

Abstract
Previously, we have shown that in vitro antidepressants modulate glucocorticoid receptor (GR) function and expression, and have suggested that these effects could be relevant for the mechanism of action of antidepressants. To further clarify the interaction between antidepressants and glucocorticoids, we evaluated the in vitro effect of the tricyclic antidepressant, clomipramine (CMI), on the GR function in 15 treatment-resistant depressed inpatients and 28 healthy controls. Diluted whole-blood cells were incubated for 24 h in the presence or absence of CMI (10 μM). Glucocorticoid function was measured by glucocorticoid inhibition of lypopolysaccharide (LPS)-stimulated interleukin-6 (IL-6) levels. The results show that glucocorticoids (dexamethasone, prednisolone, cortisol and corticosterone) caused a concentration-dependent inhibition of LPS-stimulated IL-6 levels. In healthy controls, CMI decreased glucocorticoid inhibition of LPS-stimulated IL-6 levels, while this effect was not present in depressed patients. Therefore, depressed patients, who were clinically treatment resistant, also showed a lack of effect of the antidepressant in vitro. Upcoming studies shall test whether assessing the effects of antidepressants in vitro on GR function could predict future treatment response in a clinical setting.
Keywords: antidepressive agents, endocrine system, human, cytokines, psychiatry, immune system
INTRODUCTION
Clinical studies have demonstrated an impairment of glucocorticoid receptor (GR)-mediated negative feedback on the hypothalamic–pituitary–adrenal (HPA) axis in major depression (GR resistance), and its resolution by anti-depressant treatment. Specifically, patients with major depression have shown increased concentrations of cortisol (CORT) in plasma, urine, and cerebrospinal fluid; an exaggerated CORT response to adrenocorticotropic hormone; and an enlargement of both the pituitary and the adrenal glands (Gold et al, 1988; Holsboer and Barden, 1996; Owens and Nemeroff, 1993; Nemeroff, 1996). As feedback regulation of the HPA axis is mediated by GRs, the impairment of the axis seen in patients with major depression may be due to reduced GR function, to reduced GR number, or to both (Pariante, 2006; Pariante and Miller, 2001).
If depressed patients show an impairment of GR function, are antidepressants able to counteract these alterations in vivo? There is a vast series of studies in animals demonstrating a direct effect of antidepressants on the GR, leading to increased receptor expression and increased negative feedback on the HPA axis (Mason and Pariante, 2006; Holsboer, 2000; Carvalho and Pariante, 2008). These studies complement the clinical evidence showing that successful antidepressant treatment is associated with resolution of the impairment in the HPA axis negative feedback by glucocorticoids (Linkowski et al, 1987; Ribeiro et al, 1993). Indeed, we have shown that the antidepressant citalopram increases HPA axis negative feedback by glucocorticoids (measured as CORT suppression by prednisolone, PRED) after as little as 4 days of administration in healthy controls (Pariante et al, 2004a).
In contrast to the effects of antidepressants in vivo, the studies on the effect of antidepressants on the GR function in vitro have produced conflicting results. Specifically, we and others have shown that antidepressants can both decrease and increase GR function in vitro, based on different experimental conditions. The final net effect of antidepressants on GR function, at least in cell lines, is likely to be mediated by direct inhibition of membrane steroid transporters that expel glucocorticoids from cells (Pariante et al, 1997, 2001b, 2003a, b; Carvalho and Pariante, 2008). In particular, when cells are co-incubated with antidepressants and glucocorticoids that are expelled by the transporter, such as dexamethasone (DEX) or CORT, there is an increase in GR function (Pariante et al, 1997, 2001a, 2003a, b). On the other hand, when cells are co-incubated with antidepressants and a glucocorticoid that is not expelled by the transporter, like corticosterone (CTC), there is a decrease in GR function (Pariante et al, 2001a, 2003b).
To our knowledge, only one study has examined the in vitro effects of antidepressants on GR function in human cells in a clinical population. Specifically, Yehuda et al (2006) have shown that the antidepressant sertraline reduces GR function in lymphocytes of patients with post-traumatic stress disorder (PTSD). Moreover, these authors reported that sertraline inhibition of GR function is correlated with severity of childhood trauma (Yehuda et al, 2006), possibly indicating a clinical relevance for this measure. Interestingly, GR function in PTSD patients seems to be virtually opposite to that of depressed patients—PTSD patients have increased in vivo GR function (Yehuda, 2001). Whether the effect of in vitro antidepressants on GR function is different in patients with major depression is still unknown.
The aim of the present study was, therefore, to evaluate the effect of the antidepressant, clomipramine (CMI), on GR function in depressed patients and in healthy controls by measuring glucocorticoid inhibition of lypopolysaccharide (LPS)-stimulated interleukin-6 (IL-6) levels in whole blood. We tested four different glucocorticoids: DEX, a synthetic glucocorticoid that has been used in a variety of previous studies to demonstrate decreased GR function both in vivo and in vitro; PRED, another synthetic steroid hormone that is similar to CORT in its pharmacodynamics and that we have proposed its use as a new suppressive test in vivo (Pariante et al, 2002; Pariante, 2004); CORT, the main endogenous glucocorticoid in humans; and CTC, the main endogenous glucocorticoid in rodents, which is similar to CORT in its pharmacodynamics but, different from all the other three, is not transported by transporters (Pariante et al, 2001a, 2003a, b). The use of different glucocorticoids allowed us to further investigate the involvement of transporters on the effect of antidepressants on GR function.
MATERIALS AND METHODS
The study protocol was approved by the Research Ethical Committee of the Institute of Psychiatry, King’s College London and Maudsley Hospital (London). All subjects gave their written and informed consent. The laboratory work was conducted at the Affective Disorders Laboratory, Affective Disorders Unit, Bethlem Royal Hospital.
Healthy Controls
A total of 28 healthy controls were recruited through hospital administrative staff and members of the local community. Healthy controls were free from acute infections or allergic reactions, as well as any psychotropic medications or drugs known to modify immune and endocrine functions for at least 1 month before blood sampling, including oral contraceptives. Urine tests for illicit drugs and pregnancy were conducted before the start of the study.
Patients
We examined 15 depressed inpatients at the National Affective Disorders Unit of the Bethlem Royal Hospital in London, UK. The Affective Disorders Unit receives referrals for many patients with long-standing or difficult to treat depressive illness. These are among the most severe depressed cases from all over the United Kingdom, usually with a history of not responding to pharmacotherapy and psychotherapy.
The patients had a diagnosis of recurrent unipolar major depression according to the Diagnostic and Statistical Manual of Mental Disorders, fourth edition (American Psychiatric Association, 1994). Psychiatrists reviewed referral information, conducted an in-depth clinical interview with each patient, and reviewed pertinent medical records as available.
Patients underwent detailed assessments using the tools described below to clarify the main features of their illness. Many of these measures are already part of the normal assessment process of the unit, but some additional measures were added to this research protocol. For diagnostic assessment we used the Structured Clinical Interview for DSM-IV Axis I disorders. The diagnosis was confirmed by clinical interview with the Schedule for Clinical Assessment in Neuropsychiatry (World Health Organization, 1994). The severity of depression was examined by using the 21-item Hamilton Depression Rating Scale (HAM-D) score (Hamilton, 1960), the Beck Depression Inventory (BDI; Beck et al, 1961), and the Beck Anxiety Inventory (BAI; Beck et al, 1988).
For assessment of treatment resistance we used the Antidepressant Treatment History Form (Sackeim, 2001). We also used the Thase staging criteria (Thase and Rush, 1997), which recognizes five stages of treatment resistance according to the number of treatment trials adequately delivered. All patients were treatment-resistant from a moderate to severe degree and all patients were currently receiving antidepressant medication. For practical and ethical reasons, it was not possible to withdraw the antidepressants and assess the patients in a drug-free state; however, a switch in medication was avoided for at least 7 days before conducting the experimental procedure. Exclusion criteria for patients were history of hypersensitivity to corticosteroids or steroid use; heavy smokers (ie more than 25 cigarettes per day); viral illnesses during the preceding 2 weeks; pregnant or lactating women; alcohol dependence; and significant physical illnesses (eg severe allergies, autoimmune diseases, hypertension, malignancy, hematological, endocrine, pulmonary, renal, hepatic, gastrointestinal, or neurological disease depressive disorder or an organic etiology were excluded).
Blood Collection
Subjects abstained from food, caffeine, tea, alcohol and cigarettes during the night before the study. On the study day, subjects were admitted to a research suite and blood was collected at 10 a.m. (±30 min). One 7.5 ml tube was immediately centrifuged (2000g, 10 min), the plasma was collected and frozen at −40°C for analyses of plasma CORT and plasma IL-6. A second 7.5 ml tube was immediately analyzed for glucocorticoid function assay as described below.
Glucocorticoid Function Assay Reagents
PBS Gibco, 500 ml, ref. 2012-019, sterile (Invitrogen); RPMI 1640 medium (Sigma), 500 ml, sterile, R8758; DEX (Sigma), D4902; PRED (Sigma), P-6004; hydrocortisone (Sigma), H4001; CTC (Sigma), C2505; clomipramine (Sigma), C7291; penicillin/streptomycin (Sigma), 500 ml, sterile, P4458; LPS (Gibco, catalog 20012-019, Lot L-2880).
Glucocorticoid Function Assay Protocol
The protocol was based on a published method (Rohleder et al, 2001, 2002) with modifications as described below. CMI was chosen for two reasons. First, from a clinical point of view, all of the patients in our study were resistant to tricyclic antidepressants, and therefore we were interested in the effect of a drug from this class. Second, based on our previous in vitro work in L929 fibroblasts, CMI has the strongest effects on GR function when compared to both other tricyclics and SSRIs (Pariante et al, 2001a, 2003a). Glucocorticoid function was measured by glucocorticoid inhibition of LPS-stimulated IL-6 levels. Whole blood was diluted tenfold with RPMI 1640 medium. All solutions were prepared in pyrogene-free sterile saline (NaCl 0.9%) to achieve final concentrations in the cultures of 20 ng/mg for LPS; 10 μM for CMI; 10 and 100 nM for DEX; 0.1 and 1 μM for PRED; 1 and 10 μM for CORT; and 1 and 10 μM for CTC. A total of 540 μl of diluted blood (in RPMI 1640 medium with l-glutamine supplemented with 100 IU/ml penicillin and 100 mg/ml streptomycin) was added onto 48-well cell culture plates (Falcon, No. 3078). LPS, CMI, and glucocorticoids were subsequently added to the wells. Samples were incubated for 24 h in a humidified atmosphere containing 5% CO2. After the incubation, plates were centrifuged (3000 r.p.m., 20 min) and supernatant carefully collected and kept at −40°C until analysis. Determinations were performed in duplicates and always by the same researcher (LC).
Sample Measurements
All analysis was carried out using a commercially available Immulite kit for the fully automated IMMULITE system (Diagnostics Products Corporation, Los Angeles, CA). The coefficient of variation (CV) for IL-6 analysis was 4.7% within run and 6.5% in between runs, and the detection limit was 2 pg/ml. For CORT, the CV values were 7.1% within run and 7.8% in between runs, and the detection limit was 5.5 nmol/l.
Statistical Analysis and Data Presentation
All variables were tested for normality of distribution by means of the Kolmogorov–Smirnov test. Wilcoxon test for paired samples was used to examine the within-subject effect of CMI, while Mann–Whitney was used for unpaired tests to examine between-subject differences (patients vs healthy controls). Results in Figure 1 are expressed as mean±SEM of raw IL-6 levels. In Figure 2, glucocorticoid (GC) inhibition was calculated by considering LPS-stimulated IL-6 levels in the absence of glucocorticoids as 100%. Specifically, the calculation of the percentage inhibition in glucocorticoids condition was as follows:
Figure 1.
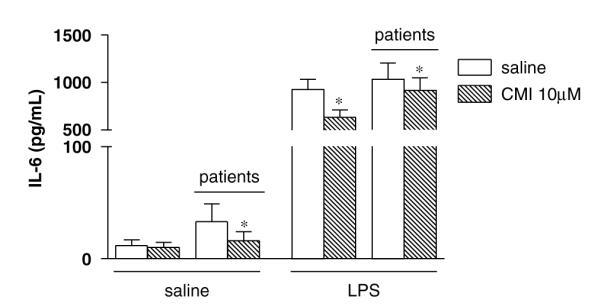
Unstimulated (saline) interleukin-6 (IL-6) levels (pg/ml) in depressed patients (n = 15) and healthy controls (n = 28) before and after 24 h clomipramine (10 μM) incubation. Data are shown as mean±SEM. *p<0.05.
Figure 2.

(a) Dexamethasone (0.01 and 0.1 μM), (b) prednisolone (0.1 and 1 μM), (c) cortisol (1 and 10 μM), and (d) corticosterone (1 and 10 μM) inhibition of lypopolysaccharide (LPS)-stimulated interleukin-6 (IL-6) levels in depressed patients (n = 15) and healthy controls (n = 28) with (hashed columns) or without (white columns) clomipramine (10 μM). Results are expressed by mean±SEM of the percent glucocorticoid inhibition (LPS-stimulated IL-6 levels with glucocorticoid divided by LPS-stimulated IL-6 levels without glucocorticoids). *p<0.05.
In the presence of the antidepressant CMI, the calculation of glucocorticoid inhibition was corrected by the presence of CMI as follows:
The significance level was set to p<0.05 (two-tailed) and computer statistical packages (GraphPad 4.1 Software Inc., San Diego, CA, USA and SPSS 15.0 Inc. Software, Chicago, Illinois, USA) were used for this study.
RESULTS
Fifteen treatment-resistant depressed inpatients and twenty-eight healthy controls participated in this study. The demographic and clinical information of all subjects is listed in Table 1. There were no differences between patients and controls according to gender distribution, age, or body mass index (BMI), confirming the groups were comparable. As expected, BDI scores were found to be very low among the controls (BDI≤2).
Table 1.
Demographic and Clinical Features of 15 Patients and 28 Controls
| Depressed inpatients mean (SEM) or n (%) | Controls mean (SEM) or n (%) | p-value (χ2/t-test) | |
|---|---|---|---|
| Gender (%) | 77 | 75 | 0.853 |
| Age (years) | 49.0±3.2 | 45.0±2.5 | 0.478 |
| BMI (kg/m2) | 28.0±1.2 | 28.6±0.9 | 0.759 |
| BDI | 31.3±2.5 | 2.0±0.6 | 0.000 |
| HAM-D | 24.7±1.6 | ||
| BAI | 23.1±3.5 | ||
| Age at onset of first MDE (years) | 30.9 (2.5) | ||
| Age at onset of the current episode (years) | 42.2 (2.9) | ||
| Duration of current MDE (months) | 47.5(8.3) | ||
| Duration of Illness (years) | 18.2 (3.2) | ||
| No. of previous hospital admissions | 3.4 (0.7) | ||
| Duration of current admission (weeks) | 20.3 (2.2) | ||
| Current medication | 9 Mood stabilizer | ||
| 10 SSRI/SNRI | |||
| 4 Benzodiazepine | |||
| 4 Atypical antipsychotic | |||
| 3 Tricyclic antidepressant | Drug free | ||
| 1 MAOI | |||
| 4 Other antipsychotic | |||
| 6 Nonpsychotropic | |||
| ECT in the past | 13 (86) | ||
| treatment resistance stage: n (%) | Stage 5: 6(40) | ||
| Stage 4: 3(20) | |||
| Stage 3: 4(26) |
Abbreviations: BAI, Beck Anxiety Inventory; BDI, Beck Depression Inventory; BMI, body mass index; ECT, electroconvulsotherapy; HAM-D, Hamilton Depression Rating Scale; MAOI, monamine oxidase inhibitor; SSRI, selective serotonin reuptake inhibitor; SNRI, serotonin and norepinephrine reuptake inhibitor; MDE, major depressive episode. Treatment resistance stage: Thase and Rush treatment resistance criteria.
The treatment-resistant depressed patients that participated in this study had marked biological disturbances, when compared to healthy controls, as shown by hypercortisolemia and evidence of inflammation. Patients had higher plasma CORT levels (429.4±55.4 vs 242.2±14.8 nmol/l, p<0.05, respectively) and higher plasma IL-6 (3.0±0.29 vs 2.4±0.1 pg/ml, p<0.05, respectively) than controls. Plasma CORT and plasma IL-6 were positively correlated in controls (Pearson’s correlation coefficient = 0.4, p = 0.035), but not in patients (Pearson’s correlation coefficient = 0.3, p = 0.14). Neither plasma CORT nor plasma IL-6 was associated with the severity of depressive symptomatology (HAM-D scores) or other clinical features (data not shown).
In the in vitro incubation experiments, healthy controls showed undetectable unstimulated IL-6 levels before or after CMI incubation. Unstimulated IL-6 levels in depressed patients, however, were detectable in seven of the fifteen patients. In these seven patients, CMI reduced the unstimulated IL-6 levels (by 51%, p = 0.03). We also analyzed these data using 2 pg/ml (the sensitivity limit of the method) as the value for those that had undetectable levels. These data are presented in Figure 1, and the statistical analysis using this approach also shows a significant effect of CMI in depressed patients.
Stimulation with LPS increased IL-6 levels in depressed patients and in healthy controls to similar levels. Moreover, in both groups CMI inhibited LPS-stimulated IL-6 levels, although the effect of CMI was more pronounced in controls (in depressed patients by 11% and in healthy controls by 32%).
Next, we investigated glucocorticoid inhibition in depressed patients and in healthy controls. Data are presented as percentage of IL-6 levels in LPS-stimulated samples. Glucocorticoids in the absence of CMI are represented by the white columns in the figures. All glucocorticoids induced a concentration-dependent inhibition of LPS-stimulated IL-6 levels in depressed patients and in healthy controls. The rank order of inhibition was DEX>PRED>CORT>CTC, and this was similar for depressed patients and healthy controls.
There was a difference on the effects of in vitro CMI coincubation with glucocorticoids between depressed patients and healthy controls (Figure 2a–d, hashed bars represent the presence of CMI). In controls, LPS-stimulated IL-6 levels were higher after CMI and glucocorticoids than after glucocorticoids alone (hashed columns vs white columns for each concentration of glucocorticoids); consequently, CMI decreased glucocorticoid function, that is, decreased glucocorticoid inhibition of LPS-stimulated IL-6 levels. This effect was present for all concentrations of DEX, PRED, and CTC, and for the highest concentration of CORT (10 μM). In contrast, this effect was not present in depressed patients (p>0.05). These data are presented in Figure 2 and described below with the appropriate statistical comparisons. For DEX (Figure 2a), LPS-stimulated IL-6 levels in the presence of DEX (0.01 μM) alone were 35.7±2.5% in controls and 34.5±3.1% in depressed; in the presence of DEX and CMI, IL-6 levels were 43.2±3.6% in controls and 36±2.9% in patients (controls: with CMI vs without CMI, p = 0.003; depressed patients: with CMI vs without CMI, p = 0.5). LPS-stimulated IL-6 levels in the presence of DEX (0.1 μM) alone were 8.9±0.9% in controls and 7.9±0.7% in depressed; in the presence of DEX and CMI, IL-6 levels were 17.4±3.2% in controls and 8.8±1.1% in patients (controls: with CMI vs without CMI, p = 0.002; depressed patients: with CMI vs without CMI, p = 0.13). For PRED (Figure 2b), LPS-stimulated IL-6 levels with PRED (0.1 μM) alone were 36.2±3% in controls and 36.1±2.9% in depressed; in the presence of PRED and CMI, IL-6 levels were 49.9±6.7% in controls and 38±2.9% in patients (controls: with CMI vs without CMI, p = 0.003; depressed patients: with CMI vs without CMI, p = 0.3). LPS-stimulated IL-6 levels with PRED (1 μM) alone were 11.7±1.1% in controls and 10.6±1.1% in depressed; in the presence of PRED and CMI, IL-6 levels were 21.3±3.8% in controls and 11.7±1.3% in patients (controls: with CMI vs without CMI, p = 0.002; depressed patients: with CMI vs without CMI, p = 0.06). For CORT (Figure 2c), LPS-stimulated IL-6 levels with CORT (1 μM) alone were 24.6±3.3% in controls and 19.7±1.9% in depressed; in the presence of CORT (1 μM) and CMI, IL-6 levels were 28.9±3.5% in controls and 22±3.5% in patients (controls: with CMI vs without CMI, p = 0.15; depressed patients: with CMI vs without CMI, p = 0.27). LPS-stimulated IL-6 levels with CORT (10 μM) alone were 14.5±1.0% in controls and 13.5±1.3% in depressed; in the presence of CORT and CMI, IL-6 levels were 20.4±3.8% in controls and 14.7±1.7% in patients (controls: with CMI vs without CMI, p = 0.002; depressed patients: with CMI vs without CMI, p= 0.09). For CTC (Figure 2d), LPS-stimulated IL-6 levels =with CTC (1 μM) alone were 68.5±3.4% in controls and 68.6±6.3% in depressed; in the presence of CTC and CMI, IL-6 levels were 75.7±3.4% in controls and 72.9±3.0% in patients (controls: with CMI vs without CMI, p = 0.003; depressed patients: with CMI vs without CMI, p = 0.46). LPS-stimulated IL-6 levels with CTC (10 μM) alone=were 53.5±2.9% in controls and 52.2±1.8% in depressed; in the presence of CTC and CMI, IL-6 levels were 59.8±3.2% in controls and 54.5±2.9% in patients (controls: with CMI vs without CMI, p = 0.002; depressed patients: with CMI vs without CMI, p = 0.5).
DISCUSSION
We have shown that 24 h in vitro incubation with CMI has no effect on GR function in diluted whole-blood cells from a group of severely depressed, treatment-resistant patients, while decreasing GR function in healthy controls. Therefore, in our study depressed patients, who are clinically resistant to the effect of antidepressants, also lack an effect of antidepressants in vitro in peripheral blood cells.
We have shown that our sample of treatment-resistant depressed patients had marked biological disturbances, as seen by hypercortisolemia and presence of inflammation. Theoretically, the patients did not show differences in GR function when compared with controls, as tested by incubation using four different steroids. However, our interpretation is that these findings do suggest glucocorticoid resistance in depressed patients: the fact that GR function was ‘normal’ in the face of such a big difference in plasma CORT in itself suggests a functional GR difference between patients and controls. Indeed, higher levels of CORT (from the plasma) would have been present in the in vitro milieu of depressed patients compared to controls. The presence of GR resistance in these patients is also supported by the fact that there was no difference between patients and controls in stimulated IL-6 levels, even in the presence of higher endogenous CORT levels in the in vitro assay of depressed patients. These results are consistent with the fact that patients with depression frequently have high CORT levels, but they do not develop the physical signs of Cushing’s syndrome (reviewed by Murphy, 1991). Other studies have examined GR function in vitro using a different technique, glucocorticoid inhibition of lymphocyte proliferation. These studies have shown both normal and reduced GR function (reviewed in Pariante and Miller, 2001; Pariante, 2004). It is important to emphasize, however, that our GR assay is not immediately comparable with that used in these studies: first, we use whole-blood cells rather than isolated lymphocytes, and it is possible that GR function is influenced by the presence of other immune cell populations; second, our assay includes endogenous glucocorticoids from the plasma, while assays using isolated lymphocytes usually allow for removal of all endogenous glucocorticoids. Nevertheless, taken together our findings are supportive of the notion that GR function in these depressed patients might be reduced.
Interestingly, we have shown that 24 h in vitro CMI decreases GR function in healthy controls. The decrease in GR function by in vitro antidepressants has been shown by us and other groups in mouse fibroblasts (Budziszewska et al, 2000, 2005; Augustyn et al, 2005; Barden, 1996; Pariante et al, 2001a, 1997). This decrease in GR function by antidepressants in vitro may be a consequence of GR downregulation, which in turn may be secondary to antidepressant-induced GR translocation to the nucleus. Specifically, we have shown that 24 h incubation with desipramine or CMI induces GR translocation and decreases GR (protein) expression in mouse fibroblasts cell line (Pariante et al, 1997, 2003). Moreover, a decrease in GR mRNA by antidepressants has also been described in a study that has measured the effect of different classes of antidepressants in human primary cell cultures (Heiske et al, 2003). On the other hand, in vitro treatment of neurons with antidepressants has produced conflicting findings, perhaps depending of the GR measure: we have shown decreased GR (protein expression after 24 h with CMI; Pariante et al, 2003), while Pepin et al (1989) found decreased GR mRNA after 24–72 h incubation with desipramine and other antidepressants. However, it is important to emphasize that antidepressant-induced GR downregulation is an in vitro phenomenon, usually occurring with incubation lasting hours (as indeed in the present study). In contrast, studies in animals undergoing chronic treatment with antidepressants have clearly shown an upregulation of the GR in the brain, possibly secondary to changes in HPA axis activity (Pariante et al, 2004b). Therefore, GR upregulation after long-term treatment with antidepressants may be more relevant to the therapeutic action of this class of drugs.
It is of note that antidepressants have also been shown to increase GR function in vitro in mouse or rats under different experimental conditions (Pariante et al, 1997, 2003b). The increased GR function is likely to be mediated by an increased intracellular concentration of glucocorticoids, following inhibition by antidepressants of steroid transporters that expel glucocorticoids from cells. Consequently, the final effect of antidepressants on GR function seems to be dependent on the balance between the amount of intracellular glucocorticoids (possibly dictated by the steroid transporters) and receptor expression. Our recent paper in mice further supports the involvement of the steroid transporters on the final effect of antidepressants on GR function. Treatment with desipramine induces GR downregulation in steroid transporter knockout mice—and therefore in a condition that is similar to the in vitro experiments that do not elicit the effect on the transporter, like the present study (as lymphocytes express low levels of the transporter); in contrast, desipramine induces GR upregulation in control mice—and therefore in a condition that is similar to the in vitro experiments that do elicit effect on the transporter (Yau et al, 2007).
We have also shown that treatment-resistant depressed patients lack an effect of CMI on GR function in vitro. The lack of effect of CMI in vitro in major depression could be related either to patients’ pathophysiology or to the previous chronic use of antidepressants (or to both). Due to the chronicity and severity of the patients studied here, it was not possible to analyze them in a drug-free or euthymic state, and this is a clear limitation of the study. However, patients who are chronic and severely depressed show the most marked HPA axis abnormalities (Nemeroff, 1996; Holsboer, 2000; Pariante et al, 1999). Moreover, other biological disturbances associated with the pathophysiology of depression might also make the GR ‘resistant’ to the effects of antidepressants in vitro. For example, depressed patients show signs of inflammation, and inflammation has been shown to reduce the ability of GR to activate and translocate (Pariante et al, 1999), an effect that has been linked to the activation of intracellular pathways like the p38 mitogen-activated protein (Wang et al, 2004). In addition to these putative effects of depression, prolonged exposure to antidepressants for clinical treatment may also alter GR function or its transduction pathways, and hence antidepressants in vitro could be unable to modulate this process any further (Carvalho and Pariante, 2008). Whether such severe treatment-resistant depressed patients would have had a different response to the in vitro effects of CMI if they had been drug free is unfortunately very difficult to examine.
Finally, would the effect of CMI on the GR in vitro be different in any other psychiatric population? Our results are consistent with the work by Yehuda et al (2006), who have also found a difference between PTSD patients and controls in the effect of antidepressants on GR function, although in the opposite direction compared with our study. Specifically, they have found that sertraline reduces GR function in patients more than in controls. It is interesting to note that GR abnormalities in PTSD are opposite to those present in depression, that is, PTSD patients have increased GR function rather than decreased (Yehuda, 2005). Therefore, the fact that the effects of antidepressants on the GR are also in opposite directions is consistent with this difference in the pathophysiology and indicates that this assessment might have a clinical significance. Upcoming studies shall test whether assessing the effects of antidepressants in vitro on GR function could predict future treatment response in a clinical setting.
ACKNOWLEDGEMENTS
Livia A Carvalho is currently funded by a grant from the UK Medical Research Council (MRC). Carmine M Pariante has been funded by the UK MRC since 1999, first as a Clinical Training Fellow, and currently as an MRC Clinician Scientist Fellow. This research was also funded by the NIHR South London and Maudsley NHS Foundation Trust and Institute of Psychiatry Specialist Biomedical Research Centre for Mental Health, the NARSAD. MF Juruena was funded by the CAPES Fellowship Award and the NARSAD Young Investigator Award. This research was conducted in the Affective Disorders Laboratory and was funded by the Medical Research Council (UK). We thank Irene Papadopoulos, from the Affective Disorders Laboratory, for assistance with laboratory techniques.
Footnotes
DISCLOSURE/CONFLICT OF INTEREST The authors have no relevant financial interest to disclose.
REFERENCES
- American Psychiatric Association . Diagnostic and Statistical Manual of Mental Disorders. American Psychiatric Press; Washington, DC: 1994. [Google Scholar]
- Augustyn M, Otczyk M, Budziszewska B, Jagla G, Nowak W, Basta-Kaim A, et al. Effects of some new antidepressant drugs on the glucocorticoid receptor-mediated gene transcription in fibroblast cells. Pharmacol Rep. 2005;57:766–773. [PubMed] [Google Scholar]
- Barden N. Modulation of glucocorticoid receptor gene expression by antidepressant drugs. Pharmacopsychiatry. 1996;29:12–22. doi: 10.1055/s-2007-979536. [DOI] [PubMed] [Google Scholar]
- Beck A, Epstein N, Brown G, Steer RA. An inventory for measuring clinical anxiety: psychometric properties. J Consult Clin Psychol. 1988;56:893–897. doi: 10.1037//0022-006x.56.6.893. [DOI] [PubMed] [Google Scholar]
- Beck A, Ward C, Mendelson M, Mock J, Erbaugh G. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:561–571. doi: 10.1001/archpsyc.1961.01710120031004. [DOI] [PubMed] [Google Scholar]
- Budziszewska B, Basta-Kaim A, Kubera M, Jaworska L, Leskiewicz M, Tetich M, et al. Effect of lipopolysaccharide and antidepressant drugs on glucocorticoid receptor-mediated gene transcription. Pharmacol Rep. 2005;57:540–544. [PubMed] [Google Scholar]
- Budziszewska B, Jaworska-Feil L, Kajta M, Lason W. Antidepressant drugs inhibit glucocorticoid receptor-mediated gene transcription—a possible mechanism. Br J Pharmacol. 2000;130:1385–1393. doi: 10.1038/sj.bjp.0703445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho LA, Pariante CM. In vitro modulation of glucocorticoid receptors by antidepressants. Stress. 2008 doi: 10.1080/10253890701850759. (in press) [DOI] [PubMed] [Google Scholar]
- Gold PW, Goodwin FK, Chrousos GP. Clinical and biochemical manifestations of depression. Relation to the neurobiology of stress (1) N Engl J Med. 1988;319:348–353. doi: 10.1056/NEJM198808113190606. [DOI] [PubMed] [Google Scholar]
- Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiske A, Jesberg J, Krieg JC, Vedder H. Differential effects of antidepressants on glucocorticoid receptors in human primary blood cells and human monocytic u-937 cells. Neuropsychopharmacology. 2003;28:807–817. doi: 10.1038/sj.npp.1300056. [DOI] [PubMed] [Google Scholar]
- Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- Holsboer F, Barden N. Antidepressants and hypothalamic–pituitary–adrenocortical regulation. Endocr Rev. 1996;17:187–205. doi: 10.1210/edrv-17-2-187. [DOI] [PubMed] [Google Scholar]
- Linkowski P, Mendlewicz J, Kerkhofs M, Leclercq R, Golstein J, Brasseur M, et al. 24-hour profiles of adrenocorticotropin, cortisol, and growth hormone in major depressive illness: effect of antidepressant treatment. J Clin Endocrinol Metab. 1987;65:141–152. doi: 10.1210/jcem-65-1-141. [DOI] [PubMed] [Google Scholar]
- Mason BL, Pariante CM. The effects of antidepressants on the hypothalamic–pituitary–adrenal axis. Drug News Perspect. 2006;19:603–608. doi: 10.1358/dnp.2006.19.10.1068007. [DOI] [PubMed] [Google Scholar]
- Murphy BE. Steroids and depression. J Steroid Biochem Mol Biol. 1991;38:537–559. doi: 10.1016/0960-0760(91)90312-s. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB. The corticotropin-releasing factor (CRF) hypothesis of depression: new findings and new directions. Mol Psychiatry. 1996;1:336–342. [PubMed] [Google Scholar]
- Owens MJ, Nemeroff CB. The role of corticotropin-releasing factor in the pathophysiology of affective and anxiety disorders: laboratory and clinical studies. Ciba Found Symp. 1993;172:296–308. doi: 10.1002/9780470514368.ch15. [DOI] [PubMed] [Google Scholar]
- Pariante CM. Glucocorticoid receptor function in vitro in patients with major depression. Stress. 2004;7:209–219. doi: 10.1080/10253890500069650. [DOI] [PubMed] [Google Scholar]
- Pariante CM. The glucocorticoid receptor: part of the solution or part of the problem? J Psychopharmacol. 2006;20:79–84. doi: 10.1177/1359786806066063. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Hye A, Williamson R, Makoff A, Lovestone S, Kerwin RW. The antidepressant clomipramine regulates cortisol intracellular concentrations and glucocorticoid receptor expression in fibroblasts and rat primary neurones. Neuropsychopharmacology. 2003a;28:1553–1561. doi: 10.1038/sj.npp.1300195. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Kim RB, Makoff A, Kerwin RW. Antidepressant fluoxetine enhances glucocorticoid receptor function in vitro by modulating membrane steroid transporters. Br J Pharmacol. 2003b;139:1111–1118. doi: 10.1038/sj.bjp.0705357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariante CM, Makoff A, Lovestone S, Feroli S, Heyden A, Miller AH, et al. Antidepressants enhance glucocorticoid receptor function in vitro by modulating the membrane steroid transporters. Br J Pharmacol. 2001a;134:1335–1343. doi: 10.1038/sj.bjp.0704368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001;49:391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Papadopoulos AS, Poon L, Checkley SA, English J, Kerwin RW, et al. A novel prednisolone suppression test for the hypothalamic–pituitary– adrenal axis. Biol Psychiatry. 2002;51:922–930. doi: 10.1016/s0006-3223(01)01314-2. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Papadopoulos AS, Poon L, Cleare AJ, Checkley SA, English J, et al. Four days of citalopram increase suppression of cortisol secretion by prednisolone in healthy volunteers. Psychopharmacology (Berl) 2004a;177:200–206. doi: 10.1007/s00213-004-1925-4. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Pearce BD, Pisell TL, Owens MJ, Miller AH. Steroid-independent translocation of the glucocorticoid receptor by the antidepressant desipramine. Mol Pharmacol. 1997;52:571–581. doi: 10.1124/mol.52.4.571. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Pearce BD, Pisell TL, Sanchez CI, Po C, Su C, et al. The proinflammatory cytokine, interleukin-1alpha, reduces glucocorticoid receptor translocation and function. Endocrinology. 1999;140:4359–4366. doi: 10.1210/endo.140.9.6986. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Pearce BD, Pisell TL, Su C, Miller AH. The steroid receptor antagonists RU40555 and RU486 activate glucocorticoid receptor translocation and are not excreted by the steroid hormones transporter in L929 cells. J Endocrinol. 2001b;169:309–320. doi: 10.1677/joe.0.1690309. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Thomas SA, Lovestone S, Makoff A, Kerwin RW. Do antidepressants regulate how cortisol affects the brain? 2003 Curt Richter Award Paper. Psychoneuroendocrinology. 2004b;29:423–447. doi: 10.1016/j.psyneuen.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Pepin MC, Beaulieu S, Barden N. Antidepressants regulate glucocorticoid receptor messenger RNA concentrations in primary neuronal cultures. Brain Res Mol Brain Res. 1989;6:77–83. doi: 10.1016/0169-328x(89)90031-4. [DOI] [PubMed] [Google Scholar]
- Ribeiro SC, Tandon R, Grunhaus L, Greden JF. The DST as a predictor of outcome in depression: a meta-analysis. Am J Psychiatry. 1993;150:1618–1629. doi: 10.1176/ajp.150.11.1618. [DOI] [PubMed] [Google Scholar]
- Rohleder N, Kudielka BM, Hellhammer DH, Wolf JM, Kirschbaum C. Age and sex steroid-related changes in glucocorticoid sensitivity of pro-inflammatory cytokine production after psychosocial stress. J Neuroimmunol. 2002;126:69–77. doi: 10.1016/s0165-5728(02)00062-0. [DOI] [PubMed] [Google Scholar]
- Rohleder N, Schommer NC, Hellhammer DH, Engel R, Kirschbaum C. Sex differences in glucocorticoid sensitivity of proinflammatory cytokine production after psychosocial stress. Psychosom Med. 2001;63:966–972. doi: 10.1097/00006842-200111000-00016. [DOI] [PubMed] [Google Scholar]
- Sackeim HA. The definition and meaning of treatment-resistant depression. J Clin Psychiatry. 2001;62(Suppl 16):10–17. [PubMed] [Google Scholar]
- Thase ME, Rush AJ. When at first you don’t succeed: sequential strategies for antidepressant nonresponders. J Clin Psychiatry. 1997;58(Suppl 13):23–29. [PubMed] [Google Scholar]
- Wang X, Wu H, Miller AH. Interleukin 1alpha (IL-1alpha) induced activation of p38 mitogen-activated protein kinase inhibits glucocorticoid receptor function. Mol Psychiatry. 2004;9:65–75. doi: 10.1038/sj.mp.4001339. [DOI] [PubMed] [Google Scholar]
- World Health Organization . Schedules for Clinical Assessment in Neuropsychiatry (SCAN) Version 2.0 World Health Organization; Geneva: 1994. [Google Scholar]
- Yau JL, Noble J, Thomas S, Kerwin R, Morgan PE, Lightman S, et al. The antidepressant desipramine requires the ABCB1 (Mdr1)-type p-glycoprotein to upregulate the glucocorticoid receptor in mice. Neuropsychopharmacology. 2007;32:2520–2529. doi: 10.1038/sj.npp.1301389. [DOI] [PubMed] [Google Scholar]
- Yehuda R. Biology of posttraumatic stress disorder. J Clin Psychiatry. 2001;62(Suppl 17):41–46. [PubMed] [Google Scholar]
- Yehuda R. Neuroendocrine aspects of PTSD. Handb Exp Pharmacol. 2005;169:371–403. doi: 10.1007/3-540-28082-0_13. [DOI] [PubMed] [Google Scholar]
- Yehuda R, Yang RK, Golier JA, Grossman RA, Bierer LM, Tischler L. Effect of sertraline on glucocorticoid sensitivity of mononuclear leukocytes in post-traumatic stress disorder. Neuropsychopharmacology. 2006;31:189–196. doi: 10.1038/sj.npp.1300862. [DOI] [PubMed] [Google Scholar]