

Abstract
Absorption of drugs from the oral cavity into the mucosal tissues is typically a fast event. Dissolved drugs partition into the mucosal membranes and within minutes will reach equilibrium with drug in solution in the oral cavity. However, this does not always equate to rapid drug appearance in the systemic circulation. This has been attributed to slow partitioning out of the mucosal tissues and into the systemic circulation. Based on information from literature, physicochemical properties of asenapine, and clinical data, we conclude that for sublingually administered asenapine, the exposure is primarily a function of rapid partitioning into the mucosal membranes. This is followed by slow partitioning out of the mucosal tissues and into the systemic circulation, leading to a Tmax value of about 1 h. The bioavailability of asenapine at doses below the saturation solubility in the mouth does not change and is controlled primarily by mass transport equilibrium. At doses above the saturation solubility, the bioavailability becomes more dependent not only on the distribution equilibrium but also on contact time in the mouth because additional variables (e.g. dissolution rate of the drug) need to be accounted for. These explanations are consistent with oral cavity absorption models from the literature and can be used to accurately describe the clinical data for asenapine.
KEY WORDS: asenapine, exposure, oral mucosal absorption, Tmax
INTRODUCTION
Oral mucosal (e.g. sub- or supralingual or buccal) administration of drugs is often the route of administration of choice when the drug shows a large first-pass effect after oral delivery. Systemic exposure of drugs after oral mucosal administration is often expected to be a route of administration with a fast onset of action. This expectation is in line with the fact that the time for absorption of drugs into the oral mucosal membranes is typically short, and the residence time of a liquid in the oral cavity is short, typically in the order of 5–10 min (1). However, rapid clearance of drug from the oral cavity does not necessarily lead to rapid systemic exposure. While there are classic examples such as nitroglycerin which do display a short time for maximum plasma concentration (Tmax) following sublingual administration, there are also examples such as lorazepam which display a (Tmax) of almost 2 h (1,2).
When comparing the residence time of the drug in the oral cavity with the rate of absorption that is derived from the plasma concentration versus time profile, a clear discrepancy becomes apparent: the rate of clearance from the oral cavity is often a much faster process than the rate at which the drug becomes visible in the systemic circulation. This aspect has been considered in a number of models that has been proposed to characterize the transport of drug from the oral cavity into the systemic circulation. More detailed descriptions of these different models are available in literature (3–11). The intent of this article is to describe a model for oral mucosal absorption that is based on time constants and to highlight aspects that relate to the bioperformance of a sublingually administered product, asenapine.
MATERIALS AND METHODS
Asenapine is an atypical antipsychotic with high affinity for serotonergic, α-adrenergic, dopaminergic, and histaminic receptors but minimal affinity for muscarinic receptors (12). Asenapine is indicated in the USA in adults for the acute treatment of schizophrenia and in the USA and Europe for treatment of adults with manic or mixed episodes associated with bipolar I disorder with or without psychotic features. A sublingual formulation was developed. Several strengths (0.01–20 mg) of asenapine maleate sublingual tablets have been manufactured and clinically evaluated using the Zydis® (Catalent Pharma Solutions, Somerset, NJ, USA) rapidly disintegrating freeze-dried technology.
All clinical trials described in this article were conducted in compliance with the current revisions of the Declaration of Helsinki, International Conference on Harmonization guidelines, Good Clinical Practice, and existing regulatory guidelines.
Physical properties of the relevant polymorphic form of asenapine maleate are depicted in Table I. Solubilities were determined by addition of an overage of the compound to the solvent of choice. Glass beads with a diameter of 2 mm were added to this suspension at a volume ratio of about 1:1. The samples were rolled overnight on a roller mixer and maintained at the specified temperature. After stirring overnight, the samples were inspected to ensure that solid API still remained. The samples were centrifuged and the supernatant was assayed using reverse phase HPLC to determine the drug solubility. The column was Waters Symmetry C18 with a particle size of 5 μm. The mobile phase was an aqueous phosphate buffer containing 0.5% triethylamine mixed with acetonitrile at a 52:48 volumetric ratio.
Table I.
Relevant Physicochemical Properties of Asenapine
| Property | Value | Source |
|---|---|---|
| Molecular weight of free base (salt) | 285.8 (401.8) g/mole | (13) |
| Melting point | 141–145°C | (13) |
| Solubility in water | ||
| Room temperature | 3 mg/mL | (13) |
| 37°C | 5.4 mg/mL | This study |
| Solubility in saliva (room temperature) | 3 mg/mL | This study |
| Intrinsic dissolution rate in water | 123 mg/m2s | This study |
| pKa of free base | 8.51 | (13) |
| pKa of maleic acid | pK1 < 3 | (13) |
| pK2 = 7.52 | ||
| Log P of free base | 6.33 | (13) |
| Caco-2 cell permeability | 0.9–2.3⋅10−5 cm/s | (14) |
For determination of the intrinsic dissolution rate, a flat-faced compact with a diameter of 5 mm was produced in a holder that was placed in a USP type 2 dissolution tester filled with 500 mL of degassed water of 37°C. The paddle rotated at 150 rpm. Samples were taken at 1-min intervals and analyzed on content by HPLC as described.
RESULTS AND DISCUSSION
Mechanistic Model for Oral Mucosal Absorption Based on Literature
Of the models proposed to study the transport kinetics from the oral cavity into the systemic circulation, the commonality between the majority of the models is an equilibrium term of drug concentration between the oral cavity and the mucosal membranes (3–11). The equilibrium term (or pseudo-equilibrium term owing to the relatively short contact time in the mouth) implies that a fixed fraction of the drug in solution will be transported into the mucosal membranes. This transport will occur until the equilibrium term is reached in the time frame that an oral solution can be held in the mouth before swallowing the dose. This equilibrium term is a distribution coefficient and thus will largely be independent of drug concentration in saliva. Fast transport of mass in solution in the mouth into the mucosal membranes does not imply that drug is immediately available to the systemic circulation. This is because the rate-limiting step can be transport from the mucosal membranes into the systemic circulation.
The considerations so far lead to a model that describes the fate of a drug starting as a drug product in the mouth and ending with drug in the systemic circulation. This model is depicted in Fig. 1, which describes the drug transport via a series of rate constants. The ratio of k2 to k−2 represents solubility of the drug in the mouth and the ratio of k3 to k−3 represents the sublingual distribution equilibrium described in the previous section.
Fig. 1.
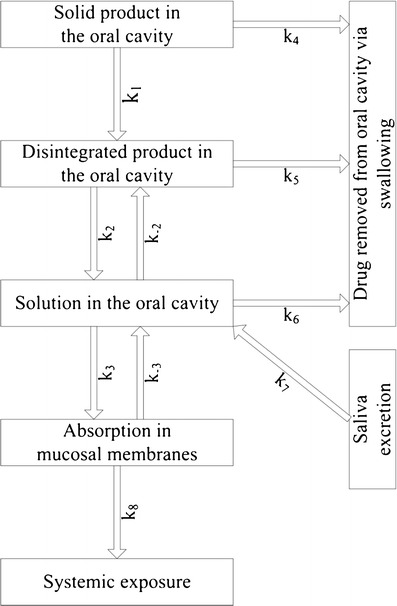
Oral mucosal absorption as a sequence of rate processes
The mucosal membranes can act as a storage compartment for drug, extracting drug from the oral cavity until either the distribution equilibrium has been reached. The drug stored in the mucosal membranes will slowly diffuse out into the systemic circulation, with rate constant k8. The implications of the model will be discussed based on results from studies using asenapine fast-disintegrating tablets.
Application of Model to Asenapine Fast-Disintegrating Tablets
The model in the previous section will be used to support the behavior of asenapine.
Swallowing Water After Sublingual Dose Administration Shows Minor Impact on the Pharmacokinetic Profiles
Time of Maximum Plasma Concentration
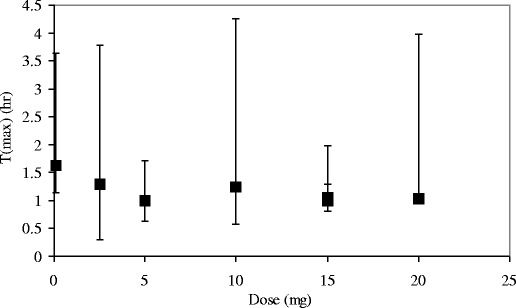
One could argue that the delayed time of maximum plasma concentration (Tmax) is simply due to prolonged residence time in the mouth by the drug product behaving as a pseudo-controlled-release system (e.g. forming a film or trapping solid particles). The results presented in Fig. 2 shows that drinking water even 2 min after sublingual administration had no visible effect on Tmax, although variations in Tmax are always large. Drinking water effectively removes drug from the oral cavity and prevents further absorption of drug by the mucosal membranes.
Fig. 2.

T max, relative C max, and AUC0-24 values after different residence times of asenapine in the oral cavity. A residence time of 30 min is taken as a reference for the relative value of C max and AUC0-24. The error bars indicate the minimum and maximum values for T max (data from (15))
The independence of drug residence time in the oral cavity on Tmax data makes clear that the time constants related to absorption of the drug into the mucosal membranes are not the controlling factors in relation to Tmax (i.e. k1 to k7 in Fig. 1). Rather, the release rate of drug from the membranes (k8) determines the Tmax values. This is consistent with relatively fast partitioning established between drug in solution in saliva and drug in the mouth tissues.
Area Under the Concentration Versus Time Curve and Maximum Plasma Concentration
Both the area under the concentration versus time curve (AUC) and the maximum plasma concentration are highly independent on the residence time of the (dissolved) drug in the oral cavity (Fig. 2). Even drinking water after only 2 min led to a modest reduction in AUC or Cmax of around 80% of the maximum values. The effects of drinking water after 5, 10, and 30 min were also investigated. Drinking water after 5 min showed an approximately 10% reduction of AUC and there was no change in bioavailability after longer times (15).
The equilibrium between drug in solution and drug in the tissue is reached fast and leads to fairly constant exposures. Fitting the buccal absorption data to a first-order equation for transport from drug in solution in the mouth transported to drug in the oral mucosa, an absorption half-life of around 0.87 min (52 s) can be approximated. The variation in Tmax values is typically large (Fig. 2). The shortest Tmax observed during any trial was 0.33 h (20 min), which is more than 20 times the absorption half-life. This underpins that Tmax values are unrelated with absorption.
The conclusion that drug absorption by the oral mucosal membranes is a fast process, while release of drug by these membranes has at least two implications for asenapine: (1) bioavailability is a result of a partitioning phenomenon which is independent on the dose and (2) Tmax does not depend on dose either. These aspects will be discussed further.
The Bioavailability After Sublingual Administration Does Not Change Significantly with Increasing Dose Until Saturation Levels in Saliva Have Been Reached
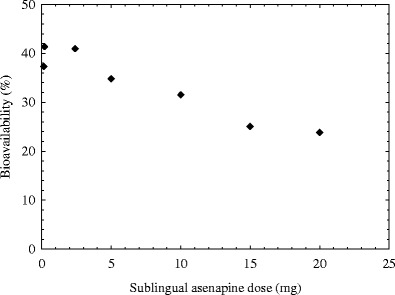
The bioavailability of a 5-mg sublingually administered dose is around 35%. The bioavailability values of other doses were calculated from clinical data (Fig. 3). The bioavailability is more or less constant at low doses and goes down when the doses exceed around 5 mg. In the non-simulated state around 1 ml saliva is present in the oral cavity (16,17). So, the dose to reach saliva saturation is about 5.4 mg. The observation that higher bioavailability is not observed at doses below the saturation solubility and that bioavailability is constant across this dose range is consistent with a rapid mass transport equilibrium being established between drug in solution in the mouth and drug in the mucosal membranes (Fig. 1). Once the mass transport equilibrium has been reached, no further drug absorption into the sublingual membranes is expected unless a shift occurs in the mass balance (e.g., sufficient drug is transported from the mucosal membranes and into the systemic circulation in the relatively short time the drug is in contact with the oral cavity). At this equilibrium point, the remaining dose, which cannot be absorbed into the mucosal tissues, will simply be swallowed over time.
Fig. 3.
Bioavailability of asenapine sublingual tablets as a function of dose
At doses below the saturation solubility, the entire dose rapidly goes into solution. The distribution equilibrium between the mass of drug in solution in the mouth and mass of drug in the mucosal tissues is reached rapidly after which point no further drug partitioning into the mucosal tissues occurs. The majority of remaining drug will not be absorbed by the mucosal tissues at this point and will be swallowed.
More variables need to be accounted for when doses exceed the saturation solubility. A dose that will not fully dissolve (e.g., 10 mg), will go into solution until the saturation solubility point is reached. From the saturated solution in the mouth, solubilized drug in the oral cavity is transported into the mucosal tissues. This lowers the concentration in the mouth below the saturation solubility point, and because solid drug is still available, the concentration in the mouth will increase until the saturation point is once again achieved. This shifts the distribution equilibrium allowing more drug to partition into the mucosal membranes. The cycle of dissolution followed by partitioning into the membranes continues until either all the solid drug remaining in the mouth is exhausted or until the solution and solids are swallowed.
Based on the mass transport model, we can conclude that the bioavailability at doses below the saturation solubility will be most heavily dictated by the distribution coefficient. This is consistent with clinical data shown in Fig. 4. Unless changes to the drug substance or drug product change the (thermodynamic) activity of the drug solution in the mouth, which would shift the distribution coefficient, the bioavailability at doses below the saturation solubility is relatively constant. At doses above the saturation solubility, the bioavailability becomes a function of not only the distribution coefficient but also on other factors such as contact time in the mouth.
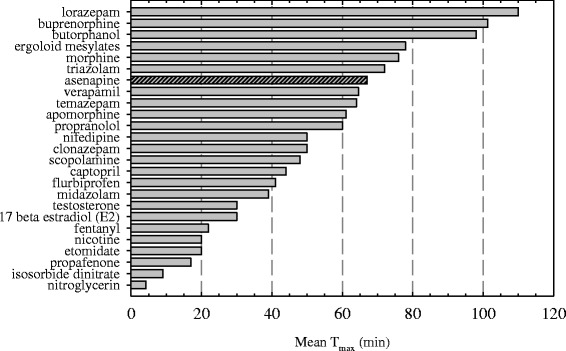
Fig. 4.
Time to reach maximum exposure (T max) as a function of dose. The error bars indicate the minimum and maximum values
The Tmax is Independent of Dose
Clinical data from asenapine sublingual tablets ranging in dose from 0.1 to 20 mg (Fig. 4) all demonstrate similar Tmax values of approximately 1 h. It is relevant to note that based on the solubility of asenapine in saliva at a temperature of 37°C (Table I), low doses such as the 0.1-mg sublingual dose would completely dissolve after administration leading to a rapid equilibrium with the mouth tissues. Therefore, if any delayed Tmax effect was coming from dissolution or solubility, one would have expected to have seen a difference across the dose range tested.
The Tmax Values of Asenapine Are in with the Range of Historical Literature Data for Other Sublingually Administered Drugs
To verify the previous conclusion, a survey of the clinical pharmacokinetic literature for studies in which drugs were administered to the oral cavity was conducted to put into context the Tmax value for asenapine. This is shown in Fig. 5, where a plot of drugs administered to the oral cavity versus the mean Tmax is shown. The data were collected from a survey of the literature (see Table II for tabular summary and references) with a careful focus on obtaining pharmacokinetic data that specifically cited administration to the oral cavity via a sublingual tablet or solution alone and prevented gastro-intestinal absorption. Dosage forms were excluded if they were orally disintegrating tablets that were not intended for absorption via the oral cavity. For compounds with multiple Tmax values reported in the literature or multiple Tmax values reported for different dosage strengths, the Tmax values were averaged and reported in the plot. Figure 5 and Table II show that the Tmax values range from 4.2 to 110 min. Sublingually administered asenapine displays a Tmax value of approximately 67 min, which falls well within the range of reported Tmax values for other compounds. This observation supports the conclusion that absorption and Tmax are independent parameters during oral mucosal absorption.
Fig. 5.
Range of reported literature T max values for sublingually administered dosage forms. Table II lists the references to the T max values of the compounds
Table II.
Summary of Clinical Pharmacokinetic T max Values for Sublingually Administered Drugs (Graphically Presented in Fig. 5)
| Compound | Mean T max (min) | Sources |
|---|---|---|
| Nitroglycerin | 4.2 | (18) |
| Isosorbide dinitrate | 9 | (19) |
| Propafenone | 17 | (20,21) |
| Etomidate | 20 | (21) |
| Nicotine | 20 | (21) |
| Fentanyl | 22 | (21) |
| 17 Beta estradiol (E2) | 30 | (21–23) |
| Testosterone | 30 | (21) |
| Midazolam | 39 | (21) |
| Flurbiprofen | 41 | (24) |
| Captopril | 44 | (21,25) |
| Scopolamine | 48 | (21) |
| Clonazepam | 50 | (21) |
| Nifedipine | 50 | (25) |
| Propranolol | 60 | (26) |
| Apomorphine | 61.1 | (27) |
| Temazepam | 64 | (25) |
| Verapamil | 64.6 | (21,28) |
| Asenapine | 67 | (29) |
| Triazolam | 72 | (21,30,31) |
| Morphine | 76 | (21,25,32) |
| Ergoloid mesylates | 78 | (33) |
| Butorphanol | 98 | (34) |
| Buprenorphine | 101.3 | (21,25,35) |
| Lorazepam | 110 | (1,2) |
CONCLUSION
The observed Tmax of ~1 h following sublingual administration of asenapine is shown to be consistent with the reported range of Tmax values following sublingual administration of other compounds found in the literature. It is also in line with a proposed model in which drug rapidly partitions into the mucosal membranes, where it is stored for extended periods and then slowly partitions out of this lipid tissue and into the systemic circulation. The bioavailability of a sublingually administered drug at doses below the saturation solubility in the mouth is constant and controlled primarily by a mass transport equilibrium. At doses above the saturation solubility, the bioavailability becomes more dependent not only on the distribution equilibrium but also on contact time in the mouth because additional variables need to be accounted for (e.g., dissolution of excess drug and re-establishing the distribution equilibrium). These explanations were shown to be consistent with oral cavity absorption models from the literature as well as the current clinical data for asenapine.
ACKNOWLEDGMENTS
The input and support from many colleagues within Pfizer and legacy Organon is highly appreciated by JB and KM.
Contributor Information
Jeremy A. Bartlett, Email: jeremy.a.bartlett@pfizer.com
Kees van der Voort Maarschalk, Email: k.van.der.voort.maarschalk@rug.nl.
REFERENCES
- 1.Wilson CG, Washington NC, Peach J, Murray GR, Kennerley J. The behaviour of fast-dissolving dosage from (Expidet) followed by γ-scintigraphy. Int J Pharm. 1987;40:119–123. doi: 10.1016/0378-5173(87)90056-1. [DOI] [Google Scholar]
- 2.Greenblatt DJ, Divoll M, Harmatz JS, Shader RI. Pharmacokinetic comparison of sublingual lorazepam with intravenous, intramuscular, and oral lorazepam. J Pharm Sci. 1982;71:248–252. doi: 10.1002/jps.2600710227. [DOI] [PubMed] [Google Scholar]
- 3.Beckett AH, Triggs EJ. Buccal absorption of basic drugs and its application as an in vivo model of passive drug transfer through lipid membranes. J Pharm Pharmacol. 1967;19:31S–41S. [PubMed] [Google Scholar]
- 4.Wagner JG, Sedman AJ. Quantitation of rate of gastrointestinal and buccal absorption of acidic and basic drugs based on extraction theory. J Pharmacokin Biopharm. 1973;1:23–50. [Google Scholar]
- 5.Beckett AH, Pickup ME. Model for steroid transport across biological membranes. J Pharm Pharmacol. 1975;27:226–234. doi: 10.1111/j.2042-7158.1975.tb10691.x. [DOI] [PubMed] [Google Scholar]
- 6.Schurmann W, Turner P. A membrane model of the human oral mucosa as derived from buccal absorption performance and physicochemical properties of the beta-blocking drugs atenolol and propranolol. J Pharm Pharmacol. 1978;30:137–147. doi: 10.1111/j.2042-7158.1978.tb13185.x. [DOI] [PubMed] [Google Scholar]
- 7.Henry JA, Ohashi K, Wadsworth J, Turner P. Drug recovery following buccal absorption of propranolol. Br J Clin Pharmacol. 1980;10:61–65. doi: 10.1111/j.1365-2125.1980.tb00502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tucker IG. A method to study the kinetics of oral mucosal drug absorption from solutions. J Pharm Pharmacol. 1988;40:679–683. doi: 10.1111/j.2042-7158.1988.tb06994.x. [DOI] [PubMed] [Google Scholar]
- 9.Rathbone MJ. Human buccal absorption. I. A method for estimating the transfer kinetics of drugs across the human buccal membrane. Int J Pharm. 1991;69:103–108. doi: 10.1016/0378-5173(91)90215-A. [DOI] [Google Scholar]
- 10.Rathbone MJ. Human buccal absorption. II. A comparative study of the buccal absorption of some parahydroxybenzoic acid derivatives using the buccal absorption test and a buccal perfusion cell. Int J Pharm. 1991;74:189–194. doi: 10.1016/0378-5173(91)90236-H. [DOI] [Google Scholar]
- 11.Rathbone MJ, Hadgraft J. Absorption of drugsfrom the human oral cavity. Int J Pharm. 1991;74:9–24. doi: 10.1016/0378-5173(91)90403-B. [DOI] [Google Scholar]
- 12.Shahid M, Walker GB, Zorn SH, Wong EH. Asenapine: a novel psychopharmacologic agent with a unique human receptor signature. J Psychopharmacol (Oxf) 2009;23:65–73. doi: 10.1177/0269881107082944. [DOI] [PubMed] [Google Scholar]
- 13.Funke CW, Hindriks H, Sam AP. Physico-chemical properties and stability of trans-5-chloro-2-methyl-2,3,3a,12b-tetrahydro-1 H- dibenz[2,3:6,7]oxepino[4,5-c]pyrrolidine maleate. Arzneimittelforschung. 1990;40:536–539. [PubMed] [Google Scholar]
- 14.Faassen F, Vogel G, Spanings H, Vromans H. Caco-2 permeability, P-glycoprotein transport ratios and brain penetration of heterocyclic drugs. Int J Pharm. 2003;263:113–122. doi: 10.1016/S0378-5173(03)00372-7. [DOI] [PubMed] [Google Scholar]
- 15.Hulskotte EGJ, Spaans E, Timmer CJ, Schrodter A, Machielsen CSM, Schnabel PG, et al. Effects of water intake and smoking on absorption of sublingually administered asenapine. Clin Pharmacol Ther. 2009;85:S86. doi: 10.1038/clpt.2008.224. [DOI] [Google Scholar]
- 16.Squier CA, Wertz PW. Permeability and the pathophysiology of oral mucosa. Adv Drug Deliv Rev. 1993;12:13–24. doi: 10.1016/0169-409X(93)90038-6. [DOI] [Google Scholar]
- 17.Lagerlof F, Dawes C. The volume of saliva in the mouth before and after swallowing. J Dent Res. 1984;63:618–621. doi: 10.1177/00220345840630050201. [DOI] [PubMed] [Google Scholar]
- 18.Jensen KM, Mikkelsen S. Studies on the bioavaialbility of glyceryl trinitrate after sublingual administration of spray and tablet. Arzneim Forsch. 1997;47:716–718. [PubMed] [Google Scholar]
- 19.Kirsten R, Nelson K, Kirsten D, Heintz B. Clinical pharmacokinetics of vasodilators. Part II. Clin Pharmacokinet. 1998;35:9–36. doi: 10.2165/00003088-199835010-00002. [DOI] [PubMed] [Google Scholar]
- 20.Sasaki S, Koumi S, Sato R, Murata M, Nagasawa K, Sakurai E, et al. Kinetics of buccal absorption of propafenone single oral loading dose in healthy humans. Gen Pharmacol. 1998;31:589–591. doi: 10.1016/S0306-3623(98)00045-7. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Zhang J, Streisand JB. Oral mucosal drug delivery: clinical pharmacokinetics and therapeutic applications. Clin Pharmacokinet. 2002;41:661–680. doi: 10.2165/00003088-200241090-00003. [DOI] [PubMed] [Google Scholar]
- 22.Burnier AM, Martin PL, Yen SS, Brooks P. Sublingual absorption of micronized 17 beta-estradiol. Am J Obstet Gynecol. 1981;140:146–150. doi: 10.1016/0002-9378(81)90101-0. [DOI] [PubMed] [Google Scholar]
- 23.Fiet J, Hermano M, Witte J, Villette JM, Haimart M, Gourmel B, et al. Post-menopausal concentrations of plasma oestradiol, oestrone, FSH and LH and of total urinary oestradiol and oestrone after a single sublingual dose of oestradiol-17 beta. Acta Endocrinol (Copenh) 1982;101:93–97. doi: 10.1530/acta.0.1010093. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez-Younes I, Wagner JG, Gaines DA, Ferry JJ, Hageman JM. Absorption of flurbiprofen through human buccal mucosa. J Pharm Sci. 1991;80:820–823. doi: 10.1002/jps.2600800903. [DOI] [PubMed] [Google Scholar]
- 25.Motwani JG, Lipworth BJ. Clinical pharmacokinetics of drug administered buccally and sublingually. Clin Pharmacokinet. 1991;21:83–94. doi: 10.2165/00003088-199121020-00001. [DOI] [PubMed] [Google Scholar]
- 26.Kates RE. Absorption kinetics of sublingually administered propranolol. J Med. 1977;8:393–402. [PubMed] [Google Scholar]
- 27.Van Laar T, Neef C, Danhof M, Roon KI, Roos R. A new sublingual formulation of apomorphine in the treatment of patients with Parkinson’s disease. Mov Disord. 1996;11:633–638. doi: 10.1002/mds.870110607. [DOI] [PubMed] [Google Scholar]
- 28.Sawicki W, Janicki S. Pharmacokinetics of verapamil and its metabolite norverapamil from a buccal drug formulation. Int J Pharm. 2002;238:181–189. doi: 10.1016/S0378-5173(02)00069-8. [DOI] [PubMed] [Google Scholar]
- 29.Dogterom P, Timmer CJ, De Greef HJMM, Spaans E, De Vries D, Peeters PAM. A phase I study to investigate the safety, tolerability and pharmacokinetics of single and multiple doses of sublingually administered asenapine in healthy male volunteers. Clin Pharmacol Ther. 2009;85:S86. doi: 10.1038/clpt.2008.224. [DOI] [Google Scholar]
- 30.Kroboth PD, McAuley JW, Kroboth FJ, Bertz RJ, Smith RB. Triazolam pharmacokinetics after intravenous, oral, and sublingual administration. J Clin Psychopharmacol. 1995;15:259–262. doi: 10.1097/00004714-199508000-00004. [DOI] [PubMed] [Google Scholar]
- 31.Scavone JM, Greenblatt DJ, Friedman H, Shader RI. Enhanced bioavailability of triazolam following sublingual versus oral administration. J Clin Pharmacol. 1986;26:208–210. doi: 10.1002/j.1552-4604.1986.tb02935.x. [DOI] [PubMed] [Google Scholar]
- 32.Watson NW, Taylor KM, Joel SP, Slevin ML, Eden OB. A pharmacokinetic study of sublingual aerosolized morphine in healthy volunteers. J Pharm Pharmacol. 1996;48(12):1256–1259. doi: 10.1111/j.2042-7158.1996.tb03932.x. [DOI] [PubMed] [Google Scholar]
- 33.Schran HF, McDonald S, Lehr R. Pharmacokinetics and bioavailability of ergoloid mesylates. Biopharm Drug Dispos. 1988;9:349–361. doi: 10.1002/bod.2510090404. [DOI] [PubMed] [Google Scholar]
- 34.Shyu WC, Mayol RF, Pfeffer M, Pittman KA, Gammans RE, Barbhaiya RH. Biopharmaceutical evaluation of transnasal, sublingual, and buccal disk dosage forms of butorphanol. Biopharm Drug Dispos. 1993;14:371–379. doi: 10.1002/bdd.2510140503. [DOI] [PubMed] [Google Scholar]
- 35.McAleer SD, Mills RJ, Polack T, Hussain T, Rolan PE, Gibbs AD, et al. Pharmacokinetics of high-dose buprenorphine following single administration of sublingual tablet formulations in opioid naive healthy male volunteers under a naltrexone block. Drug Alcohol Depend. 2003;72:75–83. doi: 10.1016/S0376-8716(03)00188-1. [DOI] [PubMed] [Google Scholar]