

Abstract
Major depression (MD) is a common psychiatric disorder with a complex and multifactor aetiology. Potential mechanisms associated with the pathogenesis of this disorder include monoamine deficits, hypothalamic-pituitary-adrenal (HPA) axis dysfunctions, inflammatory and/or neurodegenerative alterations. An increased secretion and reactivity of cortisol together with an altered feedback inhibition are the most widely observed HPA abnormalities in MD patients. Glucocorticoids, such as cortisol, are vital hormones that are released in response to stress, and regulate metabolism and immunity but also neuronal survival and neurogenesis. Interestingly depression is highly prevalent in infectious, autoimmune and neurodegenerative diseases and at the same time, depressed patients show higher levels of pro-inflammatory cytokines. Since communication occurs between the endocrine, immune and central nervous system, an activation of the inflammatory responses can affect neuroendocrine processes, and vice versa. Therefore, HPA axis hyperactivity and inflammation might be part of the same pathophysiological process: HPA axis hyperactivity is a marker of glucocorticoid resistance, implying ineffective action of glucocorticoid hormones on target tissues, which could lead to immune activation; and, equally, inflammation could stimulate HPA axis activity via both a direct action of cytokines on the brain and by inducing glucocorticoid resistance. In addition, increased levels of pro-inflammatory cytokines also induce the production of neurotoxic end products of the tryptophan–kynurenine pathway. Although the evidence for neurodegeneration in MD is controversial, depression is comorbid with many other conditions where neurodegeneration is present. Since several systems seem to be involved interacting with each other, we cannot unequivocally accept the simple model that glucocorticoids induce neurodegeneration, but rather that elevated cytokines, in the context of glucocorticoid resistance, are probably the offenders. Chronic inflammatory changes in the presence of glucocorticoid resistance may represent a common feature that could be responsible for the enhanced vulnerability of depressed patients to develop neurodegenerative changes later in life. However, further studies are needed to clarify the relative contribution of glucocorticoids and inflammatory signals to MD and other disorders.
Keywords: Cytokines, Depression, Glucocorticoid resistance, Glucocorticoids, Kynurenine pathway, Neurodegeneration
1. Introduction
Major depression (MD) is one of the most common psychiatric disorders in the Western World, and according to the World Health Organization it is predicted to be the leading cause of burden of disease by 2030 (WHO report 2004). Only a third of patients receive adequate treatment and up to half of them relapse despite the increasing number of antidepressant drugs currently available (Thase, 2006). This reflects the heterogeneity of the disorder, which has a complex and multifactorial aetiology originating from the interaction between environmental and genetic factors and presents frequent co-morbidity. Several theories have been proposed to explain its pathogenesis, including the monoamine hypothesis, based on deficiency of the biogenic amine system (particularly serotonin and norepinephrine) (Charney, 1998; Hirschfeld, 2000), the hypothalamic-pituitary-adrenal (HPA) axis dysfunction theory, based on hyperactivity of this system usually reflected in high levels of glucocorticoids (Dinan, 1994; Pariante and Lightman, 2008), cognitive and behavioural theories (Beck et al., 1979; Harmer et al., 2009), the neurogenesis hypothesis (Jacobs et al., 2005; Duman, 2004; Kempermann and Kronenberg, 2003; Sahay and Hen, 2007; Kempermann et al., 2008; Zhao et al., 2008), the inflammatory theory, also known as the malaise or cytokine theory (Smith, 1991; Ur et al., 1992; Maes et al., 2009; Miller et al., 2009), and the neurodegenerative hypothesis (Myint and Kim, 2003; Maes et al., 2009). With the aim to better understand the pathophysiology of depression, these hypotheses, which indeed have some biological features in common, are being drawn together. Specifically, the last two models, reviewed in this special issue on ‘Neuro-inflammatory and Neuro-degenerative Pathways in Major Depression’, are gaining more relevance. Most interesting is the role of glucocorticoids not only in the HPA axis dysfunction model but also in the two models stated above. Are glucocorticoids a potential link between inflammation and degeneration? The answer to this question is not clear.
Glucocorticoids, released by the adrenal gland in response to stress, are among the most potent anti-inflammatory hormones in the body (Vinson, 2009). Some studies have suggested that they contribute to the hippocampal atrophy found in depressed patients (Sapolsky, 2000; McEwen, 2005). The involvement of stress in the development of depressive symptomatology may involve several systems, including the neuroendocrine, the neurotransmitter and the immune systems, that interact with the HPA axis in complex ways (Baune, 2009). However, to date, the role of the above-mentioned systems in the pathogenesis of depression is contradictory and subject to differing interpretations. In this review we will focus the attention on glucocorticoids and inflammation processes, trying to summarize and clarify as best as we can their involvement in MD, and to understand whether they have a direct cause in the abnormalities observed in the brain of depressed patients.
2. The HPA axis in major depression
One of the characteristic features of MD that has been found over the past years is the disturbance in the HPA functionality (Pariante, 2003, 2006). HPA axis activity is governed by the secretion of corticotrophin releasing hormone (CRH) and arginine–vasopressin (AVP) from the hypothalamus, which in turn activate the secretion of adrenocorticotrophin hormone (ACTH) from the pituitary. ACTH then stimulates the secretion of glucocorticoids (cortisol in humans and corticosterone in rodents) from the adrenal cortex, and these glucocorticoids interact with their receptors in multiple target tissues including the HPA axis, where they are responsible for feedback inhibition both on CRF and AVP from the hypothalamus (Antoni, 1993). The HPA abnormalities found in MD, extensively studied also in our laboratory, include increased secretion and reactivity of cortisol (Carpenter and Bunney, 1971; McClure, 1966; Luby et al., 2003; Bhagwagar et al., 2003, 2005; Cowen, 2009; Juruena et al., 2006) elevated basal cerebrospinal fluid CRH levels (Nemeroff et al., 1984) and increased size as well as activity of the pituitary and adrenal glands (Nemeroff et al., 1992). Interestingly, some of these findings are shared with other stress-related mental disorders, such as schizophrenia (Pariante et al., 2004, 2005; Mondelli et al., 2010). It is therefore possible to speculate that some of the biological mechanisms and models described in this review with relevance to MD are also operating in other psychiatric disorders.
We will start by providing some insight into the role of glucocorticoids, including their molecular basis of action, which might contribute to the development of depressive symptoms.
2.1. Role of glucocorticoids
Glucocorticoids regulate body peripheral functions such as metabolism and immunity, and they represent the classic endocrine response to stress. Being an essential component of the homeostatic system of the body, their levels in blood and in tissues must remain within an optimal range. In fact, they have profound effects on the brain, since excess or disturbed patterns of their circulating levels can have deleterious effects. Glucocorticoids have been shown to regulate neuronal survival, neurogenesis, the acquisition of new memories, the emotional appraisal of events as well as the immune response to stress (Herbert et al., 2006). Therefore, the involvement of glucocorticoids both in stress and in brain functioning could explain, at least in part, the HPA abnormalities found in psychiatric disorders, and in particular in MD. Hyperactivity of the HPA axis in MD can be demonstrated by the altered feedback inhibition, as seen by increased circulating cortisol and non-suppression of cortisol following administration of dexamethasone (Holsboer et al., 1982; Holsboer-Trachsler et al., 1991). While dexamethasone administration significantly suppressed the mitogen-induced lymphocyte proliferation and the interleukin-1beta (IL-1beta) production in healthy controls, MD patients exhibited dexamethasone non-suppression (Maes et al, 1991). In a similar way, dexamethasone administration induced an increase in the number of neutrophils and a decrease in the number of lymphocytes in healthy controls, whereas this effect was not observed in non-suppressors. Overall, these changes in immune function occurring in depression are said to confer an immune resistance to glucocorticoids (Maes et al., 1994).
However, why exactly MD patients have this glucocorticoid resistance, or impairment of the glucocorticoid-mediated negative feedback of the HPA axis, is still unknown, even though an alteration of glucocorticoid receptor (GR) function might play a central role (Carvalho and Pariante, 2008). We will now discuss this issue in more detail.
2.2. Glucocorticoid resistance
Decreased responsiveness to glucocorticoids, a phenomenon known as glucocorticoid resistance, is believed to be related in part to an impaired function of the GR (Pariante et al., 1995; Pariante and Miller, 2001; Pariante, 2006; Carvalho and Pariante, 2008). Evidence supporting the role of glucocorticoid resistance (rather than glucocorticoid excess) in depression comes from a multitude of studies, some of them conducted in our own laboratory, showing that antidepressants in human, animal and cellular models tend to increase (rather than decrease) GR function (Pariante and Miller, 2001; Pariante, 2003; Pariante et al., 2004; Yau et al., 2007; Carvalho et al., 2008; Carvalho and Pariante, 2008).
Glucocorticoids can act on two different receptors: the already mentioned GR and the mineralocorticoid receptor (MR). The MR binds cortisol with higher affinity, hence it regulates normal HPA fluctuations, being significantly bound even at the circadian trough, and fully bound at the circadian peak and during stressful episodes. The GR, of lower affinity, gets extensively occupied in times of stress. These receptors act as transcriptional regulators (see below), targeting several genes which control neuronal and network responses that underlie behavioural adaptation (de Kloet et al., 2005). Actions mediated by both receptors are complementary and operate in different time domains of the stress response: the MR normally prevents stress-induced disturbances, but, if such disturbances occur, the GR can operate and help in the recovery process. Although it is possible that an imbalance between MR and GR could increase the vulnerability to stress-related psychiatric disorders in predisposed individuals (de Kloet et al., 2007), we have decided to focus the attention of this review on the role of the GR, due to its importance during stressful events.
2.3. Molecular basis of glucocorticoid action
Glucocorticoids act by binding to the cytosolic GR, which is subsequently activated and is thus able to translocate into the nucleus. Unbound GR is mainly kept inactive in the cytoplasm of the cell, complexed with chaperone proteins, such as heat-shock proteins and immunophilins. Upon ligand binding, GR undergoes a conformational change, resulting in its dissociation from the chaperone protein complex and causing exposure of a nuclear localization signal that allows its translocation into the nucleus. Once in the nucleus, the GR either binds to DNA and switches on the expression of anti-inflammatory genes, or acts indirectly by protein–protein interaction to repress the activity of a number of distinct signalling pathways such as nuclear factor-kappaB (NF-kappaB), involving the recruitment of co-repressor molecules. The GR can also mediate rapid nongenomic signalling events initiated in the cytoplasm (Revollo and Cidlowski, 2009).
A failure in GR function, as hypothesized to take place in depression, may therefore result from reduced glucocorticoid binding to GR, reduced GR expression, enhanced activation of inflammatory pathways, or lack of co-repressor activity (De Bosscher and Haegeman, 2009). We will describe later the potential role of cytokines in affecting GR function.
2.4. Effects of glucocorticoids on neurogenesis
During life it is common to be repeatedly exposed to brief periods of stress, most of which can normally be controlled. After any short episode of stress, excitability in limbic areas is enhanced, and stress hormones promote focused attention and alertness. Subsequently, when the hormone concentrations go back to their pre-stress levels, gene-mediated actions induced by corticosteroids reverse and normalize the enhanced excitability. However, if stress is repetitively experienced in an uncontrollable and unpredictable manner, it can cause alterations in dendrite and spine morphology in specific brain regions, and eventually suppress adult neurogenesis (Mirescu and Gould, 2006; Joels et al., 2007). Neurogenesis in the adult hippocampus has been implicated in cognitive function and is stimulated by antidepressant drugs, although its functional impact and contribution to the aetiology of depression remains unclear (Sapolsky, 2004; Surget et al., 2008; David et al., 2009). However, severe or chronic stress exposure, both in early life as well as in adulthood, can cause neural cell death and atrophy of neuronal processes, and also affect hippocampal neurogenesis and plasticity (McEwen and Seeman, 1999; Sapolsky, 2000). A reduction in neurogenesis can theoretically contribute to the cognitive symptoms of depression, even though by itself is unlikely to produce the full mood disorder (Lucassen et al., 2010). The molecular mechanisms by which stress does induce these changes are however still unclear.
3. Inflammation and depression
As stated in the Introduction, the current theories on serotonergic dysfunctions and cortisol hypersecretion, on their own, do not provide sufficient explanations for the nature of depression. Since depression is a complex disorder, it is likely that alterations in several systems, which interact and interplay in concert, underlie the pathogenesis of the disease. The evidence that cytokine-mediated inflammatory processes play an important role in the development of depression is now strong. Depression is highly prevalent in infectious, autoimmune and neurodegenerative diseases, and this co-morbidity cannot be attributed only to the psychological distress of the starting disease (Pollak and Yirmiya, 2002). At the same time, depressed patients show higher levels of pro-inflammatory cytokines, acute phase proteins, chemokines and cellular adhesion molecules (Raison et al., 2006; Howren et al., 2009). In addition, it has been demonstrated that therapeutic administration of the cytokine interferon-alpha (IFN-alpha) leads to a depressive symptomatology in up to 50% of patients, who show similar biological alterations as those found in MD and can be treated with antidepressant medication (Capuron and Miller, 2004; Raison et al., 2005; Maddock et al., 2005; Lotrich et al., 2007; Bull et al., 2009).
The cytokine hypothesis suggests that external or psychosocial stressors and internal stressors such as organic inflammatory conditions may trigger depression via inflammatory processes (Maes et al., 2009). In fact, activation of the inflammatory immune system provokes numerous neuroendocrine and neurotransmitter changes, many of which are similar to those provoked by physical or psychological stressors. Moreover it has been proposed that the brain translates an immune activation much as if it were a stressor (Anisman, 2009). For a long time, the brain was considered to be a privileged organ from an immunological point of view, but we now know that pro-inflammatory cytokines produced in response to peripheral infections act on the brain to cause what is known as ‘sickness behaviour’. If, during systemic infections, cancer or autoimmune diseases, the activation of the peripheral immune system is prolonged, the immune signalling to the brain could lead to an exacerbation of sickness behaviour and to the development of depressive symptoms in vulnerable individuals (Dantzer et al., 2008).
Pro-inflammatory cytokines also affect neuroplasticity. They influence neuronal functioning through changes in apoptosis, oxidative stress and metabolic derangement, as well as by impairing processes of neuronal branching (Hayley et al., 2005). In particular, IL-1beta has been shown to inhibit long-term potentiation, a form of neuronal plasticity believed to underlie learning and memory (Pickering and O’Connor, 2007), both of which are frequently affected in MD.
3.1. Mechanisms of cross talk between cytokines and GR
The way a cell responds to glucocorticoid and cytokines is determined by interactions between several signalling pathways. Cytokines can activate the HPA axis, causing an elevation of systemic glucocorticoid levels, and at the same time, they can inhibit GR function at multiple levels, including GR translocation and induction of GR isoforms with reduced capacity to bind ligand (Pace et al., 2007). Some interactions, such as GR binding to its DNA response element, occur in the nucleus. In the cytoplasm, interference can happen with various signalling proteins, such as mitogen-activated protein kinase (MAPK), ERK, c-Jun N-terminal kinase (JNK), Janus kinase (JAK) and IkB kinase beta (Pace and Miller, 2009). In particular, all of the MAPKs have been identified as potential targets for the anti-inflammatory actions of glucocorticoids through blockage of their activating phosphorylations. Vice versa, cytokine-activated MAPK signalling can phosphorylate the GR protein itself, thereby modulating its turnover and its transcriptional activity imposing an extra layer of regulation of GR function. This mutually antagonistic cross talk mechanism may contribute to the occurrence of steroid insensitivity.
3.2. Pro-inflammatory cytokines IL-1beta and IL-6
Among the pro-inflammatory cytokines, IL-1beta and IL-6, both of which increase during infection, have been described as playing a central role in synaptic plasticity, neurogenesis and neuromodulation (McAfoose and Baune 2009). The increase in these two cytokines, together with IFN-alpha, have received the greatest attention in the context of MD, although the decrease in anti-inflammatory cytokines such as IL-4 and IL-10 might also be of significant importance. Howren et al. (2009) conducted a meta-analysis of articles published between 1967 and 2008 and reported that both IL-1 and IL-6 were positively associated with depression (IL-6, p< .001; IL-1, p=.03). A recent meta-analysis of twenty-four studies (Dowlati et al., 2010) also reported significantly higher levels of IL-6 (p< .00001) in MD patients compared to controls. Furthermore, in another study conducted on 60 pregnant women, it has been found that post partum depressive symptoms are associated with higher levels of maternal serum IL-6 during pregnancy (Christian et al., 2009).
Animal studies have also provided some useful evidence for a putative role of these cytokines in the context of stress and depression. For example, mice subjected to chronic mild stress (CMS) showed increased IL-1beta levels in the hippocampus and in parallel a development of changes in behaviour resembling depressive symptoms, such as decreased sucrose preference and reduced social exploration. In contrast, mice with deletion of the IL-1 receptor did not display such behavioural changes (Goshen et al., 2008). In addition, it has also been demonstrated that both inhibiting the IL-1 receptor and using IL-1 receptor null mice prevented the anhedonic behaviour caused by chronic stress exposure (Koo and Duman, 2008).
Taken together, it is possible that increased levels of pro-inflammatory cytokines, especially IL-1 and IL-6, may contribute to the pathophysiology of depression inducing, as we will discuss below, glucocorticoid resistance, neurodegeneration, or indeed other yet unknown mechanism.
4. Interactions between cytokines and glucocorticoids: what comes first?
Is it possible to know if cytokines directly cause disturbances of the HPA axis and GR downregulation? Or do they maybe enhance a disturbance that was already caused by a previous stressor? Glucocorticoid resistance can underlie the HPA axis disturbances and this might be caused by cytokines. Since communication occurs between the endocrine, immune, and central nervous system, an activation of the inflammatory responses can affect neuroendocrine processes, and vice versa. In fact, stress can promote inflammatory responses through effects on sympathetic and parasympathetic nervous system pathways. Both stressors and inflammatory immune activation may produce parallel neurochemical changes that can act either independently, additively or synergistically in promoting MD (Anisman et al., 2008b; Anisman, 2009). Whereas depressed patients with increased inflammatory biomarkers are more likely to be resistant to antidepressant therapy, a response to treatment has been associated with decreased inflammatory responses (Raison et al., 2006; Miller et al., 2009). Several mechanisms may be involved. Inflammatory cytokines and their signalling pathways including MAPK, NF-kappaB, signal transducers and activators of transcription and cyclooxygenase have been found to inhibit GR function by acting on GR translocation or on GR-mediated gene transcription (Miller et al., 1999; Pariante et al., 1999; Pace et al., 2007). In addition, changes produced by stress or immune responses may affect neurogenesis and influence cellular viability, predominantly through NF-kappaB and MAPK pathways (Anisman et al., 2008b).
An interesting recent study supports the idea that chronic stress is accompanied by GR resistance and activation of pro-inflammatory pathways (Miller et al., 2008). The study, conducted in peripheral blood monocytes of caregivers of patients with malignant brain cancer, involved genome expression microarrays. The caregivers’ patterns of cortisol secretion were similar to those of matched control. However, their monocytes showed diminished expression of transcripts bearing response elements for glucocorticoids, and heightened expression of transcripts with response elements for the key pro-inflammatory transcription factor NF-kappaB (Hayden et al., 2006). These findings suggest that stress produces functional resistance to glucocorticoids, which enables activation of pro-inflammatory transcription control pathways, even in the absence of decreased GR mRNA expression or excess cortisol production.
Additional information has been provided by animal model studies. In this respect, it has been shown that mice with deletion of the IL-1 receptor did not display neuroendocrine changes when subjected to chronic stress. Interestingly, the removal of endogenous glucocorticoids abolished the depressive-like effects of CMS, indicating that the blunting of the adrenocortical activation may play a role in their resistance to develop depression. Furthermore, chronic administration of corticosterone produced depressive symptoms both in wild type and knockout mice, and the effects of CMS on behavioural depression could be mimicked by exogenous administration of IL-1beta (Goshen et al., 2008). In another study, splenocytes of mice stimulated with the bacterial cell wall component lipopolysaccharide (LPS) and also repeatedly subjected to social disruption were less sensitive to the anti-inflammatory actions of glucocorticoids, showing increased levels of pro-inflammatory cytokines. The development of functional glucocorticoid resistance was accompanied by the accumulation of glucocorticoid-insensitive cells in the spleen that exhibit impaired nuclear translocation of the GR and lack of glucocorticoid-induced suppression of NF-kappaB (Engler et al., 2008). Recurrent stressor exposure significantly increased mRNA expression and plasma protein levels of IL-1beta. Furthermore, mice lacking IL-1 receptor exhibited adrenal hypertrophy and elevated serum corticosterone levels in response to stressors and failed to develop glucocorticoid resistance.
All these experiments indicate that there is interplay between cytokine receptors, stress and depressive symptoms. In addition, there are two other factors that we have to take into account if we want to answer our question of what comes first: the duration and the timing of stress.
4.1. Effect of duration and timing of stress
Whereas an acute or short-term stressful life event can enhance immune responses, chronic or long-term stress can suppress immunity. In this respect, mice with a spared nerve injury have been used to assess the hypothesis that stress precipitates depressive symptomatology through the induction of inflammation in the brain, and that a prior exposure to stress is able to exacerbate behavioural and neuroinflammatory consequences. In fact, this model has demonstrated that exposure to chronic restraint stress prior to the injury exacerbated a depressive-like behaviour causing also an increase in IL-1beta gene expression, whereas a pre-treatment with a corticosteroid synthesis inhibitor before stress was able to eliminate the damaging effects of chronic stress. In addition, interference with IL-1beta signalling, through administration of an IL-1 receptor antagonist, ameliorated the effects of the injury on depressive-like behaviour (Norman et al., 2010). In a similar way, the length of immune activation alters behaviour and exerts a central neurochemical impact. For example, it has been shown that, in animal models, an acute injection of IL-1beta increases sickness and levels of plasma corticosterone, while these effects are attenuated after subchronic treatment (Anisman et al., 2008a).
In addition to the duration of stressful life events, also the timing of a stressor relative to the time of activation and time course of an immune response is important. Immunoenhancement is observed when acute stress is experienced at early stages of immune activation, while immunosuppression may be observed at late stages of the immune response (Dhabhar, 2009). Using an animal model, Frank et al. (2010) showed that glucocorticoids potentiate both the peripheral and central pro-inflammatory response to a peripheral immune challenge if they are administered prior to the challenge. Contrarily, when glucocorticoids are administered after the peripheral immune challenge, they suppress the pro-inflammatory response measured as IL-1beta and IL-6.
In conclusion, HPA axis hyperactivity and inflammation might be part of the same pathophysiological process: HPA axis hyperactivity is a marker of glucocorticoid resistance, implying ineffective action of glucocorticoid hormones on target tissues, which could lead to immune activation; equally, inflammation could stimulate HPA axis activity via both a direct action of cytokines on the brain and by inducing glucocorticoid resistance.
5. Neurodegeneration
Several brain regions, specially the hippocampus, undergo structural changes in depression (Sheline, 2003). In particular, neuroimaging studies have consistently shown reduced size of hippocampal volume in MD as reported by meta-analysis studies of patients with recurrent depression (Videbech and Ravnkilde, 2004; Campbell et al., 2004) and with longer duration of illness (McKinnon et al., 2009). Even though in vivo imaging studies document these significant reductions of volume, the exact underlying reasons for these changes are still unclear. The selective and persistent loss of hippocampal volume could be induced by neurodegeneration, leading to hippocampal neuronal death, and also by decreased neurogenesis (Sapolsky, 2004). Other factors like shifts in fluid balance or changes in the extracellular space could also explain this hippocampal shrinkage (Czeh and Lucassen, 2007). Considering that the actual volumetric increase that could be due to regular neurogenesis, occurring only in the adult dentate gyrus, is quite small, this is unlikely to explain on its own the hippocampal volume reduction observed in MD. It could be possible that the explanation lies in a more general neurodegenerative process rather than simply a reduction of neurogenesis. In fact, enhanced neurodegeneration in depression may be caused by the presence of persistent inflammatory processes. Multiple inflammatory cytokines, oxygen radical damage, tryptophan catabolites (see below) and neurodegenerative biomarkers have all been established in patients with MD and corroborated by animal models of depression (Maes et al., 2009). However, it has to be noticed that histopathological studies examining the hippocampus of depressed individuals seem to be contradictory. In fact, Stockmeier et al. (2004) observed that the packing density of glia, pyramidal neurons and granule cell neurons is significantly increased in all hippocampal subfields and the dentate gyrus, and pyramidal neuron soma size is significantly decreased, suggesting that a significant reduction in neuropil in MD may account for the decreased hippocampal volume detected by neuroimaging.
Other researchers found no evidence of neurodegeneration in depressed patients. More specifically, the study of MD patients done by Müller et al. (2001) did not found any proof of patterns of reactive astrogliosis, synaptic density or synaptic reorganization, all signs that characterize neurodegenerative disorders like Parkinson’s and Alzheimer’s diseases. This finding was also corroborated by Reif et al. (2006), who found normal amounts of newly formed neural stem cells in MD. Thus, despite the frequent co-morbidity of depression with Parkinson’s and Alzheimer’s diseases, it has been hypothesized that depression is not a true neurodegenerative disorder (Thompson et al., 2008) or at least it does not involve only a deficit in neurogenesis. Interestingly, however, antidepressant treatment in MD patients seems to have an effect on neurogenesis. In fact, Boldrini et al. (2009) showed increased progenitor cells in MD patients treated with antidepressants. More specifically, patients receiving these drugs had more dividing cells in the hippocampus than untreated patients and controls.
All these analyses were conducted on post-mortem tissues, where results are indicative of conditions at one time point only, but as many of the structural changes and the volume reduction appear to be reversible, clearly more studies are needed. Therefore, research on cellular systems, which would allow for controlled experiments in living human cells, would be essential to investigate molecular mechanisms.
Several studies have investigated the role of cytokines and glucocorticoids leading to neurodegeneration by a variety of mechanisms. The following paragraphs will describe some of these mechanisms.
5.1. Neurodegeneration due to imbalances in the kynurenine pathway caused by cytokines
Administration of the cytokine IFN-alpha has been shown to cause the development of depressive symptoms in humans (Capuron and Miller, 2004). A key molecular mediator in the induction of this mechanism appears to be the enzyme indoleamine 2,3-dioxygenase (IDO), which converts tryptophan into kynurenine (KYN), a precursor of 3-hydroxykynurenine, a neurotoxic metabolite, and kynurenic acid, a neuroprotective metabolite, as seen in Fig. 1. Following IDO activation, both the reduced peripheral availability of tryptophan (putatively leading to reduced serotonin synthesis in the brain) and the production of neurotoxic tryptophan metabolites are considered essential steps in the pathophysiological processes. In fact, kynurenic acid is an N-methyl d-aspartate (NMDA) receptor antagonist, and is generally considered neuroprotective, whereas 3-hydroxykynurenine and quinolinic acid are NMDA receptor agonists, potentially neurotoxic and thus potentially contributing to depression (Wichers and Maes 2004; Wichers et al., 2005; Müller and Schwarz, 2007 Hashimoto, 2009). Moreover, pro-inflammatory cytokines enhance the kynurenine-3-monooxygenase enzyme (KMO), which degrades kynurenine into 3-hydroxykynurenine and thus diverts the kynurenine pathway more into the neurotoxic path. The importance of IDO as a critical molecular mediator in the development of inflammation-induced depressive-like behaviour has been recently reinforced by studies by O’Connor and colleagues. They showed that peripheral administration of LPS in mice activates IDO to induce a depressive-like behavioural syndrome and to increase the brain serotonin turnover. On the contrary, blockade of IDO activation either indirectly with an anti-inflammatory drug that attenuates LPS-induced expression of pro-inflammatory cytokines, or directly with an IDO antagonist, prevents the development of depressive-like behaviour. Interestingly, both the anti-inflammatory drug and the IDO antagonist are able to normalize the kynurenine/tryptophan ratio. Moreover, administration of KYN to naive mice dose dependently induces depressive-like behaviour (O’Connor et al., 2009).
Fig. 1.
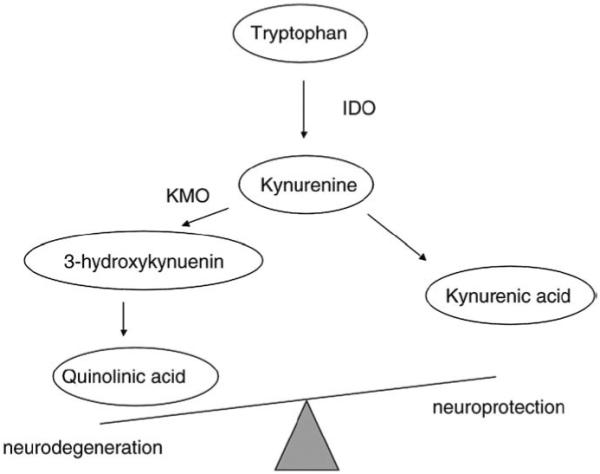
Kynurenine pathway of tryptophan metabolism. IDO, indoleamine 2,3-dioxygenase; KMO, kynurenine-3-monooxygenase.
5.2. Neurodegeneration and glucocorticoids
The development of depressive symptomatology is a frequent side effect of glucocorticoid treatment and one of the symptoms of Cushing’s syndrome. Furthermore, neuronal loss has been reported in the hippocampus of stressed or corticosteroid-treated animals. Due to an inhibitory control of the hippocampus on the HPA axis, damage to this structure is expected to disinhibit the HPA axis, causing increased glucocorticoid levels over time. This ‘glucocorticoid cascade hypothesis’ of stress and hippocampal damage was proposed to be involved in age-related accumulation of hippocampal damage in disorders like Alzheimer’s disease and depression (Sapolsky et al., 1986). Furthermore, preclinical studies in which animals are exposed to chronic stress (a paradigm used to mimic what happens in depressed patients where stressful life events represent a precipitating factor for the pathogenesis of depression) have been used to understand the hippocampal shrinkage in depressed patients. Indeed, these studies have shown that this hippocampal shrinkage is related to dendritic retraction, suppressed adult neurogenesis and neuronal death in association with elevated levels of glucocorticoids (Czeh and Lucassen, 2007).
Another significant study that supports a protective role for glucocorticoids even during stressful situations is that of animals subjected to immune activation while being treated with the GR inhibitor mifepristone (RU486). In particular, exposure to LPS produces an inflammatory reaction, which is greatly increased and accompanied by a profound neurodegeneration when mifepristrone is used (Nadeau and Rivest, 2003). In response to LPS, cells produce several cytokines, and glucocorticoids control their expression. In the absence of this negative feedback, some cytokines, in particular tumor necrosis factor alpha (TNF-alpha), become highly toxic and cause a nonselective cell death. Therefore, in controlling the cerebral innate immunity and TNF-alpha production, glucocorticoids play a major role in protecting the brain.
An additional useful model to assess the effect of glucocorticoids on neurodegeneration is Type 1 diabetes, a common metabolic disorder characterized by an increased secretion of glucocorticoids and cognitive deficits. In order to assess whether excessive stimulation of GR could be the cause of the cognitive deficits, mice with induced diabetes were treated with the GR antagonist mifepristone (RU486). Interestingly, this GR blockade attenuated the hippocampal abnormalities and rescued the diabetic mice from the cognitive deficits (Revsin et al., 2009). This model therefore supports the role of glucocorticoids in inducing cognitive (and possibly morphological) changes.
Contrary to the studies shown above where glucocorticoids exert neurodegenerative effects in the developing brain, a few studies have shown that dexamethasone may also play a neurotrophic and neuroprotective role. Dexamethasone is used in premature infants to prevent and/or treat bronchopulmonary dysplasia that can adversely affect early neurodevelopment and can probably cause a loss of cerebral volume (Grier and Halliday, 2003). Treatment of the neonatal rat brain with dexamethasone significantly decreases the gain of body and brain weight. In addition, repeated doses of dexamethasone increase caspase-3 activity (a marker of cell apoptosis). At the same time, dexamethasone causes an increase in mRNA and protein levels of vascular endothelial growth factor A (VEGF), which has neurotrophic and neurogenesis enhancing effects. Therefore, while dexamethasone can exert neurodegenerative effects in the developing brain, it can also increase VEGF levels, which, in contrast, may play a neuroprotective role (Feng et al., 2009).
5.3. Are cytokines or glucocorticoids the culprits? Final remarks
Chronic inflammation is now considered to play a central role in the pathogenesis of cardiovascular disease, multiple sclerosis, diabetes, cancer and MD. If chronic inflammatory changes represent a common feature of depression, this could predispose depressed patients to neurodegenerative changes later in life. Indeed there is clinical evidence that depression is a common antecedent of Alzheimer’s disease and may be an early manifestation of dementia before the cognitive declines become apparent. The progression from depression to dementia could result from increased levels of pro-inflammatory cytokines, which stimulate a cascade of inflammatory changes and a hypersecretion of cortisol and which also induce the production of neurotoxic end products of the tryptophan–kynurenine pathway (Leonard, 2007). Nevertheless, as mentioned before, all these changes may be also generated by a HPA axis hyperdrive. Therefore, we cannot unequivocally accept the simple model that glucocorticoids cause neurodegeneration, but rather that elevated cytokines, in the context of glucocorticoid resistance, are probably the offenders. Chronic stressors induce sustained elevations in cortisol, which, over time, cause immune cells to undergo a compensatory downregulation of GR activity. These changes may be an adaptative mechanism specifically induced by cytokines to increase immune reaction in situation of stress. The development of glucocorticoid resistance after social disruption may be such a mechanism, allowing animals to heal injuries and clear invading microbes in the presence of the anti-inflammatory stress hormones (Avitsur et al., 2009). This limits the ability of cortisol to further reduce immune responses, even in the presence of high circulating glucocorticoid levels, and as a result there is low-grade inflammation, which is hypothesized to contribute to the infectious, autoimmune, and cardiac diseases, whose risk is linked to stress.
In conclusion, chronic inflammatory changes in the presence of glucocorticoid resistance may represent a common feature configuring vulnerability that could predispose depressed patients to neurodegenerative changes later in life.
Further studies are needed to clarify the relative contribution of glucocorticoids and inflammatory signals to MD and other disorders.
Acknowledgements
Christoph Anacker is currently funded by a studentship from the NIHR BRC. Carmine M. Pariante has been funded by the UK MRC since 1999, first as a Clinical Training Fellow, and currently as an MRC Clinician Scientist Fellow. His research is also funded by the NIHR South London and Maudsley NHS Foundation Trust & Institute of Psychiatry Specialist Biomedical Research Centre for Mental Health, the NARSAD, the APIRE, the British Academy and the European Union Framework 7. Livia Carvalho is funded by the NARSAD Young Investigator Award and ECNP Young Investigator Award. The authors have no relevant financial interest to disclose.
Abbreviations
- ACTH
adrenocorticotrophin hormone
- AVP
arginine–vasopressin
- CMS
chronic mild stress
- CRH
corticotrophin releasing hormone
- GR
glucocorticoid receptor
- HPA
hypothalamic-pituitary-adrenal
- IDO
indoleamine 2,3-dioxygenase
- IFN
interferon
- IL
interleukin
- KMO
kynurenine-3-monooxygenase enzyme
- KYN
kynurenine
- LPS
lipopolysaccharide
- MAP
mitogen-activated protein
- MD
major depression
- MR
mineralocorticoid receptor
- NF-kappaB
nuclear factor-kappaB
- NMDA
N-methyl d-aspartate
- TNF
tumor necrosis factor
References
- Anisman H. Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. J Psychiatry Neurosci. 2009;34:4–20. [PMC free article] [PubMed] [Google Scholar]
- Anisman H, Gibb J, Hayley S. Influence of continuous infusion of interleukin-1beta on depression-related processes in mice: corticosterone, circulating cytokines, brain monoamines, and cytokine mRNA expression. Psychopharmacology (Berl) 2008a;199:231–44. doi: 10.1007/s00213-008-1166-z. [DOI] [PubMed] [Google Scholar]
- Anisman H, Merali Z, Hayley S. Neurotransmitter, peptide and cytokine processes in relation to depressive disorder: comorbidity between depression and neurodegenerative disorders. Prog Neurobiol. 2008b;85:1–74. doi: 10.1016/j.pneurobio.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Antoni FA. Vasopressinergic control of pituitary adrenocorticotropin secretion comes of age. Front Neuroendocrinol. 1993;14:76–122. doi: 10.1006/frne.1993.1004. [DOI] [PubMed] [Google Scholar]
- Avitsur R, Powell N, Padgett DA, Sheridan JF. Social interactions, stress, and immunity. Immunol Allergy Clin North Am. 2009 May;29(2):285–93. doi: 10.1016/j.iac.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Baune B. Conceptual challenges of a tentative model of stress-induced depression. PLoS ONE. 2009;4:e4266. doi: 10.1371/journal.pone.0004266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck AT, Rush AJ, Shaw BF, Emery G. Cognitive therapy of depression. Guilford. 1979 [Google Scholar]
- Bhagwagar Z, Hafizi S, Cowen PJ. Increase in concentration of waking salivary cortisol in recovered patients with depression. Am J Psychiatry. 2003;160:1890–1. doi: 10.1176/appi.ajp.160.10.1890. [DOI] [PubMed] [Google Scholar]
- Bhagwagar Z, Hafizi S, Cowen PJ. Increased salivary cortisol after waking in depression. Psychopharmacology (Berl) 2005;182:54–7. doi: 10.1007/s00213-005-0062-z. [DOI] [PubMed] [Google Scholar]
- Boldrini M, Underwood MD, Hen R, Rosoklija GB, Dwork AJ, John Mann J, et al. Antidepressants increase neural progenitor cells in the human hippocampus. Neuropsychopharmacology. 2009;34(11):2376–89. doi: 10.1038/npp.2009.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull SJ, Huezo-Diaz P, Binder EB, Cubells JF, Ranjith G, Maddock C, et al. Functional polymorphisms in the interleukin-6 and serotonin transporter genes, and depression and fatigue induced by interferon-alpha and ribavirin treatment. Mol Psychiatry. 2009;14(12):1145. doi: 10.1038/mp.2008.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S, Marriott M, Nahmias C, MacQueen GM. Lower hippocampal volume in patients suffering from depression: a meta-analysis. Am J Psychiatry. 2004;161(4):598–607. doi: 10.1176/appi.ajp.161.4.598. [DOI] [PubMed] [Google Scholar]
- Capuron L, Miller AH. Cytokines and psychopathology: lessons from interferon-alpha. Biol Psychiatry. 2004;56:819–24. doi: 10.1016/j.biopsych.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Carpenter WT, Jr, Bunney WE., Jr Adrenal cortical activity in depressive illness. Am J Psychiatry. 1971;128:31–40. doi: 10.1176/ajp.128.1.31. [DOI] [PubMed] [Google Scholar]
- Carvalho LA, Juruena MF, Papadopoulos AS, Poon L, Kerwin R, Cleare AJ, et al. Clomipramine in vitro reduces glucocorticoid receptor function in healthy subjects but not in patients with major depression. Neuropsychopharmacology. 2008 Dec;33(13):3182–9. doi: 10.1038/npp.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho LA, Pariante CM. In vitro modulation of the glucocorticoid receptor by antidepressants. Stress. 2008;11:411–24. doi: 10.1080/10253890701850759. [DOI] [PubMed] [Google Scholar]
- Charney DS. Monoamine dysfunction and the pathophysiology and treatment of depression. J Clin Psychiatry. 1998;59(Suppl 14):11–4. [PubMed] [Google Scholar]
- Christian LM, Franco A, Iams JD, Sheridan J, Glaser R. Depressive symptoms are associated with elevated serum proinflammatory cytokines among pregnant women. Brain Behav Immun. 2009;23(6):750–4. doi: 10.1016/j.bbi.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowen PJ. Not fade away: the HPA axis and depression. Psychol Med. 2009:1–4. doi: 10.1017/S0033291709005558. [DOI] [PubMed] [Google Scholar]
- Czeh B, Lucassen PJ. What causes the hippocampal volume decrease in depression? Are neurogenesis, glial changes and apoptosis implicated? Eur Arch Psychiatry Clin Neurosci. 2007;257:250–60. doi: 10.1007/s00406-007-0728-0. [DOI] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DJ, Samuels BA, Rainer Q, Wang JW, Marsteller D, Mendez I, et al. Neurogenesis-dependent and -independent effects of fluoxetine in an animal model of anxiety/depression. Neuron. 2009;62:479–93. doi: 10.1016/j.neuron.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bosscher K, Haegeman G. Minireview: latest perspectives on antiinflammatory actions of glucocorticoids. Mol Endocrinol. 2009;23:281–91. doi: 10.1210/me.2008-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kloet ER, Derijk RH, Meijer OC. Therapy insight: is there an imbalanced response of mineralocorticoid and glucocorticoid receptors in depression? Nat Clin Pract Endocrinol Metab. 2007;3:168–79. doi: 10.1038/ncpendmet0403. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–75. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- Dhabhar FS. Enhancing versus suppressive effects of stress on immune function: implications for immunoprotection and immunopathology. Neuroimmunomodulation. 2009;16:300–17. doi: 10.1159/000216188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinan TG. Glucocorticoids and the genesis of depressive illness. A psychobiological model. Br J Psychiatry. 1994 Mar;164(3):365–71. doi: 10.1192/bjp.164.3.365. [DOI] [PubMed] [Google Scholar]
- Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010 Mar 1;67(5):446–57. doi: 10.1016/j.biopsych.2009.09.033. [DOI] [PubMed] [Google Scholar]
- Duman RS. Depression: a case of neuronal life and death? Biol Psychiatry. 2004;56:140–5. doi: 10.1016/j.biopsych.2004.02.033. [DOI] [PubMed] [Google Scholar]
- Engler H, Bailey MT, Engler A, Stiner-Jones LM, Quan N, Sheridan JF. Interleukin-1 receptor type 1-deficient mice fail to develop social stress-associated glucocorticoid resistance in the spleen. Psychoneuroendocrinology. 2008;33:108–17. doi: 10.1016/j.psyneuen.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Rhodes PG, Liu H, Bhatt AJ. Dexamethasone induces neurodegeneration but also up-regulates vascular endothelial growth factor A in neonatal rat brains. Neuroscience. 2009;158:823–32. doi: 10.1016/j.neuroscience.2008.10.024. [DOI] [PubMed] [Google Scholar]
- Frank MG, Miguel ZD, Watkins LR, Maier SF. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain Behav Immun. 2010 Jan;24(1):19–30. doi: 10.1016/j.bbi.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Goshen I, Kreisel T, Ben-Menachem-Zidon O, Licht T, Weidenfeld J, Ben-Hur T, et al. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry. 2008;13:717–28. doi: 10.1038/sj.mp.4002055. [DOI] [PubMed] [Google Scholar]
- Grier DG, Halliday HL. Corticosteroids in the prevention and management of bronchopulmonary dysplasia. Semin Neonatol. 2003 Feb;8(1):83–91. doi: 10.1016/s1084-2756(02)00189-6. [DOI] [PubMed] [Google Scholar]
- Harmer CJ, Goodwin GM, Cowen PJ. Why do antidepressants take so long to work? A cognitive neuropsychological model of antidepressant drug action. Br J Psychiatry. 2009;195:102–8. doi: 10.1192/bjp.bp.108.051193. [DOI] [PubMed] [Google Scholar]
- Hashimoto K. Emerging role of glutamate in the pathophysiology of major depressive disorder. Brain Res Rev. 2009 Oct;61(2):105–23. doi: 10.1016/j.brainresrev.2009.05.005. [DOI] [PubMed] [Google Scholar]
- Hayden MS, West AP, Ghosh S. NF-kappaB and the immune response. Oncogene. 2006 Oct 30;25(51):6758–80. doi: 10.1038/sj.onc.1209943. [DOI] [PubMed] [Google Scholar]
- Hayley S, Poulter MO, Merali Z, Anisman H. The pathogenesis of clinical depression: stressor- and cytokine-induced alterations of neuroplasticity. Neuroscience. 2005;135(3):659–78. doi: 10.1016/j.neuroscience.2005.03.051. [DOI] [PubMed] [Google Scholar]
- Herbert J, Goodyer IM, Grossman AB, Hastings MH, de Kloet ER, Lightman SL, et al. Do corticosteroids damage the brain? J Neuroendocrinol. 2006;18(6):393–411. doi: 10.1111/j.1365-2826.2006.01429.x. [DOI] [PubMed] [Google Scholar]
- Hirschfeld RM. History and evolution of the monoamine hypothesis of depression. J Clin Psychiatry. 2000;61(Suppl 6):4–6. [PubMed] [Google Scholar]
- Holsboer-Trachsler E, Stohler R, Hatzinger M. Repeated administration of the combined dexamethasone-human corticotropin releasing hormone stimulation test during treatment of depression. Psychiatry Res. 1991;38:163–71. doi: 10.1016/0165-1781(91)90041-m. [DOI] [PubMed] [Google Scholar]
- Holsboer F, Liebl R, Hofschuster E. Repeated dexamethasone suppression test during depressive illness. Normalisation of test result compared with clinical improvement. J Affect Disord. 1982;4:93–101. doi: 10.1016/0165-0327(82)90039-8. [DOI] [PubMed] [Google Scholar]
- Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med. 2009;71(2):171–86. doi: 10.1097/PSY.0b013e3181907c1b. [DOI] [PubMed] [Google Scholar]
- Jacobs BL, Praag H, Gage FH. Adult brain neurogenesis and psychiatry: a novel theory of depression. Mol Psychiatry. 2005:262–9. doi: 10.1038/sj.mp.4000712. [DOI] [PubMed] [Google Scholar]
- Joels M, Karst H. Chronic stress: implications for neuronal morphology, function and neurogenesis. Front Neuroendocrinol. 2007;28(2–3):72–96. doi: 10.1016/j.yfrne.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Juruena MF, Cleare AJ, Papadopoulos AS, Poon L, Lightman S, Pariante CM. Different responses to dexamethasone and prednisolone in the same depressed patients. Psychopharmacology (Berl) 2006;189:225–35. doi: 10.1007/s00213-006-0555-4. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Krebs J, Fabel K. The contribution of failing adult hippocampal neurogenesis to psychiatric disorders. Curr Opin Psychiatry. 2008;21:290–5. doi: 10.1097/YCO.0b013e3282fad375. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Kronenberg G. Depressed new neurons—adult hippocampal neurogenesis and a cellular plasticity hypothesis of major depression. Biol Psychiatry. 2003;54:499–503. doi: 10.1016/s0006-3223(03)00319-6. [DOI] [PubMed] [Google Scholar]
- Koo JW, Duman RS. IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc Natl Acad Sci U S A. 2008;105:751–6. doi: 10.1073/pnas.0708092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard BE. Inflammation, depression and dementia: are they connected? Neurochem Res. 2007;32:1749–56. doi: 10.1007/s11064-007-9385-y. [DOI] [PubMed] [Google Scholar]
- Lotrich FE, Rabinovitz M, Gironda P, Pollock BG. Depression following pegylated interferon-alpha: characteristics and vulnerability. J Psychosom Res. 2007;63:131–5. doi: 10.1016/j.jpsychores.2007.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luby JL, Heffelfinger A, Mrakotsky C, Brown K, Hessler M, Spitznagel E. Alterations in stress cortisol reactivity in depressed preschoolers relative to psychiatric and no-disorder comparison groups. Arch Gen Psychiatry. 2003;60:1248–55. doi: 10.1001/archpsyc.60.12.1248. [DOI] [PubMed] [Google Scholar]
- Lucassen PJ, Meerlo P, Naylor AS, van Dam AM, Dayer AG, Fuchs E, et al. Regulation of adult neurogenesis by stress, sleep disruption, exercise and inflammation: implications for depression and antidepressant action. Eur Neuropsychopharmacol. 2010 Jan;20(1):1–17. doi: 10.1016/j.euroneuro.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Maddock C, Landau S, Barry K, Maulayah P, Hotopf M, Cleare AJ, et al. Psychopathological symptoms during interferon-alpha and ribavirin treatment: effects on virologic response. Mol Psychiatry. 2005;10(4):332–3. doi: 10.1038/sj.mp.4001634. [DOI] [PubMed] [Google Scholar]
- Maes M, Bosmans E, Suy E, Vandervorst C, DeJonckheere C, Raus J. Depression-related disturbances in mitogen-induced lymphocyte responses and interleukin-1 beta and soluble interleukin-2 receptor production. Acta Psychiatr Scand. 1991;84(4):379–86. doi: 10.1111/j.1600-0447.1991.tb03163.x. Oc. [DOI] [PubMed] [Google Scholar]
- Maes M, Meltzer HY, Stevens W, Cosyns P, Blockx P. Multiple reciprocal relationships between in vivo cellular immunity and hypothalamic-pituitary-adrenal axis in depression. Psychol Med. 1994 Feb;24(1):167–77. doi: 10.1017/s0033291700026933. [DOI] [PubMed] [Google Scholar]
- Maes M, Yirmyia R, Noraberg J, Brene S, Hibbeln J, Perini G, et al. The inflammatory & neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab Brain Dis. 2009;24:27–53. doi: 10.1007/s11011-008-9118-1. [DOI] [PubMed] [Google Scholar]
- McAfoose J, Baune BT. Evidence for a cytokine model of cognitive function. Neurosci Biobehav Rev. 2009;33(3):355–66. doi: 10.1016/j.neubiorev.2008.10.005. [DOI] [PubMed] [Google Scholar]
- McClure DJ. The diurnal variation of plasma cortisol levels in depression. J Psychosom Res. 1966;10:189–95. doi: 10.1016/0022-3999(66)90062-6. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Glucocorticoids, depression, and mood disorders: structural remodeling in the brain. Metabolism. 2005;54:20–3. doi: 10.1016/j.metabol.2005.01.008. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Seeman T. Protective and damaging effects of mediators of stress. Elaborating and testing the concepts of allostasis and allostatic load. Ann N Y Acad Sci. 1999;896:30–47. doi: 10.1111/j.1749-6632.1999.tb08103.x. [DOI] [PubMed] [Google Scholar]
- McKinnon MC, Yucel K, Nazarov A, MacQueen GM. A meta-analysis examining clinical predictors of hippocampal volume in patients with major depressive disorder. J Psychiatry Neurosci. 2009;34(1):41–54. [PMC free article] [PubMed] [Google Scholar]
- Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65:732–41. doi: 10.1016/j.biopsych.2008.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AH, Pariante CM, Pearce BD. Effects of cytokines on glucocorticoid receptor expression and function. Glucocorticoid resistance and relevance to depression. Adv Exp Med Biol. 1999;461:107–16. doi: 10.1007/978-0-585-37970-8_7. [DOI] [PubMed] [Google Scholar]
- Miller GE, Chen E, Sze J, Marin T, Arevalo JM, Doll R, et al. A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry. 2008;64:266–72. doi: 10.1016/j.biopsych.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirescu C, Gould E. Stress and adult neurogenesis. Hippocampus. 2006;16(3):233–8. doi: 10.1002/hipo.20155. [DOI] [PubMed] [Google Scholar]
- Mondelli V, Dazzan P, Hepgul N, Di Forti M, Aas M, D’Albenzio A, et al. Abnormal cortisol levels during the day and cortisol awakening response in first-episode psychosis: the role of stress and of antipsychotic treatment. Schizophr Res. 2010 Feb;116(2–3):234–42. doi: 10.1016/j.schres.2009.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller MB, Lucassen PJ, Yassouridis A, Hoogendijk WJ, Holsboer F, Swaab DF. Neither major depression nor glucocorticoid treatment affects the cellular integrity of the human hippocampus. Eur J NeuroSci. 2001;14(10):1603–12. doi: 10.1046/j.0953-816x.2001.01784.x. [DOI] [PubMed] [Google Scholar]
- Müller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry. 2007 Nov;12(11):988–1000. doi: 10.1038/sj.mp.4002006. [DOI] [PubMed] [Google Scholar]
- Myint AM, Kim YK. Cytokine–serotonin interaction through IDO: a neurodegeneration hypothesis of depression. Med Hypotheses. 2003;61:519–25. doi: 10.1016/s0306-9877(03)00207-x. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Glucocorticoids play a fundamental role in protecting the brain during innate immune response. J Neurosci. 2003 Jul 2;23(13):5536–44. doi: 10.1523/JNEUROSCI.23-13-05536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeroff CB, Krishnan KR, Reed D, Leder R, Beam C, Dunnick NR. Adrenal gland enlargement in major depression. A computed tomographic study. Arch Gen Psychiatry. 1992;49:384–7. doi: 10.1001/archpsyc.1992.01820050048008. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB, Widerlov E, Bissette G, Walleus H, Karlsson I, Eklund K, et al. Elevated concentrations of CSF corticotropin-releasing factor-like immunoreactivity in depressed patients. Science. 1984;226:1342–4. doi: 10.1126/science.6334362. [DOI] [PubMed] [Google Scholar]
- Norman GJ, Karelina K, Zhang N, Walton JC, Morris JS, Devries AC. Stress and IL-1beta contribute to the development of depressive-like behavior following peripheral nerve injury. Mol Psychiatry. 2010 Apr;15(4):404–14. doi: 10.1038/mp.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor JC, Lawson MA, Andre C, Moreau M, Lestage J, Castanon N, et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2, 3-dioxygenase activation in mice. Mol Psychiatry. 2009;14:511–22. doi: 10.1038/sj.mp.4002148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace TW, Hu F, Miller AH. Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun. 2007;21:9–19. doi: 10.1016/j.bbi.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace TW, Miller AH. Cytokines and glucocorticoid receptor signaling. Relevance to major depression. Ann N Y Acad Sci. 2009 Oct;1179:86–105. doi: 10.1111/j.1749-6632.2009.04984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariante CM. Depression, stress and the adrenal axis. J Neuroendocrinol. 2003;15:811–2. doi: 10.1046/j.1365-2826.2003.01058.x. [DOI] [PubMed] [Google Scholar]
- Pariante CM. The glucocorticoid receptor: part of the solution or part of the problem? J Psychopharmacol. 2006;20:79–84. doi: 10.1177/1359786806066063. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Dazzan P, Danese A, Morgan KD, Brudaglio F, Morgan C, et al. Increased pituitary volume in antipsychotic-free and antipsychotic-treated patients of the AEsop first-onset psychosis study. Neuropsychopharmacology. 2005;30:1923–31. doi: 10.1038/sj.npp.1300766. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Lightman SL. The HPA axis in major depression: classical theories and new developments. Trends Neurosci. 2008;31:464–8. doi: 10.1016/j.tins.2008.06.006. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001;49:391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Nemeroff CB, Miller AH. Glucocorticoid receptors in depression. Isr J Med Sci. 1995;31:705–12. [PubMed] [Google Scholar]
- Pariante CM, Pearce BD, Pisell TL, Sanchez CI, Po C, Su C, et al. The proinflammatory cytokine, interleukin-1alpha, reduces glucocorticoid receptor translocation and function. Endocrinology. 1999 Sep;140(9):4359–66. doi: 10.1210/endo.140.9.6986. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Vassilopoulou K, Velakoulis D, Phillips L, Soulsby B, Wood SJ, et al. Pituitary volume in psychosis. Br J Psychiatry. 2004;185:5–10. doi: 10.1192/bjp.185.1.5. [DOI] [PubMed] [Google Scholar]
- Pickering M, O’Connor JJ. Pro-inflammatory cytokines and their effects in the dentate gyrus. Prog Brain Res. 2007;163:339–54. doi: 10.1016/S0079-6123(07)63020-9. [DOI] [PubMed] [Google Scholar]
- Pollak Y, Yirmiya R. Cytokine-induced changes in mood and behaviour: implications for ‘depression due to a general medical condition’, immunotherapy and antidepressive treatment. Int J Neuropsychopharmacol. 2002;5:389–99. doi: 10.1017/S1461145702003152. [DOI] [PubMed] [Google Scholar]
- Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27:24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raison CL, Demetrashvili M, Capuron L, Miller AH. Neuropsychiatric adverse effects of interferon-alpha: recognition and management. CNS Drugs. 2005;19:105–23. doi: 10.2165/00023210-200519020-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif A, Fritzen S, Finger M, Strobel A, Lauer M, Schmitt A, et al. Neural stem cell proliferation is decreased in schizophrenia, but not in depression. Mol Psychiatry. 2006;11(5):514–22. doi: 10.1038/sj.mp.4001791. [DOI] [PubMed] [Google Scholar]
- Revollo JR, Cidlowski JA. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann N Y Acad Sci. 2009 Oct;1179:167–78. doi: 10.1111/j.1749-6632.2009.04986.x. [DOI] [PubMed] [Google Scholar]
- Revsin Y, Rekers NV, Louwe MC, Saravia FE, De Nicola AF, de Kloet ER, et al. Glucocorticoid receptor blockade normalizes hippocampal alterations and cognitive impairment in streptozotocin-induced type 1 diabetes mice. Neuropsychopharmacology. 2009;34:747–58. doi: 10.1038/npp.2008.136. [DOI] [PubMed] [Google Scholar]
- Sahay A, Hen R. Adult hippocampal neurogenesis in depression. Nat Neurosci. 2007;10:1110–5. doi: 10.1038/nn1969. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM, Krey LC, McEwen BS. The neuroendocrinology of stress and aging: the glucocorticoid cascade hypothesis. Endocr Rev. 1986 Aug;7(3):284–301. doi: 10.1210/edrv-7-3-284. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry. 2000;57(10):925–35. doi: 10.1001/archpsyc.57.10.925. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Is impaired neurogenesis relevant to the affective symptoms of depression? Biol Psychiatry. 2004;56:137–9. doi: 10.1016/j.biopsych.2004.04.012. [DOI] [PubMed] [Google Scholar]
- Sheline YI. Neuroimaging studies of mood disorder effects on the brain. Biol Psychiatry. 2003;54:338–52. doi: 10.1016/s0006-3223(03)00347-0. [DOI] [PubMed] [Google Scholar]
- Smith RS. The macrophage theory of depression. Med Hypotheses. 1991 Aug;35(4):298–306. doi: 10.1016/0306-9877(91)90272-z. [DOI] [PubMed] [Google Scholar]
- Stockmeier CA, Mahajan GJ, Konick LC, Overholser JC, Jurjus GJ, Meltzer HY, et al. Cellular changes in the postmortem hippocampus in major depression. Biol Psychiatry. 2004 Nov 1;56(9):640–50. doi: 10.1016/j.biopsych.2004.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surget A, Saxe M, Leman S, Ibarguen-Vargas Y, Chalon S, Griebel G, et al. Drug-dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol Psychiatry. 2008;64:293–301. doi: 10.1016/j.biopsych.2008.02.022. [DOI] [PubMed] [Google Scholar]
- Thase ME. Preventing relapse and recurrence of depression: a brief review of therapeutic options. CNS Spectr. 2006;11:12–21. doi: 10.1017/s1092852900015212. [DOI] [PubMed] [Google Scholar]
- Thompson A, Boekhoorn K, Van Dam AM, Lucassen PJ. Changes in adult neurogenesis in neurodegenerative diseases: cause or consequence? Genes Brain Behav. 2008;7(Suppl 1):28–42. doi: 10.1111/j.1601-183X.2007.00379.x. [DOI] [PubMed] [Google Scholar]
- Ur E, White PD, Grossman A. Hypothesis: cytokines may be activated to cause depressive illness and chronic fatigue syndrome. Eur Arch Psychiatry Clin Neurosci. 1992;241:317–22. doi: 10.1007/BF02195983. [DOI] [PubMed] [Google Scholar]
- Videbech P, Ravnkilde B. Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry. 2004;161(11):1957–66. doi: 10.1176/appi.ajp.161.11.1957. [DOI] [PubMed] [Google Scholar]
- Vinson GP. The adrenal cortex and life. Mol Cell Endocrinol. 2009;300:2–6. doi: 10.1016/j.mce.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Wichers MC, Koek GH, Robaeys G, Verkerk R, Scharpe S, Maes M. IDO and interferon-alpha-induced depressive symptoms: a shift in hypothesis from tryptophan depletion to neurotoxicity. Mol Psychiatry. 2005;10:538–44. doi: 10.1038/sj.mp.4001600. [DOI] [PubMed] [Google Scholar]
- Wichers MC, Maes M. The role of indoleamine 2,3-dioxygenase (IDO) in the pathophysiology of interferon-alpha-induced depression. J Psychiatry Neurosci. 2004 Jan;29(1):11–7. [PMC free article] [PubMed] [Google Scholar]
- WHO global report accessed from http://www.who.int/healthinfo/global_burden_disease/GBD_report_2004update_part4.pdf.
- Yau JL, Noble J, Thomas S, Kerwin R, Morgan PE, Lightman S, et al. The antidepressant desipramine requires the ABCB1 (Mdr1)-type p-glycoprotein to upregulate the glucocorticoid receptor in mice. Neuropsychopharmacology. 2007 Dec;32(12):2520–9. doi: 10.1038/sj.npp.1301389. [DOI] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–60. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]