

Abstract
The aim was to develop niosomal gel as a transdermal nanocarrier for improved systemic availability of lopinavir. Niosomes were prepared using thin-film hydration method and optimized for molar quantities of Span 40 and cholesterol to impart desirable characteristics. Comparative evaluation with ethosomes was performed using ex vivo skin permeation, fluorescence microscopy, and histopathology studies. Clinical utility via transdermal route was acknowledged using in vivo bioavailability study in male Wistar rats. The niosomal formulation containing lopinavir, Span 40, and cholesterol in a molar ratio of 1:0.9:0.6 possessed optimally high percentage of drug entrapment with minimum mean vesicular diameter. Ex vivo skin permeation studies of lopinavir as well as fluorescent probe coumarin revealed a better deposition of ethosomal carriers but a better release with niosomal carriers. Histopathological studies indicated the better safety profile of niosomes over ethosomes. In vivo bioavailability study in male Wistar rats showed a significantly higher extent of absorption (AUC0→∞, 72.87 h × μg/ml) of lopinavir via transdermally applied niosomal gel as compared with its oral suspension. Taken together, these findings suggested that niosomal gel holds a great potential of being utilized as novel, nanosized drug delivery vehicle for transdermal lopinavir delivery.
KEY WORDS: ethosomes, lopinavir, niosomes, transdermal
INTRODUCTION
Acquired immune deficiency syndrome (AIDS), the advanced stage of the disease caused by human immunodeficiency virus (HIV) infection, is one of the most serious infectious diseases that challenges public health globally (1). Interventions such as AIDS counseling, educational tools, and antiretroviral drug therapy have contributed to transforming HIV infection from a fatal to a manageable chronic infectious disease. However, as per the global report by UNAIDS in 2010 (2), the number of people receiving therapy has grown 13-fold since 2004, including more than five million people in low- and middle-income countries. Hence, despite available preventive measures, much remains to be accomplished as the number of newly reported HIV infections still remains unacceptably high.
Lopinavir, (2S)-N-[(2S,4S,5S)-5-[2-(2,6-dimethylphenoxy)-acetamido]-4-hydroxy-1,6-diphenylhexan-2-yl]-3-methyl-2-(2-oxo-1,3-diazinan-1-yl)-butanamide, a specific reversible inhibitors of the HIV protease, exerts its effect against HIV infection by blocking the ability of the protease to cleave the Gag-Pol polyprotein, resulting in the production of immature, noninfectious viral particles. However, the systemic availability of lopinavir via oral route is severely limited (3) by its sensitivity towards cytochrome P450 3A4, susceptibility for P-glycoprotein efflux transporters, poor aqueous solubility (~0.01 mg/ml), moderately high molecular weight (~628 Da), and high log P value (~4.56). Though the marketed tablet and capsule formulations of lopinavir are generally combined with Ritonavir, a potent inhibitor of cytochrome P450 3A4, to minimize presystemic metabolism of lopinavir (4), other challenges contributing to poor oral absorption remain unanswered. Hence, to overcome all the limitations associated with oral administration of lopinavir and to promote single drug administration, utilization of vesicular nanocarriers through transdermal route could prove to be effective, as the approach combines the inherent advantages of transdermal route and the drug carrying potential of vesicular nanocarriers across the tough and otherwise impervious skin barrier layer, i.e., stratum corneum (SC).
Among several nanovesicular carriers, niosome is selected here as a carrier of choice owing to its dominance over conventional liposomes with respect to stability and cost-effectiveness. Niosome contains several concentric bilayer membrane mainly composed of nonionic surfactants and cholesterol enclosing aqueous phase in the core. Niosomes are known to improve the solubility, bioavailability, and stability of some poorly soluble drugs (5–8) along with an ability to provide sustained release for prolonged drug action (9). Surfactants contribute to the overall penetration enhancement of compounds primarily by adsorption at interfaces, by interacting with biological membranes and by alteration of the barrier function of the SC, as a result of reversible lipid modification (10). Ethosomes, soft, malleable lipid vesicles with high ethanol content tailored for enhanced transdermal delivery of active agents (11), are used for comparative study wherever required.
So, the aim of the present work was to develop lopinavir-loaded niosomal gel and characterize it for its usefulness as a transdermal nanocarrier in delivering therapeutically sufficient quantity of lopinavir to combat against AIDS. The work encompasses its comparative evaluation with lopinavir-loaded ethosomes for drug-carrying potential through full-thickness rat’s abdominal skin ex vivo and the bioavailability assessment of lopinavir via both oral suspension (OS) as well as transdermal F3c gel in vivo.
MATERIALS AND METHODS
Materials
Lopinavir was received as a gift sample from Aurobindo Pharma Ltd. Phospholipid (Phospholipon 90 H) was a gift sample from Lipoid, GmbH, UK. Span 40, cholesterol, carbopol, coumarin, chloroform, and methanol were purchased from S. D. Fine Chemicals, India. Nuclepore polycarbonate membrane 0.2 μm 25 mm was purchased from Whattman, USA. Water (distilled) prepared in laboratory by distillation. All the chemicals and reagents used were of analytical grade.
Preparation of Lopinavir-Loaded Niosomes
Different niosomal formulations of lopinavir were prepared by changing the proportions of drug surfactant and surfactant cholesterol (Table I) using thin-film hydration technique as described by Agarwal et al. with slight modification (12). Briefly, the Span 40, cholesterol, and lopinavir were dissolved in a mixture of chloroform and methanol (ratio of 7:3, v/v) in a 250-ml round bottom flask. The solvent was evaporated in the rotary flash evaporator until thin, dry, and uniform film is formed. The thin dry lipid film thus formed was hydrated using phosphate-buffered saline (pH 7.4) at a temperature slightly above the Tg of Span 40 (49 ± 1°C). The formed niosomal dispersion was first sonicated in an ice bath using probe sonicator (four cycles of 30 s each) to convert multilamellar vesicles into desired size unilamellar vesicles and then subjected to centrifugation at 4,000 rpm and 4°C for 15 min using laboratory centrifuge (Remi, India) to effect sedimentation of unentrapped drug as pellet at the bottom of the centrifugation tube (13). Niosomal dispersion (supernatant) was then decanted and characterized for vesicle size and percentage of drug entrapment (PDE) while the drug pellet (sediment) was used to measure unentrapped drug in order to ascertain mass balance. The formulation process parameters were optimized to achieve maximum possible drug entrapment with desirable size range (14).
Table I.
Composition and Characterization of Lopinavir-Loaded Niosomal Dispersions
| S. No. | Formulation code | Composition (molar quantities) | Vesicular sizea (nm) | PDIa | % entrapment efficiencya | ||
|---|---|---|---|---|---|---|---|
| Lopinavir | Span 40 | Cholesterol | |||||
| 1 | F1 | 1 | 0.5 | – | 354.94 ± 9.16 | 0.228 ± 0.012 | 45.3 ± 1.87 |
| 2 | F2 | 1 | 1.0 | – | 276.38 ± 8.22 | 0.194 ± 0.014 | 52.9 ± 3.39 |
| 3 | F3 | 1 | 1.5 | – | 196.45 ± 6.23 | 0.165 ± 0.011 | 58.9 ± 1.98 |
| 4 | F4 | 1 | 2.0 | – | 203.52 ± 3.97 | 0.216 ± 0.021 | 57.4 ± 2.76 |
| 5 | F3a | 1 | 1.3 | 0.2 | 169.34 ± 1.24 | 0.212 ± 0.022 | 62.5 ± 1.84 |
| 6 | F3b | 1 | 1.1 | 0.4 | 132.86 ± 2.01 | 0.189 ± 0.014 | 68.1 ± 0.37 |
| 7 | F3c | 1 | 0.9 | 0.6 | 105.22 ± 1.96 | 0.127 ± 0.011 | 75.5 ± 0.50 |
| 8 | F3d | 1 | 0.7 | 0.8 | 114.65 ± 2.21 | 0.133 ± 0.009 | 74.5 ± 0.86 |
PDI polydispersity index
aValues represented as mean ± SD (n = 3)
Preparation of Lopinavir-Loaded Ethosomes
Ethosomes of lopinavir consisting of soya phosphatidylcholin (PC), propylene glycol and ethanol were prepared by cold method (15). Briefly, lopinavir with varying proportions of phospholipid were dissolved in varying amount of ethanol (Table II) in a covered vessel at room temperature by vigorous stirring. Propylene glycol was added during stirring. The mixture was then heated to 47°C in a water bath. The water, previously heated to 47°C in a separate vessel, was added slowly to the mixture till a white suspension was formed and stirred for 10 min. Resultant dispersion of ethosomes was then sonicated in an ice bath using probe sonicator (four cycles of 30 s each) to get desired sized unilamellar vesicles. Unentrapped drug was separated by centrifugation at 4,000 rpm and 4°C for 15 min using laboratory centrifuge (Remi, India) and estimated for drug content (same procedure as mentioned for niosomes).
Table II.
Composition and Characterization of Lopinavir-Loaded Ethosomal Dispersions
| S. No. | Formulation code | Composition (% w/w) | Vesicular sizea (nm) | PDIa | % entrapment efficiencya | ||
|---|---|---|---|---|---|---|---|
| Lopinavir | Soya PC | Ethanol | |||||
| 1 | E1 | 0.5 | 1 | 30 | 225.6 ± 11.0 | 0.264 ± 0.016 | 48.3 ± 3.7 |
| 2 | E2 | 0.5 | 1 | 45 | 157.2 ± 07.5 | 0.212 ± 0.027 | 63.4 ± 4.1 |
| 3 | E3 | 0.5 | 1 | 60 | 178.2 ± 09.2 | 0.118 ± 0.006 | 39.2 ± 4.3 |
| 4 | E4 | 0.5 | 2 | 30 | 176.7 ± 18.6 | 0.146 ± 0.013 | 61.7 ± 3.8 |
| 5 | E5 | 0.5 | 2 | 45 | 112.8 ± 12.4 | 0.131 ± 0.008 | 79.6 ± 4.1 |
| 6 | E6 | 0.5 | 2 | 60 | 132.7 ± 07.6 | 0.223 ± 0.017 | 43.6 ± 4.7 |
| 7 | E7 | 0.5 | 3 | 30 | 164.5 ± 06.1 | 0.283 ± 0.023 | 50.3 ± 3.4 |
| 8 | E8 | 0.5 | 3 | 45 | 105.3 ± 03.9 | 0.244 ± 0.018 | 70.7 ± 4.4 |
| 9 | E9 | 0.5 | 3 | 60 | 123.8 ± 11.2 | 0.198 ± 0.011 | 33.7 ± 4.2 |
PDI polydispersity index, PC phosphatidylcholin
aValues represented as mean ± SD (n = 3)
Preparation of Gel
The gel of optimized niosomal (F3c) and ethosomal (E5) dispersions were prepared by dispersing 0.8% (w/v) carbopol 934 in it and allowing it to hydrate for 24 h. Finally, neutralization of gel was done by adding triethanolamine that made it transparent. Plain drug gel was also prepared by using same procedure with hydroethanolic solution (15% (v/v) ethanol) of lopinavir.
Characterization of Niosomes and Ethosomes
Vesicular Shape and Surface Morphology
Photomicrographs taken by Olympus BX 40 microscope (at ×40) were used for initial visualization of niosomes and ethosomes before sonication. Scanning electron microscope (JSM-5610LV, JEOL, Japan) was used later to determine surface morphology of F3c and E5 dispersions. Samples were attached to sample stubs, silver coated, and then viewed using an accelerating voltage (15 kV) at the magnification of ×100,000.
Vesicular Size and Zeta Potential
Vesicular dispersions were diluted ten times with distilled water and then it was analyzed for vesicular size and zeta potential by Malvern Zetasizer Nanoseries-ZS (Malvern Instruments, Malvern, UK).
Percentage of Drug Entrapment
Amount of lopinavir entrapped within niosomes and ethosomes was estimated using first derivative UV–visible spectroscopic technique (UV-1700, Shimadzu). In brief, an appropriate quantity of drug-loaded formulation and unentrapped fraction (separation method is described earlier in preparation section) of lopinavir were dissolved separately in a mixture of aetonitrile/methanol (7:3 ratio) and absorbance difference (dA/dλ) were recorded at a wavelength of 219 nm. The corresponding concentrations were obtained from first derivative standard calibration curve of lopinavir prepared at 219 nm. The method was found to obey Beer’s law between concentration range of 10 to 35 μg/ml with limit of detection (LOD) and limit of quantification (LOQ) values as 0.844 and 2.558 μg/ml, respectively. The PDE was finally calculated using the formula:
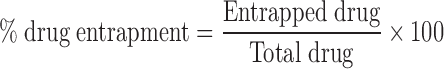 |
Stability Study
The lopinavir-loaded F3c and E5 dispersions were stored in a sealed glass vials and subjected to stability study in triplicate. The vials were kept at two different storage conditions, i.e., 4 ± 1°C with ambient humidity and 30 ± 2°C with 65 ± 5% RH, and the samples were withdrawn periodically at an interval of 1 month for 2 months, suitably diluted with water and analyzed for vesicular size and percent drug retention.
Ex vivo Studies
Drug Release Study
Ex vivo study was carried out using full-thickness rat abdominal skin (16). Rat was killed by cervical dislocation method; the abdominal skin was removed and dipped into phosphate buffer solution (pH 6.8). Hairs were gently removed using electric clipper. The hairless skin was then dipped in hot water and subcutaneous fat was removed with scalpel. The skin was mounted on receptor compartment of the Franz diffusion cell (effective surface area, 3.14 cm2) in such a way that the SC was facing upwards and then the donor chamber was clamped in place. The excess skin was trimmed off and the receptor compartment was filled with 24 ml of diffusion media consisting of phosphate buffer (pH 6.8)/isopropyl alcohol mixture in a ratio of 3:1. The whole assembly was put on magnetic stirrer for gentle stirring and the temperature of the diffusion media was maintained at 32 ± 0.5°C. Various gel formulations selected for the study (Table III) were applied to the skin. The samples were collected over 24 h at predetermined time intervals and analyzed by HPLC method. Two milliliters of sample was withdrawn each time from the receptor compartment, and the volume was then maintained by adding 2 ml of fresh diffusion media. On completion of 24 h, the skin surface was washed thrice with diffusion medium and the washings were filtered under vacuum using 0.22 μm polycarbonate membrane filter. These filtered samples were then suitably diluted and analyzed for drug retained on skin surface. To extract out the drug deposited into the skin, the skin was chopped into small pieces using sharp knife and collected in diffusion medium. It was then subjected to bath sonication for three cycles each of 5 min, and the extracted drug was analyzed by HPLC method.
Table III.
Ex vivo Drug Release and Skin Deposition Profile of Lopinavir from Various Formulations
| S. No. | Parameters | Plain gel | Niosomal dispersion | Niosomal gel | Ethosomal gel |
|---|---|---|---|---|---|
| 1 | % drug releasea | 6.43 ± 0.21 | 18.32 ± 0.18 | 21.24 ± 0.23 | 11.15 ± 0.15 |
| 2 | % skin depositiona | 10.21 ± 0.22 | 22.65 ± 0.35 | 24.45 ± 0.64 | 54.63 ± 0.29 |
| 3 | % drug retained on the surfacea | 83.42 ± 0.77 | 58.35 ± 1.23 | 53.82 ± 0.79 | 33.42 ± 0.45 |
aValues represented as mean ± SD (n = 3)
Fluorescence Microscopic Study
For the purpose of visualizing skin penetration behavior of F3c as well as E5 formulations, coumarin was added to the formulation as a fluorescent marker and the formulations were prepared using the same method as described earlier for F3c gel preparation by replacing the drug with coumarin. Hairs from the dorsal portion of rat’s skin were gently removed using electrical clipper and the formulations were applied on hairless portion of skin. After 6 h, the rats were killed by cervical dislocation and the skin was wiped with cotton wool wetted with PBS to remove any formulation left onto the skin surface. The skin was then removed, embedded in paraffin block and 5-μm thick sections were cut using microtome. These sections were investigated by fluorescence microscopy at 40-fold magnification.
Histopathological Study
F3c and E5 gel were applied on hairless rat’s abdominal skin obtained using the method as described in previous section. After 6 h, the rat was killed and the skin was excised. The excised skin was immediately immersed in 10% buffered formalin, dehydrated in graded concentrations of ethanol, immersed in xylene, and then embedded in paraffin block. The 5-μm thick sections of skin were cut using microtome and were mounted on slide using commercial glycerol’s mounting fluid. The paraffin wax was removed by warming the slide gently, until the wax melted, and then was washed with xylene followed by washings with absolute alcohol and water. The sections were stained with hematoxylin–eosin to determine gross histopathology and collagen deposition, respectively. The slides were analyzed at 40-fold magnification using optical microscope. Phosphate-buffered saline was used as negative control.
In vivo Bioavailability Study
The experimental protocol of the study was approved by the Institutional Animal Ethics Committee of MS University of Baroda and was in accordance with the guidelines of Committee for Purpose of Control and Supervision of Experiments on Animals, Ministry of Social Justice and Empowerment, Government of India. Healthy male Wistar rats weighing around 240–270 g were selected for the study and divided into two groups of six animals each. One group served as control receiving oral lopinavir suspension while another group kept as test group receiving F3c gel topically. For the control group, after overnight fasting the animals were administered with 7.2 mg/ml of lopinavir suspension in distilled water (dose calculated as per USFDA guidelines). For the test group, hairs from abdominal area were removed using electrical clipper followed by application of F3c gel (an equivalent amount containing 7.2 mg of lopinavir) on abdominal skin of anaesthetized rat. Serial blood sampling (0.5 ml) was then done from the tail vein at predetermined time intervals. Plasma was separated by centrifugation at 3,000 rpm, 4°C, for 15 min and 4 ml methanol was added to 200 μl plasma samples for deproteination and for extraction of drug. The mixture was then vortexed for 2 min, followed by centrifugation for 5 min at 3,200 rpm using a tabletop centrifuge (Remi Instruments, Mumbai, India). The organic layer was separated and filtered using 0.2-μm membrane syringe filter. About 20 μl of the filtrate was injected into the HPLC for estimation of lopinavir concentrations. The pharmacokinetic parameters were calculated using Kinetica software (version 4.4).
HPLC Analysis of Lopinavir
For quantitative estimation of lopinavir in samples obtained from ex vivo and in vivo studies, a more sensitive Shimadzu HPLC system equipped with a LC 20AT pump, a SPD-20A UV visible detector, a Thermosil® C-18 column (250 × 4.6 × 10 μ) and a guard column (4.5 mm internal diameter) was used. 10 mM of ammonium acetate buffer (pH 6.0) mixed with acetonitrile in a ratio of 40:60 was used as mobile phase at a flow rate of 1.5 ml/min (for ex vivo study) and at 1.0 ml/min (for in vivo study). Column eluant was monitored at 220 nm as λmax and concentrations of lopinavir were compared against a standard calibration curve of lopinavir in mobile phase. The method was found to obey Beer’s law between concentration range of 100 ng/ml to 20 μg/ml with LOD and LOQ values as 10 and 50 ng/ml, respectively.
Statistical Analysis
Data analysis was carried out using Microsoft Excel (version 2007), and results are expressed as mean ± standard deviation (n = 3 independent samples). Statistical analysis was performed using GraphPad InStat software (version 5.00) using one way ANOVA followed by Tukey’s multiple comparison test with P < 0.05 as a minimal level of significance.
RESULT AND DISCUSSION
Enormous surface area of skin and advantages such as noninvasive nature, bypassing first-pass metabolism, reduced dosing frequency, controlled delivery of medicament, improved patient compliance have established the transdermal route as a better alternative to drugs with limited oral bioavailability. Several investigations demonstrating the potential of various nanoconstructs in combating the barrier nature of SC further raised the interest of researchers in exploring the newer ways to maximize the drug delivery through transdermal route. Among these nanocarriers, niosomes were selected in the present study for their better stability, less toxicity, and cost-effectiveness with an aim to enhance the systemic availability of lopinavir, and the results were compared with ethosomes, lipid vesicles containing a well-known penetration enhancer ethanol.
Formation of uniform, tiny and predominantly spherical systems was ascertained from photomicrographs taken using Olympus BX-40 microscope at a magnification of ×40 before ultrasonication. Scanning electron photomicrograph and polydispersity index of niosomal and ethosomal dispersions further confirmed the formation of smooth surfaced nanoconstructs possessing vesicular characteristics and uniformity in size distribution (Fig. 1a–d).
Fig. 1.
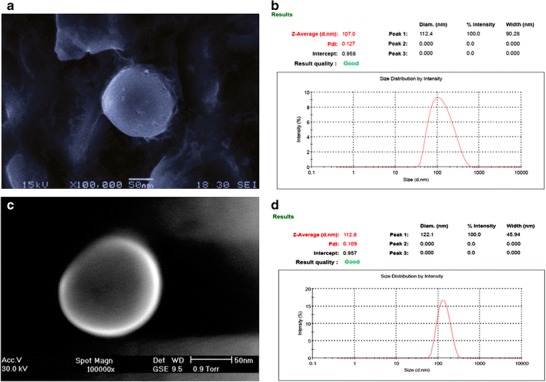
Scanning electron photomicrograph and size distribution graph of lopinavir-loaded F3c (a, b) and E5 dispersion (c, d)
To decide the optimum amount of Span 40 and cholesterol in niosomal formulation, vesicular size and PDE was estimated. As shown in Table I and Fig. 2a, increase in molar fraction of Span 40 resulted in a decreased vesicular size and increased entrapment efficiency of niosomes. The result may be attributed to the saturation of Span 40 bilayers with drug at initial stages causing the precipitation of drug and poor entrapment efficiency (17) which later on improved as the amount of Span 40 was increased. F3c vesicles with a 1:1.5 molar ratio of lopinavir: Span 40 (formulation code, F3) possessed minimum size (196.45 ± 6.23 nm) and maximum entrapment efficiency (58.9 ± 1.98%). A further increase in Span 40 level adversely affected both the parameters. Hence, further optimization of cholesterol content was performed by replacing moles of Span 40 with similar moles of cholesterol while keeping the moles of lopinavir constant. It was observed that increasing the cholesterol level at first reduced the vesicle size and enhanced the drug entrapment capacity to their optimum level (105.22 ± 1.96 nm and 75.5 ± 0.5%, respectively) at a 0.9:0.6 molar ratio of Span 40/cholesterol indicating an improvement in overall bilayer-forming ability of Span 40. Unfavourable results beyond this point led us to consider 0.9:0.6 as an optimum ratio of surfactant-to-cholesterol (formulation code, F3c) to produce vesicles with desirable characteristics. An initial decrease in vesicular size could be an indicative of close packing of surfactant monomers by cholesterol molecule providing suitable molecular geometry and hydrophobicity for bilayer vesicle formation (18). Beyond Span 40/cholesterol molar ratio of 0.9:0.6, a further increase in cholesterol level resulted in increased vesicle size that may be due to disturbance imparted in the vesicular membrane by increased hydrophobicity and thereby formation of larger vesicles with more thermodynamic stability taken place (19). An improvement in entrapment efficiency of lopinavir with increasing cholesterol level may be attributed to a better lipophilic behavior and stability of lipid bilayer (20) with decreased permeability (21) in presence of higher cholesterol content. However, on higher cholesterol level a reduction in entrapment efficiency resulted probably due to the competition between cholesterol and drug for packing space within the bilayer (6).
Fig. 2.
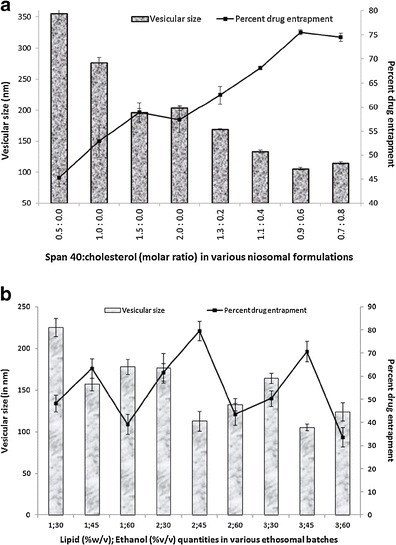
Effect of a Span 40 and cholesterol; b soya PC and ethanol on vesicular size and PDE of lopinavir-loaded niosomal and ethosomal formulations, respectively
As the amount of lipid and ethanol are known to affect vesicular size as well as PDE, the optimization of ethosomal system were based on these parameters and the results obtained are shown in Table II and Fig. 2b. On decreasing the drug-to-lipid ratio by increasing the lipid fraction of bilayer, the vesicular size was found to decrease while the PDE was found to increase probably due to a better packing of drug molecule within lipid bilayer improving the overall geometry and integrity of vesicle membrane. A decrease in PDE at 3% (w/w) lipid level could be a result of low drug-to-lipid ratio causing poor association of lopinavir with bilayer. Increasing concentrations of ethanol also contributed in size reduction as well as PDE enhancement may be owing to its ability to impart fluidity to bilayer (22). However, beyond optimum level, a further increase in ethanol concentration may have rendered the bilayer leaky causing a severe decrease in PDE while slightly increasing the vesicular size. Ethosomal formulation with 2% (w/v) of soya PC and 45% (v/v) of ethanol had maximum drug entrapment with desirable size and was considered to be the optimized composition.
The stability study of F3c and E5 dispersions were conducted by subjecting the formulations to aging for a period of 2 months at two different environmental conditions and observing the changes in vesicular size and drug retentive potential were selected as key indicators for stability. As shown in Table IV, the storage of F3c formulations at 4 ± 1°C/ambient RH for 2 months did not affect the mean vesicular size (115.31 ± 1.87 nm) as well as percent drug retention (91.18 ± 0.759) significantly while the formulations stored at 30 ± 2°C/65 ± 5% RH have managed to retain only 39.32 ± 0.346% of the drug with a significant rise in mean vesicular size (186.51 ± 2.66 nm) at the end of 2 months. During 2 months of storage, a significant rise in mean vesicular size of ethosomes along with a significant fall in percentage of drug retained within these systems, even at 4 ± 1°C/ambient RH, clearly indicated the poor stability of lipidic systems when compared with niosomal carriers. The results provided compelling evidence of better stability profile of niosomes over ethosomal systems and supported 4 ± 1°C/ambient RH as an optimal storage condition for carriers under study as compared with storage condition of 30 ± 2°C/65 ± 5%RH.
Table IV.
Stability Study of Optimized Niosomal (F3c) and Ethosomal (E5) Dispersions
| S. No. | Formulation | Time (in months) | Vesicular sizea (nm) | % drug retaineda | ||
|---|---|---|---|---|---|---|
| 2–8°C | 30 ± 2°C | 2–8°C | 30 ± 2°C | |||
| 1 | F3c dispersion | 0 | 105.76 ± 1.24 | 105.76 ± 1.24 | 100 | 100 |
| 2 | 1 | 109.24 ± 2.16 | 138.18 ± 3.75 | 96.23 ± 0.458 | 54.43 ± 0.538 | |
| 3 | 2 | 115.31 ± 1.87 | 186.51 ± 2.66 | 91.18 ± 0.759 | 39.32 ± 0.346 | |
| 4 | E5 dispersion | 0 | 113.3 ± 5.43 | 113.3 ± 5.43 | 100 | 100 |
| 5 | 1 | 143.6 ± 1.09 | 198.72 ± 2.13 | 85.79 ± 2.983 | 43.12 ± 5.789 | |
| 6 | 2 | 178.2 ± 6.83 | 301.68 ± 7.69 | 69.52 ± 4.191 | 23.73 ± 2.443 | |
aValues represented as mean ± SD (n = 3)
In order to understand the ability of the F3c gel to aid in lopinavir permeation through the skin and also to study its skin deposition and toxicity, ex vivo studies were conducted and compared with the conventional plain drug gel, F3c dispersion and E5 gel.
Skin permeation profiles of lopinavir under nonocclusive condition was studied from plain gel, F3c dispersion, F3c gel, and E5 gel using rat’s abdominal skin ex vivo. Interesting results were observed when ex vivo skin deposition and release pattern of lopinavir via F3c gel was compared with E5 gel (Fig. 3; Table III). In terms of overall skin permeation (including percent deposited within skin and percent released into diffusion media), E5 gel appeared to be more proficient than F3c gel (Table III). However, it is also evident that major fraction of lopinavir delivered via E5 gel remained deposited within the skin (54.63 ± 0.29) while lopinavir in F3c gel efficiently delivered deeper into the skin and released 21.24 ± 0.23% of drug in 24 h, an amount significantly greater than that released via E5 gel (11.15 ± 0.15%). A less skin deposition and better permeation of F3c carriers across more hydrophilic dermal region may be attributed to greater hydrophilicity of niosomes as compared with ethosomal carriers. The percent release and skin deposition of lopinavir obtained from F3c gel was better than that obtained from F3c dispersion (18.32 ± 0.18%). This may be due to sufficient porosity of gel matrix as well as the occlusive condition provided by the gel that helps in holding the water and thus keeping the hydration level of skin at a higher level for a longer duration as compared with dispersed system where water starts evaporating at a higher rate soon after applying the formulation (23). Highest amount of drug retained on the surface was observed in case of plain drug loaded gels (83.42 ± 0.77) indicating the limited permeability of the drug alone across the SC layer that may be attributed to its moderately high molecular weight.
Fig. 3.
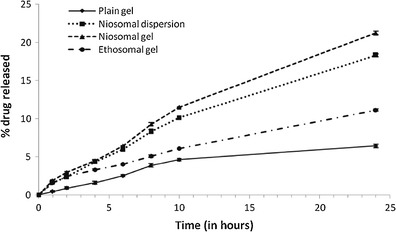
Cumulative percentage of drug release via full-thickness rat abdominal skin versus time profile of various lopinavir formulations
Figure 4a, b represents the fluorescence microscopic images of rat skin sections after 6 h application of coumarin-loaded E5 gel and F3c gel, respectively. Relatively high fluorescence intensity is evident in cases of ethosomes demonstrating a better skin permeation profile of ethosomes over niosomes. However, the presence of fluorescence at greater skin depths in case of niosomes suggests its potential as a better transdermal drug delivery module over ethosomes. The deeper penetration of niosomes than ethosomes could be because of the fact that sub-epidermal tissues contain a higher proportion of water supporting the permeation of more hydrophilic molecules across it while causing difficulty in passage of molecules with relatively high lipophilicity.
Fig. 4.
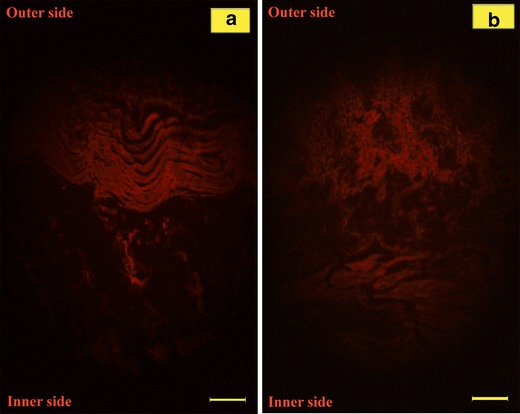
Photomicrograph showing deposition of fluorescence probe coumarin within rat skin after 6 h via a E5 gel and b F3c gel. Scale bar, 100 μm
The clinical usefulness of prepared F3c gel was further examined by staining rat’s skin, previously treated with formulations, using hematoxylin–eosin, and microscopic investigation was performed to observe any pathological changes as a sign of skin irritation or toxicity. The histopathological images were then compared with the images of skin treated with PBS used as negative control (Fig. 5). On comparing the extent of epidermal and subepidermal cellular damage or presence of prominent blood vessels, the formulations can be arranged in an order of PBS < F3c gel < E5 gel representing niosomes as a better drug delivery tool than ethosomes with respect to skin irritancy. Hence, it can be stated that the result provided an undeniable evidence of better safety profile of Span 40 over ethanol
Fig. 5.
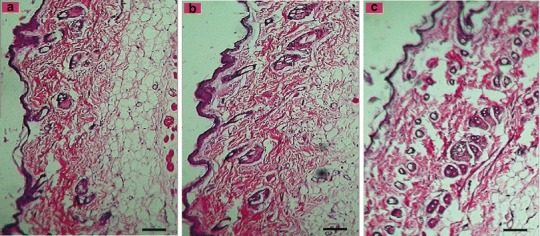
Photomicrograph of hematoxylin–eosin stained rat skin for histopathological evaluation of a PBS as negative control, b F3c gel and c E5 gel. Scale bar, 50 μm
To demonstrate the effect of route of administration as well as F3c gel on lopinavir bioavailability, in vivo bioavailability study in a group of six male Wistar rats after single transdermal application of F3c gel was performed and compared with orally administered lopinavir suspension to another group of six male Wistar rats. The plasma drug concentration (PDC) at various time points were estimated and various important pharmacokinetic parameters (such as peak plasma concentration (Cmax), peak time (Tmax), and complete area under the PDC versus time curve (AUC0→∞)) useful in establishing the overall bioavailability of lopinavir from its formulation were calculated. As represented in Table V and Fig. 6, the significant differences in Cmax (7.9 and 5.8 μg/ml, respectively) and AUC0→∞ value (72.87 and 52.94 h μg/ml, respectively) of transdermally and orally administered lopinavir evidently advocate the enhanced bioavailability via F3c gel. The results were in accordance with the two facts that (a) extensive presystemic metabolism observed in oral drug administration can be bypassed by selecting transdermal route and (b) F3c carriers can efficiently carry the drug across skin layers.
Table V.
In vivo Pharmacokinetic Parameters of Lopinavir-Loaded Niosomal (F3c) Gel Compared with Its Oral Suspension (OS)
| Formulations | Pharmacokinetic parameters | |||
|---|---|---|---|---|
| C max a (μg/ml) | T max (h) | AUC0→12 (h × μg/ml) | AUC0→∞ (h × μg/ml) | |
| OS | 5.8 ± 0.2 | 4 | 16.19 | 52.94 |
| F3c gel | 7.9 ± 0.2 | 4 | 24.34 | 72.87 |
C max peak plasma concentration, T max peak time, AUC 0→∞ complete area under the PDC versus time curve
aValues represented as mean ± SD (n = 6)
Fig. 6.
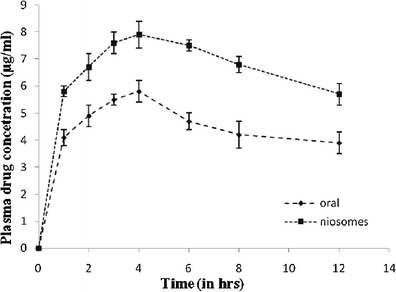
PDC-time profile of lopinavir achieved via F3c gel and OS
CONCLUSIONS
Lopinavir, highly potent protease inhibitor used in the treatment of AIDS, needs to be co-administered with ritonavir because of its high presystemic metabolism resulting in poor systemic bioavailability. In the current study, an attempt was made to develop niosomal gel for improved systemic availability of lopinavir via transdermal route. As suggested by the results, niosomes with a 1:0.9:0.6 molar ratio of lopinavir, Span 40, and cholesterol, respectively possess optimum characteristics with respect to vesicular size and percentage of entrapment efficiency along with sufficient stability at 4 ± 1°C/ambient RH. Ex vivo skin permeation studies showing 18.32 ± 0.18% of drug release in 24 h suggested a better systemic availability of lopinavir via F3c gel as compared with E5 gel showing significantly less drug release in same duration (11.15 ± 0.15%). Presence of fluorescence throughout the skin thickness as well as insignificant alteration in skin histopathology further aided in considering niosomes as better permeating and non-irritant transdermal drug delivery tool in comparison to ethosomes. Systemic availability of lopinavir at a better rate (Cmax, 7.9 μg/ml) and to a greater extent (AUC0→∞, 72.87 h μg/ml) was also observed with transdermally administered F3c gel during in vivo studies. Encouraging results of niosomal stability, skin penetrability, non-irritancy and ability to improve overall bioavailability of lopinavir have led us to conclude that niosomal gel holds a great potential as a novel transdermal nanocarriers in safely carrying lopinavir to systemic circulation and thus surmounting the need of adding Ritonavir in formulations to hoist the lopinavir bioavailability.
References
- 1.Ojewole E, Mackraj I, Naidoo P, Govender T. Exploring the use of novel drug delivery systems for antiretroviral drugs. Eur J Pharm Biopharm. 2008;70(3):697–710. doi: 10.1016/j.ejpb.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 2.WHO/UNAIDS. UNAIDS report on the global AIDS epidemic. Switzerland; 2010.
- 3.Abbott L. Clinical Pharmacology and Biopharmaceutics review of Kaletra oral solution (NDA#021251). U.S. Food and Drug Administration (updated 20 November 2001). Available from: http://www.accessdata.fda.gov/drugsatfda_docs/nda/2000/21-226_Kaletra_biopharmr_P1.pdf. Accessed 23 August 2012
- 4.ter Heine R, Van Waterschoot RA, Keizer RJ, Beijnen JH, Schinkel AH, Huitema AD. An integrated pharmacokinetic model for the influence of CYP3A4 expression on the in vivo disposition of lopinavir and its modulation by ritonavir. J Pharm Sci. 2011;100(6):2508–15. doi: 10.1002/jps.22457. [DOI] [PubMed] [Google Scholar]
- 5.Attia IA, El-Gizawy SA, Fouda MA, Donia AM. Influence of a niosomal formulation on the oral bioavailability of acyclovir in rabbits. AAPS PharmSciTech. 2007;8(4):E106. doi: 10.1208/pt0804106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balakrishnan P, Shanmugam S, Lee WS, Lee WM, Kim JO, Oh DH, et al. Formulation and in vitro assessment of minoxidil niosomes for enhanced skin delivery. Int J Pharm. 2009;377(1–2):1–8. doi: 10.1016/j.ijpharm.2009.04.020. [DOI] [PubMed] [Google Scholar]
- 7.Cable C. An examination of the effects of surface modifications on the physicochemical and biological properties of non-ionic surfactant vesicles. Ph.D. thesis, Glasgow University of Strathclyde; 1989.
- 8.Jadon PS, Gajbhiye V, Jadon RS, Gajbhiye KR, Ganesh N. Enhanced oral bioavailability of griseofulvin via niosomes. AAPS PharmSciTech. 2009;10(4):1186–92. doi: 10.1208/s12249-009-9325-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solanki AB, Parikh JR, Parikh RH, Patel MR. Evaluation of different compositions of niosomes to optimize aceclofenac transdermal delivery. Asian J Pharm Sci. 2010;5(3):87–95. [Google Scholar]
- 10.Kushla GP, Zatz JL, Mills OH, Jr, Berger RS. Noninvasive assessment of anesthetic activity of topical lidocaine formulations. J Pharm Sci. 1993;82(11):1118–22. doi: 10.1002/jps.2600821110. [DOI] [PubMed] [Google Scholar]
- 11.Godin B, Touitou E. Ethosomes: new prospects in transdermal delivery. Crit Rev Ther Drug Carrier Syst. 2003;20(1):63–102. doi: 10.1615/CritRevTherDrugCarrierSyst.v20.i1.20. [DOI] [PubMed] [Google Scholar]
- 12.Agarwal R, Katare OP, Vyas SP. Preparation and in vitro evaluation of liposomal/niosomal delivery systems for antipsoriatic drug dithranol. Int J Pharm. 2001;228(1–2):43–52. doi: 10.1016/S0378-5173(01)00810-9. [DOI] [PubMed] [Google Scholar]
- 13.Karki R, Mamatha GC, Subramanya G, Udupa N. Preparation, characterization and tissue disposition of niosomes containing isoniazid. Rasayan J Chem. 2008;1(2):224–7. [Google Scholar]
- 14.Verma DD, Verma S, Blume G, Fahr A. Particle size of liposomes influences dermal delivery of substances into skin. Int J Pharm. 2003;258(1–2):141–51. doi: 10.1016/S0378-5173(03)00183-2. [DOI] [PubMed] [Google Scholar]
- 15.Touitou E, Dayan N, Bergelson L, Godin B, Eliaz M. Ethosomes—novel vesicular carriers for enhanced delivery: characterization and skin penetration properties. J Control Release. 2000;65(3):403–18. doi: 10.1016/S0168-3659(99)00222-9. [DOI] [PubMed] [Google Scholar]
- 16.Bhalaria MK, Naik S, Misra AN. Ethosomes: a novel delivery system for antifungal drugs in the treatment of topical fungal diseases. Indian J Exp Biol. 2009;47(5):368–75. [PubMed] [Google Scholar]
- 17.Mokhtar M, Sammour OA, Hammad MA, Megrab NA. Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int J Pharm. 2008;361(1–2):104–11. doi: 10.1016/j.ijpharm.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 18.Manosroi A, Wongtrakul P, Manosroi J, Sakai H, Sugawara F, Yuasa M, et al. Characterization of vesicles prepared with various non-ionic surfactants mixed with cholesterol. Colloids Surf B. 2003;30(1):129–38. doi: 10.1016/S0927-7765(03)00080-8. [DOI] [Google Scholar]
- 19.Essa EA. Effect of formulation and processing variables on the particle size of sorbitan monopalmitate niosomes. Asian J Pharm. 2010;4(4):227–33. doi: 10.4103/0973-8398.76752. [DOI] [Google Scholar]
- 20.Bernsdorff C, Wolf A, Winter R, Gratton E. Effect of hydrostatic pressure on water penetration and rotational dynamics in phospholipid–cholesterol bilayers. Biophys J. 1997;72(3):1264–77. doi: 10.1016/S0006-3495(97)78773-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirby C, Clarke J, Gregoriadis G. Effect of the cholesterol content of small unilamellar liposomes on their stability in vivo and in vitro. Biochem J. 1980;186(2):591–8. doi: 10.1042/bj1860591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paolino D, Lucania G, Mardente D, Alhaique F, Fresta M. Ethosomes for skin delivery of ammonium glycyrrhizinate: in vitro percutaneous permeation through human skin and in vivo anti-inflammatory activity on human volunteers. J Control Release. 2005;106:99–110. doi: 10.1016/j.jconrel.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 23.Morgan CJ, Renwick AG, Friedmann PS. The role of stratum corneum and dermal microvascular perfusion in penetration and tissue levels of water-soluble drugs investigated by microdialysis. Br J Dermatol. 2003;148(3):434–43. doi: 10.1046/j.1365-2133.2003.05163.x. [DOI] [PubMed] [Google Scholar]