual guanine on which modification occurred is unknown. Experimental masses are in black, while theoretical masses are in grey.
Genetic Knockdown of Nucleotide Excision Repair
Although An-Hq and its derivatives can modify double stranded DNA, it was not known if DNA modification occurs within a cellular context. Therefore, we turned to D. melanogaster, a powerful whole-animal genetic model. Because of the DNA lesion identified in vitro, we reasoned that nucleotide excision repair should be an important means to repair this adduct if it was occurring in vivo. Thus, our prediction was that the knockdown of Ercc1, a gene in the nucleotide excision repair pathway, would sensitize Drosophila to oxidatively-activated agents. This sensitization would result as limited avenues for repair would remain, even at low basal levels of An-Hq activation. Furthermore, we predicted we would see an expedient response to DNA modifying agents. DNA damaging agents, due to their reactive nature, are rapidly metabolized in cells and plasma. For example, cisplatin is hydrolyzed in blood within minutes to hours to its active forms.45 The active agents quickly react with DNA, glutathione, thioredoxin, or protein to produce irreversible adducts.46,47 Similar profiles are observed for chlorambucil, which rapidly produces DNA lesions in minutes.48
As a first step in testing our hypotheses, we began by validating Drosophila as a suitable model for the study of agents with known mechanisms of action. The effects of three agents, cisplatin, chlorambucil, and 5-fluorouracil, were examined. Specifically, we conducted a targeted transgenic RNAi knockdown experiment using the GAL4/UAS system in which expression of Ercc1 was silenced. We confirmed silencing of Ercc1 expression under the da-GAL4 driver using RT-PCR (Figure S7). Because of the rapid induction of these agents’ mechanisms of action, we monitored the tolerated doses of these agents in Drosophila over the course of 7 days. To do so, we recorded differences in survival relative to the control, following delivery of either the DNA modifying agent or vehicle control.35 In the majority of injections with healthy animals with the just the vehicle alone, death was noted in approximately 20%. This was attributed to the stress of the injection process and was expected.
First, we examined the effects of cisplatin, which cross-links proximal guanines to create helix distorting adducts. When cross-linking occurs, nucleotide excision repair is required to correct this adduct. In fact, ERCC1 activity can be used as a prognostic marker of cisplatin-based cancer treatments.49,50 As predicted, when Drosophila lacking Ercc1 expression (da-GAL4/UAS-Ercc1RNAi) were injected with cisplatin, a rapid reduction in survival was observed relative to the control line (da-GAL4/+) by Day 1 (X2 = 5.0, P < 0.025; Figure 6A). On Day 1, 40% of flies expressing Ercc1RNAi had died in comparison to none of the control flies. The 40% difference in Drosophila lacking Ercc1 is significant when compared to injection of vehicle by Day 1 (X2 = 5.0, P < 0.025). No significant difference in survival was observed between vehicle and cisplatin injected Drosophila when Ercc1 was not silenced. Finally, over the seven day period, we observed reduced survival in both genotypes with agent delivery consistent with the highly toxic nature of cisplatin. Hence, our Ercc1 results in D. melanogaster accurately replicate the observations of cisplatin and Ercc1 in cancer.
Figure 6. Drosophila with RNAi Silencing of Ercc1 Display Expected Sensitivity Towards DNA-Modifying Agents with Known Modes of Action.

Time dependant survival of D. melanogaster after microinjection with vehicle or one of the following agents: cisplatin (panel A), chlorambucil (panel B), and 5-fluorouracil (panel C). Cisplatin and chlorambucil serve as positive verification for our assay, while 5-fluorouracil acts as a negative control. Each agent was injected into D. melanogaster in which Ercc1 expression was silenced (da-GAL4/UAS-Ercc1RNAi) and compared to flies with wild-type Ercc1 expression (da-GAL4/+). For each genotype, survival with injection of vehicle alone was also assessed.
To further confirm that the Drosophila-based assay is sensitive to DNA modifying agents producing bulky lesions, we examined the response of Drosophila lacking Ercc1 expression towards chlorambucil. Chlorambucil is a nitrogen mustard that will produce bulky DNA-DNA cross-links, which require Ercc1 for repair. In the case of chlorambucil, a significant difference in response to the agent was observed on Day 1 between Drosophila lacking Ercc1 expression and the control line(X2 = 3.8, P < 0.05; Figure 6B). This suggests that flies with wild-type Ercc1 expression levels can tolerate the formation of cross-links and that Drosophila lacking Ercc1 have much higher sensitivity, with 50% death on Day 1. All the Drosophila lacking Ercc1 expression that received chlorambucil died after four days compared to 80% survival in wild-type Drosophila. Additionally, a significant difference in the survival of flies lacking Ercc1 expression was observed when the flies were injected with either chlorambucil or the vehicle (X2 = 6.7, P < 0.001). However, no significant survival difference was observed between agent and vehicle in wild-type flies. This further supports our model by demonstrating that Ercc1 is vital to the repair of bulky lesions induced from chlorambucil.
We investigated a second agent to provide additional validation of our model system. The second agent, 5-fluorouracil, is mainly repaired through base excision repair, as it signals the incorrect incorporation of uracil. Consequently, we did not expect downregulation of Ercc1 to affect its mechanism of action, and, therefore, it can be considered a negative control. When similar experiments were conducted with 5-fluorouracil, we found no significant discrimination between the flies with Ercc1 silencing and controls (Figure 6C). On Day 1, none of the injected control Drosophila had died. Similarly, in the Ercc1 knockdown only 20% had died upon 5-fluorouracil injection, after which no more Drosophila perished. Survival after agent delivery did not significantly differ from injection of the vehicle alone for either genotype. Thus, little change in mortality is observed upon treatment with 5-fluorouracil and is indicative of DNA modification occurring and being repaired. Therefore, we have validated that Drosophila, as a model organism, can be used to determine if an agent is inducing DNA damage in vivo.
Finally, we investigated if modification of DNA by our oxidatively activated agents was being repaired in the Drosophila model by a pathway involving Ercc1. Experiments similar to those using other DNA-modifying agents were accomplished, using An-Hq and An-Hq2. Because of the bulky lesion produced upon DNA modification, it is unlikely that Drosophila lacking Ercc1 expression would be able to repair the damage. Therefore, we expected that these flies would exhibit greater sensitivity towards An-Hq and An-Hq2. We first explored An-Hq. Upon injection of An-Hq, we observed that only 10% of Drosophila had died on Day 1 in the controls (Figure 7A). In contrast, Drosophila lacking Ercc1 expression showed 60% death on Day 1. This result was significant at a P < 0.002 and X2 = 5.5. Drosophila lacking Ercc1 expression also had a significant reduction in survival with agent delivery relative to injection with the vehicle (X2 = 8.5 P < 0.003). These results were not unique to An-Hq; An-Hq2 showed similar trends with flies lacking Ercc1 showing a significant reduction in survival (50% death) on Day 1 compared to the control line (X2 = 3.8, P < 0.05; Figure 7B) and compared to the delivery of vehicle alone (X2 = 6.7, P < 0.01). Wild-type Drosophila did not differ significantly when treated with An-Hq2 versus vehicle on Day 1. This data emphasizes that our novel oxidatively activated agents induce DNA modification in vivo and strongly supports their role as DNA damaging agents with highly selective cytotoxicity.
Figure 7. Ercc1 Silencing in D. melanogaster Results in Sensitivity Towards Oxidatively-Activated Agents.

(Panel A) An-Hq or (Panel B) An-Hq2 was injected into D. melanogaster in which Ercc1 expression was silenced (da-GAL4/UAS-Ercc1RNAi) and compared to flies with wild-type Ercc1 expression (da-GAL4/+). For each genotype, survival with injection of vehicle alone was also assessed.
Potency Against Renal Carcinoma Cells
We then sought to elucidate a target cell type for these novel agents. In our previous work, an MTT assay was used to quantify potency against several types of cancer cell lines. Interestingly, many of the cancer cells tested displayed weak potency against when treated with An-Hq2.24 Two renal cancer cells displayed a low IC50 value. We, therefore, assessed whether renal cells were sensitive to An-Hq2 treatment. An-Hq2 was evaluated for effects on viability using a sulforhodamine B total protein content assay at the NCI Developmental Therapeutics Program. Seven renal cancer cell lines were examined: 786-0, A498, ACHN, CAKI-1, RXF 393, SN12C, and UO-31. Data at the NCI was then fit to sigmoid and the IC50 and fitting error determined. Most renal cancer cells tested were sensitive to An-Hq2 with a median IC50 value was 1.8 μM. Importantly, some cell lines display high sensitivity since the IC50 values in ACHN and CAKI-1 cells were 360 ± 90 and 370 ± 40 nM, respectively. One cell line, SN12C, displayed low potency with an IC50 value of 21 ± 1.2 μM. Since six of the seven renal carcinoma cell lines had IC50 values below 5 μM, we infer that this cancer is targeted by oxidatively-activated agents.
CONCLUSION
DNA modifying agents are highly used in anticancer treatments, even though many are non-selective. We have designed agents in which activation occurs by the elevated reactive oxygen species that is present within some cancers. Previously, we demonstrated that our designed agents were selective for certain cancer cell types. Expansion of these studies in renal cell carcinoma cell lines shows that these agents can have potency in the mid-nanomolar range. This is an important finding because several research teams are utilizing ROS-activated agents.7,51 For example, boronic esters limit off-target effects of nitrogen mustards and protease inhibitors.52–55 Our work shows renal cancer cells as a natural target for these approaches. In this manuscript, we explored the mechanism of action of these agents through evaluation of the DNA lesion produced. These novel oxidatively activated agents behave much differently than current DNA modifying agents. They are not nitrogen mustards, nor are they a simple quinone. Instead, what occurs is the aniline ring tethered to the quinone induces nucleophilic addition to the 4″-carbon by DNA. DNA addition occurs on any exocylic amine bearing residue, which means they are capable of modifying three out of the four DNA bases. Our primer extension experiment revealed that that modification is occurring, likely at the minor groove for guanine and in the major groove for adenine and cytosine. The end product is a bulky hydroxy-N2,3-benzetheno-2′-deoxyguanosine addition that is apt to be a strong replication stop. By making such an array of lesions, these oxidatively activated agents are expected to impart a strong need for DNA repair. Thus, DNA repair, which is altered in many cancers, is an essential component to the cytotoxic mechanism of action of our novel anticancer agents.
Our Drosophila model has shown that loss of Ercc1 function plays a major role in the repair of these bulky DNA lesions. It should be noted that to assess the cellular DNA modifying ability of our novel agents, we have validated a rapid assay. In fact, the detection of DNA lesions formed from anticancer agents is a challenging process. This difficulty is due to the fact that only a few lesions need to form to impart cytotoxicity. The RNAi-mediated silencing of Ercc1 in Drosophila can serve as a simple means to determine if a therapeutic is a DNA damage agent. This study illustrates that DNA modifying agents can be selective, even if they form highly disruptive lesions.
Supplementary Material
Table 1.
IC50 of An-Hq2 against Renal Cancer Cells.
| Renal Cancer Cell | IC50, μM |
|---|---|
| 786-0 | 1.8 ± 0.4 |
| A498 | 5.0 ± 0.5 |
| ACHN | 0.36 ± 0.09 |
| CAKI-1 | 0.37 ± 0.04 |
| RXF 393 | 2.6 ± 0.6 |
| SN12C | 21.0 ± 1.2 |
| UO-31 | 1.5 ± 0.3 |
note: IC50 measured using sulforhodamine B total protein assay
Acknowledgments
FUNDING SUPPORT
Funding came from the University of Cincinnati in the form of startup funding and a faculty research award to E.J.M. as well as a graduate student research fellowship to T.R.B.H. Furthermore, we acknowledge the NIH (Grant NIHRR19900) for acquisition of the LTQ-MS used in these studies. Drosophila research was supported by a grant from the National Institute of Health to S.M.R. (GM080592).
We would like to acknowledge Jim Deddens, John Layne, and Larry Sallans for technical assistance and/or helpful discussions. We also thank the Vienna Drosophila RNAi center for providing Drosophila stocks.
ABBREVIATIONS
- ROS
reactive oxygen species
- dG
2′-deoxyguanosine
- An-Hq
4-(2-(ethyl(phenyl)amino)ethoxy)phenol
- An-Hq2
4,4′-(2,2′-(phenylazanediyl)bis(ethane-2,1-diyl)bis(oxy))diphenol
- NMR
nuclear magnetic resonance
References
- 1.Chabner B, Roberts T. Timeline - Chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5:65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 2.Dhar S, Kolishetti N, Lippard S, Farokhzad O. Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc Natl Acad Sci USA. 2011;108:1850–1855. doi: 10.1073/pnas.1011379108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lovejoy K, Lippard S. Dalton Trans. 2009. Non-traditional platinum compounds for improved accumulation, oral bioavailability, and tumor targeting; pp. 10651–10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Puckett C, Barton J. Targeting a ruthenium complex to the nucleus with short peptides. Bioorg Med Chem. 2010;18:3564–3569. doi: 10.1016/j.bmc.2010.03.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhilina Z, Ziemba A, Nielsen P, Ebbinghaus S. PNA-nitrogen mustard conjugates are effective suppressors of HER-2/neu and biological tools for recognition of PNA/DNA interactions. Bioconj Chem. 2006;17:214–222. doi: 10.1021/bc0502964. [DOI] [PubMed] [Google Scholar]
- 6.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 7.Pelicano H, Carney D, Huang P. ROS stress in cancer cells and therapeutic implications. Drug Resist Updat. 2004;7:97–110. doi: 10.1016/j.drup.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 8.Sudarshan S, Sourbier C, Kong HS, Block K, Romero VAV, Yang Y, Galindo C, Mollapour M, Scroggins B, Goode N, Lee MJ, Gourlay CW, Trepel J, Linehan WM, Neckers L. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1 stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol. 2009;29:4080–4090. doi: 10.1128/MCB.00483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wittgen H, van Kempen L. Reactive oxygen species in melanoma and its therapeutic implications. Melanoma Res. 2007;17:400–409. doi: 10.1097/CMR.0b013e3282f1d312. [DOI] [PubMed] [Google Scholar]
- 10.Hole P, Darley R, Tonks A. Do reactive oxygen species play a role in myeloid leukemias? Blood. 2011;117:5816–5826. doi: 10.1182/blood-2011-01-326025. [DOI] [PubMed] [Google Scholar]
- 11.Cadet J, Douki T, Ravanat J. Oxidatively generated base damage to cellular DNA. Free Radic Biol Med. 2010;49:9–21. doi: 10.1016/j.freeradbiomed.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 12.Solivio MJ, Nemera DB, Sallans L, Merino EJ. Biologically relevant oxidants cause bound proteins to readily oxidatively cross-link at guanine. Chem Res Toxicol. 2012;25:326–336. doi: 10.1021/tx200376e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alp O, Zhang Y, Merino EJ, Caruso JA. Selenium effects on arsenic cytotoxicity and protein phosphorylation in human kidney cells using chip-based nanoLC-MS/MS. Metallomics. 2011;3:482–490. doi: 10.1039/c0mt00110d. [DOI] [PubMed] [Google Scholar]
- 14.Valko M, Rhodes C, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 2006;160:1–40. doi: 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 15.Adhikary A, Becker D, Collins S, Koppen J, Sevilla M. C5′ - and C3′-sugar radicals produced via photo-excitation of one-electron oxidized adenine in 2′-deoxyadenosine and its derivatives. Nucleic Acids Res. 2006;34:1501–1511. doi: 10.1093/nar/gkl026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murtas D, Piras F, Minerba L, Ugalde J, Floris C, Maxia C, Demurtas P, Perra M, Sirigu P. Nuclear 8-hydroxy-2′-deoxyguanosine as survival biomarker in patients with cutaneous melanoma. Oncol Rep. 2010;23:329–335. [PubMed] [Google Scholar]
- 17.Loft S, Poulsen H. Cancer risk and oxidative DNA damage in man. J Mol Med. 1996;74:297–312. doi: 10.1007/BF00207507. [DOI] [PubMed] [Google Scholar]
- 18.Vafa O, Wade M, Kern S, Beeche M, Pandita T, Hampton G, Wahl G. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol Cell. 2002;9:1031–1044. doi: 10.1016/s1097-2765(02)00520-8. [DOI] [PubMed] [Google Scholar]
- 19.Hu Y, Lu W, Chen G, Wang P, Chen Z, Zhou Y, Ogasawara M, Trachootham D, Feng L, Pelicano H, Chiao PJ, Keating MJ, Garcia-Manero G, Huang P. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012;22:399–412. doi: 10.1038/cr.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hole PS, Pearn L, Tonks AJ, James PE, Burnett AK, Darley RL, Tonks A. Ras-induced reactive oxygen species promote growth factor-independent proliferation in human CD34(+) hematopoietic progenitor cells. Blood. 2010;115:1238–1246. doi: 10.1182/blood-2009-06-222869. [DOI] [PubMed] [Google Scholar]
- 21.Jourden JLM, Cohen SM. Hydrogen peroxide activated matrix metalloproteinase inhibitors: A Prodrug Approach. Angew Chem Int Edit. 2010;49:6795–6797. doi: 10.1002/anie.201003819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuang Y, Balakrishnan K, Gandhi V, Peng X. Hydrogen peroxide inducible DNA cross-linking agents: targeted anticancer prodrugs. J Am Chem Soc. 2011;133:19278–19281. doi: 10.1021/ja2073824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagen H, Marzenell P, Jentzsch E, Wenz F, Veldwijk MR, Mokhir A. Aminoferrocene-based prodrugs activated by reactive oxygen species. J Med Chem. 2012;55:924–934. doi: 10.1021/jm2014937. [DOI] [PubMed] [Google Scholar]
- 24.Li G, Bell T, Merino EJ. Oxidatively activated DNA-modifying agents for selective cytotoxicity. ChemMedChem. 2011;6:869–875. doi: 10.1002/cmdc.201100014. [DOI] [PubMed] [Google Scholar]
- 25.Richgels PK, Rollmann SM. Genetic variation in odorant receptors contributes to variation in olfactory behavior in a natural population of Drosophila melanogaster. Chem Senses. 2012;37:229–240. doi: 10.1093/chemse/bjr097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bier E. Drosophila, the golden bug, emerges as a tool for human genetics. Nat Rev Gen. 2005;6:9–23. doi: 10.1038/nrg1503. [DOI] [PubMed] [Google Scholar]
- 27.Bell A, McBride S, Dockendorff T. Flies as the ointment Drosophila modeling to enhance drug discovery. Fly. 2009;3:39–49. doi: 10.4161/fly.3.1.7774. [DOI] [PubMed] [Google Scholar]
- 28.Das T, Cagan R. Drosophila as a novel therapeutic discovery tool for thyroid cancer. Thyroid. 2010;20:689–695. doi: 10.1089/thy.2010.1637. [DOI] [PubMed] [Google Scholar]
- 29.Kislukhin G, Murphy ML, Jafari M, Long AD. Chemotherapy-induced toxicity is highly heritable in Drosophila melanogaster. Pharmacogenet Genom. 2012;22:285–289. doi: 10.1097/FPC.0b013e3283514395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sekelsky J, Brodsky M, Burtis K. DNA repair in Drosophila: Insights from the Drosophila genome sequence. J Cell Biol. 2000;150:F31–F36. doi: 10.1083/jcb.150.2.f31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pandey UB, Nichols CD. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev. 2011;63:411–436. doi: 10.1124/pr.110.003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reiter L, Potocki L, Chien S, Gribskov M, Bier E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001;11:1114–1125. doi: 10.1101/gr.169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pagliarini R, Quinones A, Xu T. Analyzing the function of tumor suppressor genes using a Drosophila model. Meth Mol Biol. 2003;223:349–382. doi: 10.1385/1-59259-329-1:349. [DOI] [PubMed] [Google Scholar]
- 34.de Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13:768–785. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- 35.Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, Couto A, Marra V, Keleman K, Dickson BJ. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- 36.Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protić M, Hübscher U, Egly JM, Wood RD. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell. 1995;80:859–868. doi: 10.1016/0092-8674(95)90289-9. [DOI] [PubMed] [Google Scholar]
- 37.Solivio MJ, Joy TJ, Sallans L, Merino EJ. Copper generated reactive oxygen leads to formation of lysine-DNA adducts. J Inorg Biochem. 2010;104:1000–1005. [PubMed] [Google Scholar]
- 38.Martin I, Jones MA, Grotewiel M. Manipulation of Sod1 expression ubiquitously, but not in the nervous system or muscle, impacts age-related parameters in Drosophila. FEBS Lett. 2009;583:2308–2314. doi: 10.1016/j.febslet.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monks A, Scudiero D, Skehan P, Shoemaker R, Paull K, Vistica D, Hose C, Langley J, Cronise P, Vaigro-Wolff A. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J Natl Cancer Inst. 1991;83:757–766. doi: 10.1093/jnci/83.11.757. [DOI] [PubMed] [Google Scholar]
- 40.Cooke MS, Loft S, Olinski R, Evans MD, Bialkowski K, Wagner JR, Dedon PC, Møller P, Greenberg MM, Cadet J. Recommendations for standardized description of and nomenclature concerning oxidatively damaged nucleobases in DNA. Chem Res Toxicol. 2010;23:705–707. doi: 10.1021/tx1000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chenna A, Singer B. Synthesis of a benzene metabolite adduct, 3″-Hydroxy-1, N2-benzetheno-2′-deoxyguanosine, and Its site-specific incorporation into DNA oligonucleotides. Chem Res Toxicol. 1997;10:165–171. doi: 10.1021/tx960168r. [DOI] [PubMed] [Google Scholar]
- 42.Munk BH, Burrows CJ, Schlegel HB. Exploration of mechanisms for the transformation of 8-hydroxy guanine radical to FAPyG by density functional theory. Chem Res Toxicol. 2007;20:432–444. doi: 10.1021/tx060187t. [DOI] [PubMed] [Google Scholar]
- 43.Jowa L, Witz G, Snyder R, Winkle S, Kalf GF. Synthesis and characterization of deoxyguanosine-benzoquinone adducts. J Appl Toxicol. 1990;10:47–54. doi: 10.1002/jat.2550100109. [DOI] [PubMed] [Google Scholar]
- 44.Khazanchi R, Yu P, Johnson F. N-2,3-Etheno-2″-deoxyguanosine [8,9-dihydro-9-oxo-2-”deoxy-3-beta-D-ribofuranosylimidazo[2,1-B]purine] - a practical synthesis and characterization. J Org Chem. 1993;58:2552–2556. [Google Scholar]
- 45.Verschraagen M, van der Born K, Zwiers T, van der Vijgh W. Simultaneous determination of intact cisplatin and its metabolite monohydrated cisplatin in human plasma. J Chromatog B-Analyt Technol Biomed Life Sci. 2002;772:273–281. doi: 10.1016/s1570-0232(02)00108-3. [DOI] [PubMed] [Google Scholar]
- 46.Vermorken JB, van der Vijgh WJ, Klein I, Hart AA, Gall HE, Pinedo HM. Pharmacokinetics of free and total platinum species after short-term infusion of cisplatin. Cancer Treat Rep. 1984;68:505–513. [PubMed] [Google Scholar]
- 47.Paz MM, Zhang X, Lu J, Holmgren A. A new mechanism of action for the anticancer drug mitomycin C: mechanism-based inhibition of thioredoxin reductase. Chem Res Toxicol. 2012;25:1502–1511. doi: 10.1021/tx3002065. [DOI] [PubMed] [Google Scholar]
- 48.Bank BB, Kanganis D, Liebes LF, Silber R. Chlorambucil pharmacokinetics and DNA binding in chronic lymphocytic leukemia lymphocytes. Cancer Res. 1989;49:554–559. [PubMed] [Google Scholar]
- 49.Besse B, Massard C, Haddad V, Andre F, Dunant A, Pirker R, Olaussen KA, Brambilla E, Fouret P, Soria JC. ERCC1 influence on the incidence of brain metastases in patients with non-squamous NSCLC treated with adjuvant cisplatin-based chemotherapy. Ann Oncol. 2011;22:575–581. doi: 10.1093/annonc/mdq407. [DOI] [PubMed] [Google Scholar]
- 50.Arora S, Kothandapani A, Tillison K, Kalman-Maltese V, Patrick SM. Downregulation of XPF-ERCC1 enhances cisplatin efficacy in cancer cells. DNA Repair (Amst) 2010;9:745–753. doi: 10.1016/j.dnarep.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peng X, Gandhi V. ROS-activated anticancer prodrugs: a new strategy for tumor-specific damage. Ther Del. 2012;3:823–833. doi: 10.4155/tde.12.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuang Y, Balakrishnan K, Gandhi V, Peng X. Hydrogen Peroxide Inducible DNA Cross-Linking Agents: Targeted Anticancer Prodrugs. J Am Chem Soc. 2011;133:19278–19281. doi: 10.1021/ja2073824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Major Jourden JL, Cohen SM. Hydrogen Peroxide Activated Matrix Metalloproteinase Inhibitors: A Prodrug Approach. Angew Chem Int Ed Engl. 2010;49:6795–6797. doi: 10.1002/anie.201003819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hagen H, Marzenell P, Jentzsch E, Wenz F, Veldwijk MR, Mokhir A. Aminoferrocene-Based Prodrugs Activated by Reactive Oxygen Species. J Med Chem. 2012;55:924–934. doi: 10.1021/jm2014937. [DOI] [PubMed] [Google Scholar]
- 55.Cao S, Wang Y, Peng X. ROS-Inducible DNA Cross-Linking Agent as a New Anticancer Prodrug Building Block. Chem Eur J. 2012;18:3850–3854. doi: 10.1002/chem.201200075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.