

Abstract
Vaginal administration is a promising alternative to the per-oral route in achieving systemic or local therapeutic effects, when intestinal drug absorption is hindered by problematic biopharmaceutical drug properties. The aim of this study was to establish an in vitro vaginal model and use it to characterize biopharmaceutical properties of liposomally associated curcumin destined for vaginal delivery. The in vitro permeability, metabolism, and tissue retention of high/low permeable compounds were assessed on cow vaginal mucosa and compared to the permeabilities determined through Caco-2 cells and rat jejunum in vitro. The results showed that the intestinal mucosa was superior to the vaginal one in categorizing drugs based on their permeabilities in high/low permeable classes. Passive diffusion was found to be the main mechanism of drug penetration through vaginal mucosa and it was not affected by transporter–enzyme alliance, as their expression/activity was significantly reduced compared to the intestinal tract. Curcumin permeability from the solution form was the lowest of all tested substances due to its significant tissue retention and curcumin–mucus interactions. The permeability of liposomally associated curcumin was even lower but the binding of liposomally associated curcumin to the vaginal tissue was significantly higher. The permeability and tissue retention of liposomal curcumin were vesicle size dependent. Vaginal application of liposomally associated curcumin provides relatively high levels of curcumin in vaginal tissue, with limited systemic absorption.
KEY WORDS: curcumin, intestinal models, liposomes, permeability, vaginal mucosa
INTRODUCTION
Up to 1918, vagina was considered as an organ incapable of drug absorption (1). Subsequent studies revoked this misconception by showing systemic therapeutic effectiveness after vaginal drug administration (2). Although this route offers a gender-specific treatment of systemic or local, female-related conditions (2,3), the merits of vaginal administration such as the avoidance of hurdles associated with per-oral or parenteral application (i.e., drug hepatotoxicity, first-pass drug metabolism, drug plasma concentrations fluctuations, side effects, inconvenience, irritation of gastrointestinal mucosa; 2) enable prolonged dosing with lower daily doses of drugs applied in controlled-release delivery systems (1). Achieving comparable pharmacodynamic effects by vaginal route as by per-oral drug delivery provides indirect proof of effective drug absorption through vaginal mucosa owing to its high absorptive surface area, rich blood supply, significantly diminished expression of phase I and II metabolizing enzymes and transporters, and sufficient permeability to drugs (4–10).
Drug release, dissolution in the vaginal discharge, and permeability through the mucosa limit systemic drug delivery or access to locally diseased targets. To identify rate-limiting steps encountered during formulation development, several models have been suggested in pursue for the most optimal one. Therefore, in vitro studies have used fresh and frozen human (7) and animal vaginal mucosa from rat, mouse, guinea pig, rabbit, cow, sheep, and rhesus monkey (11). Since similar drug permeabilities were established through vaginal and oral pig and human mucosa (4), van der Bijl even suggested replacing vaginal mucosa, which suffers from estrogen-dependent cyclic changes in mucosal thickness, histology, pH, volume, and composition of vaginal secretions (1), with the oral one. The literature regarding in situ and in vivo animal studies is scarce, as models involve additional time- and cost-associated expenditure related to surgical procedures to obtain the animal mucosa, histologically similar to human noncornified, stratified squamous epithelium, and housing in the controlled environment (11). There is also no consensus on which in situ/in vivo animal model should be accepted as the most relevant and thus, several have been recommended (rabbit, rhesus monkey, and sheep vaginal mucosa; 11). Ethical issues limit the number of in vivo human studies for research and development purposes, while clinical studies have been concentrating mostly on different vaginal formulations intended for hormonal replacement therapy, contraception, and infertility treatment (1).
Considering the aforementioned facts, this study attempted to establish in vitro vaginal model using freshly excised cow vaginal mucosa mounted in Franz diffusion chambers. Bovine vaginal mucosa was selected as surrogate for human tissue owing to anatomical, physiological, and functional similarities (12). Namely, human and bovine reproductive organs show identical anatomy (e.g., vulva, vagina, cervix, uterus, oviducts, ovaries), differing only in the shape of the uterus (13). Also, both types of vaginal tissues are under constant cyclic hormonal regulation by plasma estrogen and progesterone levels, which influence the thickness of vaginal epithelia, the rate of mucus secretion and its physiochemical properties (14–16). Additionally, bovine vaginal epithelia like the human one consist of serosa, outer longitudinal and inner circular muscle layers, submucosa, and mucosa. However, bovine epithelium is composed of cylindrical cells, while noncornified, stratified squamous epithelium is characteristic for humans (17). Apart from the difference in the cell type forming the vaginal lining, studies have shown that the composition and function of vaginal mucus in bovine and human vagina are similar. Namely, sialomucins and sulfomucins represent the main glycoproteins in the mucus (18), whose pH simulates the pH in the vaginal tract of post-menopausal women (pH is 6–7.5 in cow and as high as 7 in post-menopausal women; 2,19). The important role of bovine mucosa and mucus as in vitro models have been illustrated in several studies addressing topical and systemic drug delivery, adhesion of Lactobacillus to the epithelium, sperm transport through the mucus, and immunization by vaginal route (19–26). More frequent use of bovine tissue is not just the consequence of the above stated similarities, but also results from the ease of obtaining multiple and large samples on regular basis at relatively low cost. The aim of this work was to determine the in vitro permeabilities of high and low permeable compounds and compare them to the permeabilities obtained with Transwell grown Caco-2 cell monolayers and rat jejunum in side-by-side diffusion chambers. Ultimately, our goal was to use this model as a prognostic tool enabling us the development of suitable liposomes for vaginal delivery of curcumin, which exerts numerous health-beneficial properties (27–29) but suffers from many pharmacokinetic shortcomings if applied orally (i.e., poor solubility and stability in gastrointestinal fluids, low permeability and extensive presystemic metabolism in the gut and liver; 30,31). Therefore, we prepared liposomes of different sizes containing curcumin, evaluated its permeability from liposomes and characterized potential rate-limiting steps in curcumin vaginal delivery (mechanism of absorption, metabolism, tissue binding, and affinity for mucus).
MATERIALS AND METHODS
Materials
Curcumin, atenolol, ranitidine, propranolol, furosemide, l-phosphatidylcholine, pig gastric mucin, and salts for the incubation saline were from Sigma Aldrich (Diesenhofen, Germany). Human albumins were purchased at Slovenian transfusion agency. Ketoconazole was from Sequoia Research LTD. All chemicals used in this study were of the highest grade available. Phosphatidylcholine (Lipoid S100) was a generous gift from Lipoid (Ludwigshafen, Germany).
Methods
The Preparation and Characterization of Liposomes
Liposomes containing curcumin were prepared by the modified film method as described by Basnet et al. (30). Photon correlation spectroscopy technique was employed to determine the vesicle size and size distribution using a Nicomp model 380 particle sizing system (Nicomp Particle Sizing Systems, Santa Barbara, CA, USA; 31). The entrapment efficiencies were determined by high-performance liquid chromatography (93.5 ± 3.1% for multilamellar vesicles (MLVs), 78.2 ± 6.1% for small unilamellar vesicles (SUVs; 30)).
The In Vitro Permeability of Drugs Through Cow Vaginal Mucosa
Cow vaginal tissue was obtained immediately after animal sacrifice in a local slaughterhouse and preserved in the isotonic NaCl solution. Tissue was rinsed three times with saline solution, whipped, stripped from the underneath connective tissues and muscles, and mounted into Franz’s diffusion chambers. Simulated vaginal fluid (SVF), composed of NaCl (3.51 g/L), KOH (1.4 g/L), Ca(OH)2 (0.222 g/L), acetic acid (1 g/L), lactic acid (2 g/L), glycerol (0.16 g/L), urea (0.4 g/L), and glucose (5 g/L; 32), with pH adjusted to 4.5 was added on the mucosal bovine surface (2.5 ml) with tested compounds (10 or 50 μM) and the acceptor solution (7.5 ml, oxygenated Ringer buffer at pH 7.4) (31), containing 10 mM glucose, 0.3 g/L L-glutamine, and 2% albumin (vol/vol) was sampled every 30 min up to 4 h. The samples were replaced by the same volume of acceptor solution containing all necessary ingredients at appropriate concentrations. Franz’s diffusion chambers were maintained at 32°C and lightly shaken throughout the experiments. To determine mass balance and drug recovery, donor solutions were sampled before and after the experiments and tissue was extracted at the end. All the samples were prepared for Liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis.
The In Vitro Penetration of Drugs into the Vaginal Tissue
Unstripped vaginal mucosa was cut into 5 cm2 segments and placed between two custom-made plastic diffusion chambers (developed at the Faculty of Pharmacy, University of Ljubljana), which allowed the addition of 2.5 ml of SVF (pH 4.5) containing drugs (10 μM) on the vaginal mucosa, while the lower plastic part of the diffusion chambers served as tissue support. Mounted tissue was kept at 32°C for 4 h. Then, donor solution was sampled and removed. Tissue was rinsed under running tap water two times and frozen in liquid nitrogen. Afterwards, tissue was cut with Cryotom at −25°C to 20 μm thick slices. Three tissue slices were joined and prepared for LC-MS/MS analysis.
The In Vitro Permeability of Drugs Through Rat Jejunum and Transwell Grown Caco-2 Cell Monolayers
The experiments conducted with rat intestinal tissue were fully abided to the Law for the Protection of Animals (Republic of Slovenia) and were registered at the Veterinary Administration of the Republic of Slovenia. Rat small intestine was obtained from male Sprague–Dawley rats (250–320 g) fasted 18 h prior to the experiments. After euthanasia and laparotomy, intestine was rinsed with ice-cold 10 mM glucose Ringer solution and jejunum, located 25–60 cm distally from the pyloric sphincter, cut into 3 cm long segments, excluding visible Peyer’s patches, was used. Intestinal segments were opened along the mesenteric border, stretched onto inserts and placed between two compartments of EasyMount side-by-side diffusion chambers (Physiologic Instruments, San Diego, USA). Ringer buffer (2.5 ml) of pH 7.4 or 6.5 containing 10 mM mannitol, and the same volume of Ringer buffer pH 7.4 with 10 mM glucose was applied on the mucosal and serosal side, respectively. Albumin (2 vol/vol.%) was added to transport buffer on both sides to increase curcumin stability, solubility, and to avoid precipitation and decomposition. The system was maintained at 37°C and continuously oxygenated with carbogen (95% O2, 5% CO2). Drugs were applied to the mucosal side and the samples were removed every 20 min for up to 2 h from the serosal sides. The withdrawn samples were prepared for LC-MS/MS analysis.
The diffusion chambers were equipped with two pairs of Ag/AgCl electrodes for measuring transepithelial potential difference, short circuit current, and transepithelial electrical resistance (TEER) with a multichannel voltage–current clamp (model VCC MC6, Physiologic Instruments) to assess the viability and integrity as described in Berginc et al. (31).
Caco-2 cells were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen ACC 169, lot 12 and were grown on Transwell Costar culture inserts with a polycarbonate membrane (diameter of 12 mm and pore size of 0.4 μm). Used for seeding was 65,000 cells/filter membranes; the medium was changed every 2 days. At day 15, TEER was measured for each filter with Caco-2 cell monolayers. If the TEER values were in the range of 300–400 Ω cm2, the Caco-2 cell monolayers were used for the subsequent testing of permeability at day 21.
The Caco-2 cells grown on Transwell Costar culture inserts were incubated in 1.5 and 0.5 ml of bathing solution (Ringer buffer) on basolateral (pH 7.4) and apical (pH 6.5 or 7.4) sides, respectively, maintained at 37°C, and continuously oxygenated with carbogen (95% O2, 5% CO2) and lightly shaken during experiment, which was performed as described for rat jejunum. Only Caco-2 cell monolayers with constant TEER during the experiment were used.
LC-MS/MS Quantification of Drugs
Samples obtained during experiments were precipitated with ice cold MeOH (1:3 vol/vol), vortexed for 10 s, and left at −20°C for 48 h. Afterwards, they were centrifuged (1,300×g for 2 h at 4°C), and the supernatants were analyzed. LC-MS/MS apparatus consisted of an Agilent 6460 triple quadrupole mass spectrometer equipped with a JetStream interface and connected to an Agilent 1290 Infinity UPLC (Agilent Techologies, Santa Clara, USA). The following LC-MS/MS conditions were maintained: Phenomenex Kinetex 50 × 2.0 mm C18 column (2.6-mm particles) with an in-line 0.5 μm filter KrudKatcher (Phenomenex, Torrance, USA), injection volume 0.5 μL, column temperature 50°C, mobile phase A 0.1% formic acid in water, mobile phase B 100% acetonitrile, flow rate 0.6 mL/min with the following linear gradient of mobile phase B: 2%, 10%, 20%, 95%, 95%, 2% in the corresponding time points: 0, 1, 1.5, 2.2, 2.9, 3 min, respectively. Total run time was 3.5 min. Mass spectrometry parameters were set as follows: the drying gas temperature, 300°C; drying gas flow rate, 5 L/min; nebulizer, 45 PSI (3.1 × 105 Pa); sheath gas temperature, 320°C; sheath gas flow, 11 L/min; capillary entrance voltage, 3,500 V for positive ions and 4,000 V for negative ions; nozzle voltage, 1,000 V; delta EMV, 200 V. For quantification, the following MRM m/z transitions and collision energies were used: for ketoconazole, 531.2 → 489.1 at 32 eV; for curcumin, 369.1 → 177.0 at 16 eV; for ranitidine, 315.2 → 175.9 at 12 eV; for atenolol, 267.2 → 145.0 at 24 eV; for propranolol, 260.2 → 116.0 at 12 eV; and for furosemide, 329.0 → 284.8 at 4 eV. Instrument control, data acquisition and quantification were performed by MassHunter Workstation software B.03.01 (Agilent Technologies, Torrance, USA).
The limits of quantitation were determined according to the accuracy and precision criteria (bias less than 20%) and signal/noise ratio greater than 10 (peak-to-peak). The limits of quantitation for all analytes were 50 nM or lower except for furosemid, where it was 500 nM. The reproducibility of the assay was better than 4.25% relative standard deviation for all analytes.
Affinity of Curcumin for Glycoproteins and Albumin
The affinity was determined with equilibrium dialysis using curcumin (20 μM) and SVF (pH 4.5) without proteins on one side and SVF with proteins (1.5 g/L gastric pig mucin or 0.018 g/L albumin) on the other after 6 h incubation at 32°C and subsequent sample preparation for LC-MS/MS analysis. Affinity constants and the number of binding sites were determined from Scatchard plots.
Affinity for proteins was also assessed by monitoring the amount of diffused curcumin (20 μM) through polycarbonate membranes of Transwell filters at 32°C by 20-min sampling intervals during 3 h experiment. SVF (pH 4.5) containing either albumins (0.018 g/L) or glycoproteins (1.5 g/L) was used on both sides. Withdrawn samples were replaced by empty SVF with albumin/glycoproteins. The handling of samples prior to LC-MS/MS analysis was identical as described. The amount of diffused curcumin was calculated.
Data Analysis
The apparent permeability coefficient (Papp) of curcumin and drugs were calculated according Eq. (1)
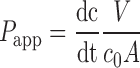 |
1 |
where dc/dt represent changes in concentration of the examined substance in the acceptor compartment per unit time under steady state conditions, V is the volume of the acceptor compartment, A the exposed surface area, and c0 the initial concentration of compounds. The permeabilities were determined only in the absorptive direction.
Results are presented as means ± SD of six measurements. Data were evaluated statistically using SPSS 16.0 for Windows. F test for testing the equality of variances and, afterwards, two-tailed Student’s t test (p = 0.05) were used.
RESULTS
In Vitro Permeabilities Through Cow Vaginal Mucosa, Caco-2 Cells, and Rat Jejunum
The Papp (Table I) were determined for high (propranolol, ketoconazole) and low permeable compounds (ranitidine, furosemide, atenolol, curcumin) at 10 μM (32). Ketoconazole was assayed only through vaginal mucosa because it was completely metabolized in intestinal cells (data not shown). Although the pH of the apical incubation buffer simulated physiological conditions and thus varied depending on the model (pH 4.5 for vaginal mucosa, pH 6.5 and 7.4 for intestinal models), tested compounds were almost completely ionized at all pH except curcumin and furosemide (at pH 4.5). Ketoconazole, propranolol, furosemide, and curcumin were more lipophilic (logP > 2) than atenolol and ranitidine (logP < 1). However, regardless of the pH, the degree of drug ionization and drug lipophilicity, clear differences in Papp values were observed between low and high permeable drugs on intestinal models; the Papp was higher than 1 × 10−5 cm/s for propranolol and below this limit for other compounds (31,32). Apical pH significantly influenced the permeability of atenolol and furosemide through Caco-2 cell and modestly through rat intestine, which is in accordance with the literature (33,34). Curcumin, although classified as low permeable phytochemical (31), exerted higher Papp through Caco-2 cells than rat jejunum, which was shown to be caused by curcumin binding to the mucus above the rat jejunum and more profound first-pass metabolism (31).
Table I.
In Vitro Permeabilities of Compounds Through Cow Vaginal Mucosa, Transwell Grown Caco-2 Cells and Rat Jejunum
| Drug | Physiochemical drug properties | Apparent drug permeabilities (×10−6 cm/s) | ||||||
|---|---|---|---|---|---|---|---|---|
| pK a | LogP | F abs (%) | Cow vagina | Caco-2 cells monolayers | Rat jejunum | |||
| pH 4.5 (%) | pH 6.5 (%) | pH 7.4 (%) | pH 6.5 | pH 7.4 | ||||
| Ketoconazole | 6.2 (base) | 4.3 | 90 | 5.5 ± 0.5 (98)a | – | – | – | – |
| Propranolol | 9.0 (base) | 3.5 | 100 | 7.8 ± 1.2 (100)a | 18.1 ± 3.5 (99.7)a | 20.6 ± 1.4 (97.5) | 13.4 ± 2.1 | 12.0 ± 1.0 |
| Furosemide | 3.9; 9.8 (acid) | 2.0 | 65 | 2.3 ± 0.8 (38.7)a | 5.2 ± 0.6 (98.4)a | 1.7 ± 0.6 (99.8)a | 5.1 ± 0.6 | 3.0 ± 0.6 |
| Atenolol | 9.6 (base) | 0.2 | 50 | 6.8 ± 0.4 (100)a | 5.7 ± 0.3 (99.9)a | 2.9 ± 0.3 (99.4)a | 5.7 ± 0.3 | 4.4 ± 0.3 |
| Ranitidine | 8.3 (base) | 0.2 | 50 | 5.6 ± 0.7 (100)a | 4.9 ± 0.5 (98.4)a | 3.9 ± 0.3 (100)a | 4.9 ± 0.5 | 4.0 ± 1.1 |
| Curcumin | 8.5; 9.9; 10.5 (acid) | >3 | nd | 1.5 ± 0.3 (0.0001)a | 13.9 ± 0.5 (0.01)a | 6.1 ± 1.9 (0.08)a | 1.4 ± 0.3 | 0.3 ± 0.2 |
However, the differences in Papp values between high and low permeable drugs were not as clearly defined when experimenting with vaginal mucosa. Although propranolol and ketoconazole Papp values were among the highest measured (Table I), they were not significantly different from Papp values for atenolol and ranitidine. Only furosemide and curcumin exerted significantly lower Papp values. As the mass balance and the drug recovery were above 85% (between 85% and 92%), stability issues were not the cause for inability of vaginal mucosa to discriminate between high and low permeable compounds.
Mechanisms Involved in Drug Permeation Through Vaginal Mucosa
In vitro absorptive Papp of tested compounds applied as 10 or 50 μM solutions were determined through vaginal mucosa. According to the results (Fig. 1), there were no significant differences between compound’s permeability values determined at both donor concentrations, indicating that the passive diffusion was most probably the predominant permeability pathway, although some of tested compounds are known substrates for efflux transporters in intestine (i.e., curcumin and furosemide are Pgp and MPR-2 substrates (31,35); ketoconazole and ranitidine are Pgp substrates (37,38)).
Fig. 1.
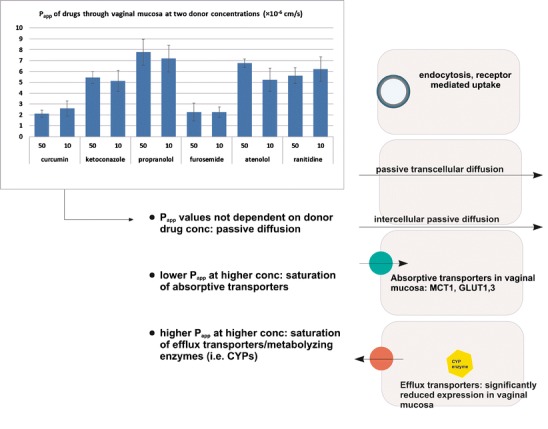
Absorptive in vitro permeabilities of tested compounds through cow vaginal mucosa at 10 and 50 μM donor concentrations
In Vitro Penetration of Compounds into Vaginal Tissue
To investigate whether high binding affinity of drugs (i.e., curcumin) for vaginal tissue could lead to low in vitro permeability, the penetration of tested substances into vaginal tissue was monitored (Fig. 2). The retention of curcumin in the upper 480 μm of vaginal mucosa was the highest (p < 0.05) and there were no significant differences between penetrated amounts of other drugs. The binding of free curcumin to cells layers in vaginal tissue would therefore diminish the amount of curcumin, diffusing into the acceptor compartment of Franz’s diffusion cells, which could explain its low permeability.
Fig. 2.
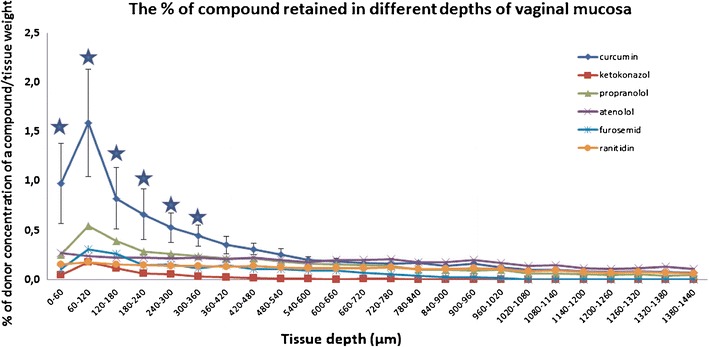
The penetration of tested compounds into vaginal tissue. Starred points significantly higher tissue retention of curcumin in different depths of vaginal tissue compared to other drugs
Drug Metabolism in Vaginal Tissue
No curcumin or ketoconazole CYP3A metabolites were determined in the incubations buffers used for permeability experiments or in tissue extract after penetration experiments (data not shown).
Curcumin Affinity for Glycoproteins and Albumin
Curcumin affinity constants ((1.1 ± 0.2) × 107 L/mol for albumin and (1.5 ± 0.1) × 107 L/mol for glycoproteins) and the number of binding places (0.87 ± 0.2 on average on albumin and 1.2 ± 0.3 on glycoproteins) on glycoproteins and albumin were determined from Scatchard plots. Diffusion of curcumin in SVF containing albumin (0.018 g/L) or glycoproteins (1.5 g/L) are shown in Fig. 3. Significantly more curcumin diffused into acceptor compartment after the initial 20 min when SVF with albumins was used (p < 0.05). Based on equilibrium dialysis and diffusion experiments, curcumin exerted high affinity for both macromolecules, with slightly higher values for glycoproteins.
Fig. 3.
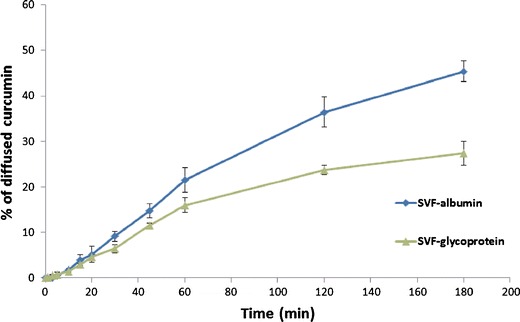
The diffusion of curcumin in simulated vaginal fluid containing either albumins or glycoproteins
In Vitro Permeability and Binding to Vaginal Tissue of Curcumin from Liposomes and Solution
The permeability of liposomally associated curcumin (50 μM) as well as its tissue penetration (Fig. 4) was evaluated and compared to the permeability and tissue penetration of curcumin solution. The in vitro permeability from liposomes was significantly lower (p < 0.05) than from the solution with identical curcumin donor concentration. MLV-associated curcumin showed the lowest permeability, while the permeability of SUV-associated curcumin was significantly higher (p < 0.05) than that of MLV; but still nearly half that of curcumin from solution. MLV formulation was also the one providing the highest amount of curcumin penetrating into the vaginal tissue (significantly higher retention of curcumin in the upper 660 μm of vaginal tissue than SUV and solution, p < 0.05). The retention of curcumin from SUV was the lowest of all three tested formulations in the first 480 μm of vaginal tissue.
Fig. 4.
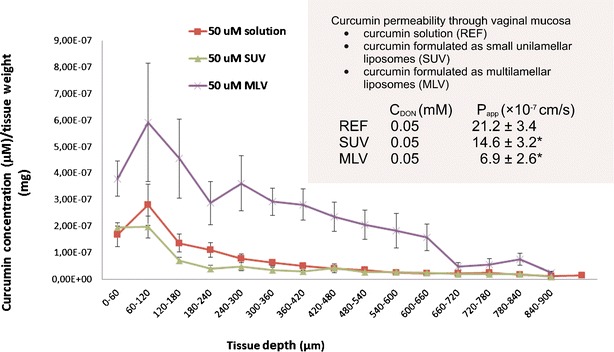
The in vitro permeability and tissue penetration of liposomally associated curcumin and curcumin solution
DISCUSSION
Human vaginal mucosa is supposedly more permeable than the intestinal one (36). That has been established based on drug permeability measurements through vaginal mucosa, but drugs selected for the tests were not suitable for per-oral application because of their profound intraluminal enzymatic degradation by proteases/peptidases (i.e., polypeptide hormones—vasopressin, oxytocin, calcitonin), extensive first-pass metabolism (i.e., sex hormones—17 β-estradiol, progesterone, prostaglandin E2) or problematic physiochemical properties (i.e., extremely low danazol water solubility (37)). Up to date, no studies were performed with “un-problematic” drugs (i.e., drugs without extreme biopharmaceutical issues) to undoubtedly ascertain good permeability of vaginal mucosa as a rule. Thus, we performed a mini-scale study, providing the evidence that intestinal mucosa is superior to the vaginal one as far as permeability is concerned, although tissues of different species (human Caco-2 cells, rat jejunum, cow vaginal mucosa) were used. Namely, the data from this study indicated that it is less straightforward to distinguish between low and high permeable drugs using cow vaginal tissue, because of comparable permeability values for low and highly permeable compounds and because the permeabilities of highly permeable drugs were found to be lower through vaginal than through intestinal models (p < 0.05). Since we did not encounter any stability or solubility issues, and the ionization state of tested drugs was similar in all situations, the incapacity of vaginal mucosa to rank drugs was most probably caused by physiologically inherent characteristics of this mucosa (1,2). Nevertheless, we proceeded to evaluate other potential barriers, which could limit local/systemic drug delivery (transporters, enzymes, and tissue binding). The hypothetical involvement of transporters in drug delivery to vaginal epithelium was evaluated by applying drugs, known substrates for various transporters (i.e., ketoconazole, curcumin, furosemide; 31,35,38,39), at two different concentrations to the bovine vaginal mucosa. Owing to the absence of significant differences between the absorptive permeabilities, passive diffusion was recognized as the most probable permeability pathway. Namely, if a compound had been subjected to an active uptake into mucosa, we would have observed lower contribution of uptake transporters to compounds overall permeability at higher concentration and we would have noticed lower permeabilities. If the active transport would have occurred in the secretory direction (i.e., drug extrusion from vaginal mucosa into the donor compartment), lower drug permeabilities would have been observed at lower drug concentration, because then efflux transporters would not have been saturated, leading to more drug being extruded back into the donor compartment. These results are also in accordance with the studies on the expression of membrane transporters in vaginal mucosa that reported significant expression of uptake monocarboxylate transporter 1 and glucose transporters GLUT1 and 3 in human vaginal epithelium (40), while the expression of other uptake or efflux transporters important in drug delivery and CYP enzymes is significantly diminished (9,10). Therefore, passive transcellular or paracellular/intercellular diffusion represents the predominant transport mechanism besides endocytosis and receptor-mediated uptake of sex hormones as depicted in Fig. 1 (11). As no curcumin or ketoconazole metabolites (CYP3A4, UDP-glucuronosyltransferase substrate) were detected, it could also be ascertained that the coordinated alliance between metabolizing enzymes and efflux transporters would also not hinder vaginal drug absorption, which is in accordance with the reported underexpression of metabolizing enzymes (2,9,10).
In further attempts to identify potential barriers preventing sufficient drug permeability through vaginal tissue, drug binding to vaginal mucus/tissue was assessed (Fig. 2). The retention of drugs in the vaginal tissue was similar for all compounds except for curcumin, which showed the lowest permeability. Based on tissue binding results, the lowest permeability of curcumin determined in our study could be explained at least partially by its high affinity to the vaginal mucosa.
Although bovine vaginal tissue has not yet been applied for such purposes as described here, we have shown that in spite of its inability in ranking drugs based on their permeabilities, it can be used to identify compounds with profound permeability limited absorption and to understand local drug delivery. Compared to other two intestinal in vitro permeability models (rat jejunum and Caco-2 cells), vaginal mucosa cannot be used to rank compounds based on drug permeabilities, because physiochemical drug properties (i.e., logP, pH, ionization) seem not to have significant impact on the permeability. Furthermore, substantial physiological differences between models limit the use of vaginal model for prediction of systemic drug absorption. Namely, the vaginal lining consists of cylindrical cells, whose morphology, structure, and function depends on cyclic changes in plasma levels of sex hormones. Additionally, underexpression of influx/efflux transporters and metabolizing enzymes significantly limits its use. This is further complicated by deviations in pH (acidic in vaginal tract and neutral/slightly acidic in the gut) that is acceptable for individual model to sustain its function, integrity, and viability. Differences notwithstanding, vaginal mucosa could be used to characterize local, topical drug delivery, because of many anatomical, physiological, and functional similarities between human and bovine vaginal mucosa, as explained in the “Introduction” section. Therefore, in the second part of this paper, an attempt was made to identify biopharmaceutical barriers in vaginal delivery of curcumin, formulated as solution and as liposomes.
As stated above, curcumin permeability applied in solution remained among the lowest measured, regardless of favorably low degree of ionization and lipophilicity, which is most probably the reason for profound binding of free curcumin to the cells in vaginal tissue that would most probably diminish the amount of diffusing molecule and explain its low in vitro permeability. Curcumin binding affinity for proteins in plasma has been demonstrated as a way to increase curcumin stability and prevent its hydrolysis and nonspecific binding to the plastics (31,41). Although the stability and solubility of curcumin in acidic pH of 4.5 are much higher than in neutral and basic environment even without the presence of proteins (41) the affinity of curcumin towards cellular proteins is obviously still high enough to prevent sufficient quantities of curcumin to be absorbed from vagina into the systemic circulation. Based on this, vaginal mucosa seems to have limited potential for systemic curcumin delivery, although it does not participate in curcumin extensive first-pass metabolism as in the intestine (31). The affinity of this phytochemical for macromolecules in SVF (pH 4.5) was therefore quantified and profound affinity of curcumin for glycoproteins and albumin was demonstrated (Fig. 3, the results of equilibrium dialysis), with slightly higher affinity for glycoproteins than for albumins as corroborated in other published articles (42). Interactions between curcumin and vaginal mucus could thus additionally reduce curcumin accessibility to systemic circulation via vaginal mucosa. Curcumin could thus only be exploited for the treatment of local conditions without posing any safety issues (i.e., drug–drug interactions) for patients concomitantly prescribed other drugs.
However, solutions lack desirable biopharmaceutical properties (sufficient residence time on target site) carry, risk of potential irritancy due to solvent and limit patient compliance (inconvenience, discomfort) (1), therefore curcumin was formulated into phospatidylcholine liposomes of two sizes (SUV, MLV) to serve as delivery systems destined for vaginal therapy (30,43). Liposomally associated curcumin exerted lower permeability than the solution. Interestingly, MLV liposomes exhibited the lowest permeability and the highest retention of curcumin in the upper layers of vaginal tissue (Fig. 4). This would make this type of liposomes the formulation of choice for local curcumin delivery, based on the fact that the lowest curcumin delivery into systemic circulation could be expected while assuring the highest local concentration at the same time. However, in the case SUVs, the correlation between the permeability and tissue retention was not as straightforward as observed for MLV. Namely, the permeability of curcumin from SUV was significantly lower than that of the solution, while curcumin tissue binding was similar to the one determined for the solution. To explain this anomaly, more factors are expected to interrelate. First, curcumin incorporation into outer/inner liposomal membrane could be different in MLV than in SUV, thus influencing curcumin partitioning from liposomal bilayer to the membranes of vaginal cells. Secondly, it is known that lipidic liposomal components readily dissolve in the skin; therefore, similar effect could also be expected in the vaginal mucosa (445). Since MLV carry higher amounts of phosphatidylcholine, more pronounced effect on curcumin tissue binding could be expected with MLV than with SUV formulation as indeed observed in this study. Different behavior of MLV and SUV phosphatidylcholine liposomes prepared with the same methods were also observed in the case of loading liposomes with mupirocin. It was noted that the antimicrobial effect of MLV containing mupirocin was better than that of SUV because MLV ensured higher depot effect than SUV (44). Additionally, tissue binding and permeability could be partially affected by different curcumin–phosphatidylcholine ratios in SUV and MLV and, more likely, by different kinetics of curcumin partitioning from liposomes to simulated vaginal fluid and further to the tissue. Based on the above premises, better phosphatidylcholine dissolution in the mucosal cells and potentially different place of curcumin loading in the MLV liposomes could have contributed to the effects presented in this research. All these facts should be considered in further formulation strategies, aiming to increase the binding of curcumin to the different depths of vaginal tissue.
CONCLUSION
The transport of tested drugs through cow vaginal mucosa was found to be mediated only by passive diffusion. The transporters and metabolizing enzymes did not influence the in vitro permeability, while tissue binding and drug–mucus interactions were shown to be able to hinder drug absorption. The ability of the utilized model to discriminate between high and low permeable compounds was rather low, however, low in vitro permeability detected with this model, would indicate that a compound exerts poor systemic in vivo permeability (i.e., curcumin). Curcumin permeability through vaginal mucosa was the lowest and it was caused by more pronounced binding to the mucosal tissue and mucus.
The liposomes composed of phospatidylcholine can be used as novel drug delivery systems for curcumin destined for vaginal therapy. They were able to ensure lower tissue permeability of curcumin and higher tissue retention as compared to curcumin in solution and could thus be used for topical vaginal curcumin delivery. The attention should be given to vesicle characteristics, in particular to the vesicle size.
Contributor Information
Katja Berginc, Phone: +386-1-4769503, FAX: +386-1-4258031, Email: katja.berginc@ffa.uni-lj.si.
Nataša Škalko-Basnet, Phone: +47-77-646640, FAX: +47-77-646151.
Purusotam Basnet, Phone: +47-77-646640, FAX: +47-77-646151.
Albin Kristl, Phone: +386-1-4769500, FAX: +386-1-4258031.
References
- 1.Alexander NJ, Baker E, Kaptein M, Karck U, Miller L, Zampaglione E. Why consider vaginal drug administration? Fertil Steril. 2004;82:1–12. doi: 10.1016/j.fertnstert.2004.01.025. [DOI] [PubMed] [Google Scholar]
- 2.Hani U, Bhat RS, Sisodiya R, Hosakote SG. Novel vaginal drug delivery systems: a review. Curr Drug Ther. 2010;5:95–104. doi: 10.2174/157488510791065067. [DOI] [Google Scholar]
- 3.Valenta C. The use of mucoadhesive polymers for vaginal delivery. Adv Drug Deliv Rev. 2005;57:1692–1712. doi: 10.1016/j.addr.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 4.van der Bijl P, Thompson IOC, Squier CA. Comparative permeability of human vaginal and bucal mucosa to water. Eur J Oral Sci. 1997;105:571–575. doi: 10.1111/j.1600-0722.1997.tb00219.x. [DOI] [PubMed] [Google Scholar]
- 5.van der Bijl P, van Eyk AD, Thompson IOC. Permeation of 17β-estradiol through human vaginal and bucal mucosa. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1998;85:393–398. doi: 10.1016/S1079-2104(98)90063-4. [DOI] [PubMed] [Google Scholar]
- 6.van der Bijl P, van Eyk AD, Thompson IOC, Stander IA. Diffusion rates of vasopressin through human vaginal and buccal mucosa. Eur J Oral Sci. 1998;106:958–962. doi: 10.1046/j.0909-8836.1998.eos106509.x. [DOI] [PubMed] [Google Scholar]
- 7.van der Bijl P, Penkler L, van Eyk AD. Permeation of sumatriptan through human vaginal and buccal mucosa. Headache. 2000;40:137–141. doi: 10.1046/j.1526-4610.2000.00019.x. [DOI] [PubMed] [Google Scholar]
- 8.Değim IT, Tuğcu-Demiröz F, Tamer-İlbasmış S, Acartürk F. Development of controlled release sildenafil formulations for vaginal administration. Drug Deliv. 2008;15:259–265. doi: 10.1080/10717540802006781. [DOI] [PubMed] [Google Scholar]
- 9.Pauletti GM, Liu JH, Benet LZ, Ritschel WA, inventors; UMD Inc., assignee. Vaginal delivery of chemotherapeutic agents and inhibitors of membrane efflux systems for cancer therapy. United States Patent US 6982091 B1, 2006 Jan 3.
- 10.Pauletti GM, Harrison DC, Desai KJ, inventors. Method for augmentation of intraepithelial and systemic exposure of therapeutic agents having substrate activity for cytochrome P450 enzymes and membrane efflux systems following vaginal or oral cavity administration. United States Patent US 2007/0036834 A1, 2007 Feb 15.
- 11.Wu SJ, Robinson JR. Vaginal epithelial models. In: Borchardt RT, Smith PL, Wilson G, editors. Models for assessing drug absorption and metabolism. New York: Plenum Press; 1996. pp. 409–424. [Google Scholar]
- 12.das Neves J, Amaral MH, Bahia MF. Performance of an in vitro mucoadhesion testing method for vaginal semisolids: influence of different testing conditions and instrumental parameters. Eur J Pharm Biopharm. 2008;69:622–632. doi: 10.1016/j.ejpb.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 13.Deutscher GH. G80-537 Reproductive trace anatomy and physiology of the cow. In: Historical materials from the University of Nebrasca-Lincoln extension, University of Nebrasca. 1980. http://digitalcommons.unl.edu/cgi/viewcontent.cgi?article=1316&context=extensionhist. Accessed 17 May 2012.
- 14.Bondurant RH. Inflammation in the bovine female reproductive tract. J Anim Sci. 1999;77:101–110. doi: 10.2527/1999.77suppl_2101x. [DOI] [PubMed] [Google Scholar]
- 15.Lamond DR, Shanahan AG. Chemical changes in cervical mucus from normal and ovariectomized cows treated with hormones. Biol Reprod. 1969;1:335–343. doi: 10.1095/biolreprod1.4.335. [DOI] [PubMed] [Google Scholar]
- 16.Roberts GP. Structural studies on the glycoproteins from bovine cervical mucus. Biochem J. 1978;173:941–947. doi: 10.1042/bj1730941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wrobel KH, Laun G, Hees H, Zwack M. Histologische und ultrastrukturelle Untersuchungen am Vaginalepithel des Rindes. Anat Histol Embryol. 1986;15:303–328. doi: 10.1111/j.1439-0264.1986.tb00543.x. [DOI] [PubMed] [Google Scholar]
- 18.Sergin NP, Kuznecov MP, Kozlova VM, Nesmejanova TN. Physico-chemical conditions in the genital tract of the cow and survival of spermatozoa. 1940;15:24–8. http://www.cabdirect.org/abstracts/19410100104.html. Accessed 17 May 2012.
- 19.Heydon RA, Adams NR. Comparative morphology and mucus histochemistry of the ruminant cervix: differences between crypt and surface epithelium. Biol Reprod. 1979;21:557–562. doi: 10.1095/biolreprod21.3.557. [DOI] [PubMed] [Google Scholar]
- 20.Baloğlu E, Özyazıcı M, Hızarcıoğlu SY, Karavana HA. An in vitro investigation for vaginal bioadhesive formulations: bioadhesive properties and swelling states of polymer mixtures. Farmacol. 2003;58:391–396. doi: 10.1016/S0014-827X(03)00044-2. [DOI] [PubMed] [Google Scholar]
- 21.Baloğlu E, Ozyazici M, Yaprak Hizarcioğlu S, Senyiğit T, Ozyurt D, Pekçetin C. Bioadhesive controlled release systems of ornidazole for vaginal delivery. Pharm Dev Technol. 2006;11:477–484. doi: 10.1080/10837450600939784. [DOI] [PubMed] [Google Scholar]
- 22.Hombach J, Palmberger TF, Bernkop-Schnürch A. Development and in vitro evaluation of a mucoadhesive vaginal delivery system for nystatin. J Pharm Sci. 2009;98:555–564. doi: 10.1002/jps.21457. [DOI] [PubMed] [Google Scholar]
- 23.Otero CM, Nader-Macias ME. Lactobacillus adhesion to epithelial cells from bovine vagina. In: Mendez-Vilas A, editors. Communicating current research and education topics and trends applied microbiology. http://www.formatex.org/microbio/pdf/pages749-757.pdf. Accessed 17 May 2012.
- 24.Rutllant J, López-Béjar M, López-Gatius F. Ultrastructural and rheological properties of bovine vaginal fluid and its relation to sperm motility and fertilization: a review. Reprod Domest Anim. 2005;40:79–86. doi: 10.1111/j.1439-0531.2004.00510.x. [DOI] [PubMed] [Google Scholar]
- 25.Corbeil LB, Munson L, Campero C, BonDurant RH. Bovine Trichomoniasis as a model for development of vaccines against sexually-transmitted disease. Am J Reprod Immunol. 2001;45:310–319. doi: 10.1111/j.8755-8920.2001.450507.x. [DOI] [PubMed] [Google Scholar]
- 26.Mariano RN, Turino LN, Cabrera MI, Scándolo DE, Maciel MG, Grau RJ. A simple pharmacokinetic model linking plasma progesterone concentrations with the hormone release from bovine intravaginal inserts. Res Vet Sci. 2010;89:250–256. doi: 10.1016/j.rvsc.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Garcea G, Berry DP, Jones DJL, Singh R, Dennison AR, Farmer PB, et al. Consumption of the putative chemopreventive agent curcumin by cancer patients: assessment of curcumin levels in the colorectum and their pharmacodynamic consequences. Cancer Epidemiol Biomarkers Prev. 2005;14:120–125. [PubMed] [Google Scholar]
- 28.Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm. 2007;4:817–818. doi: 10.1021/mp700113r. [DOI] [PubMed] [Google Scholar]
- 29.Dhillon N, Aggarwal BB, Newman RA, Wolff RA, Kunnumakkara AB, Abbruzzese JL, et al. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res. 2008;14:4491–4499. doi: 10.1158/1078-0432.CCR-08-0024. [DOI] [PubMed] [Google Scholar]
- 30.Basnet P, Hussain H, Tho I, Skalko-Basnet N. Liposomal delivery system enhances anti-inflammatory properties of curcumin. J Pharm Sci. 2012. doi:101:598–609.doi.10.1002/jps.22785. [DOI] [PubMed]
- 31.Berginc K, Trontelj J, Skalko-Basnet N, Kristl B. The physiological barriers to oral delivery of curcumin. Die Pharm. 2012;67:1–7. [PubMed] [Google Scholar]
- 32.Yu LX, Amidon GL, Polli JE, Zhao H, Mehta MV, Conner DP, et al. Biopharmaceutical classification system: the scientific basis for biowaiver extensions. Pharm Res. 2002;19:921–925. doi: 10.1023/A:1016473601633. [DOI] [PubMed] [Google Scholar]
- 33.Chungi VS, Dittert LW, Smith RB. Gastrointestinal sites of furosemide absorption in rats. Int J Pharm. 1979;4:27–38. doi: 10.1016/0378-5173(79)90095-4. [DOI] [Google Scholar]
- 34.Taylor DC, Rownall R, Burke W. The absorption of β-adrenoreceptor antagonists in rat in-situ small intestine: the effect of lipophilicity. J Pharm Pharmacol. 1985;37:280–283. doi: 10.1111/j.2042-7158.1985.tb05064.x. [DOI] [PubMed] [Google Scholar]
- 35.Zvonar A, Berginc K, Kristl A, Gašperlin M. Microencapsulation of self-microemulsifying system: improving solubility and permeability of furosemide. Int J Pharm. 2010;388:151–158. doi: 10.1016/j.ijpharm.2009.12.055. [DOI] [PubMed] [Google Scholar]
- 36.van der Bijl P, van Eyk AD. Comparative in vitro permeability of human vaginal, small intestinal and colonic mucosa. Int J Pharm. 2003;261:147–152. doi: 10.1016/S0378-5173(03)00298-9. [DOI] [PubMed] [Google Scholar]
- 37.Takano R, Furumoto K, Shiraki K, Takana N, Hayashi Y, Y A, et al. Rate-limiting steps of oral absorption for poorly-water soluble drugs in dogs; prediction from a miniscale dissolution test and physiologically-based computer simulation. Pharm Res. 2008;25:2334–2344. doi: 10.1007/s11095-008-9637-9. [DOI] [PubMed] [Google Scholar]
- 38.Bourdet DL, Pollack GM, Thakker DR. Intestinal absorptive transport of the hydrophilic cation ranitidine: a kinetic modeling approach to elucidate the role of uptake and efflux transporters and paracellular vs. transcellular transport in Caco-2 cells. Pharm Res. 2006;23:1178–1187. doi: 10.1007/s11095-006-0204-y. [DOI] [PubMed] [Google Scholar]
- 39.Takano M, Hasegawa R, Fukuda T, Yumoto R, Nagai J, Murakami T. Interaction with P-glycoprotein and transport of erythromycin, midazolam and ketoconazole in Caco-2 cells. Eur J Pharmacol. 1998;358:289–294. doi: 10.1016/S0014-2999(98)00607-4. [DOI] [PubMed] [Google Scholar]
- 40.Kuchiiwa T, Nio-Kobayashi J, Takahashi-Iwanaga H, Yajima T, Iwanaga T. Cellular expression of monocarboxylate transporters in the female reproductive organ of mice: implications for the genital lactate shuttle. Histochem Cell Biol. 2011;135:351–360. doi: 10.1007/s00418-011-0794-2. [DOI] [PubMed] [Google Scholar]
- 41.Hatcher H, Planalp R, Cho J, Torti FM, Torti SV. Curcumin: from ancient medicine to current clinical trials. Cell Mol Life Sci. 2008;65:1631–1652. doi: 10.1007/s00018-008-7452-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gupta SC, Prasad S, Kim JH, Patchva S, Webb LJ, Priyadarsini IK, et al. Multitargeting by curcumin as revealed by moledular interaction studies. Nat Prod Rep. 2011;28:1937–1955. doi: 10.1039/c1np00051a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pavelić Ž, Škalko-Basnet N, Jalšenjak I. Characterization and in vitro evaluation of bioadhesive liposome gels for local therapy of vaginitis. Int J Pharm. 2005;301:140–148. doi: 10.1016/j.ijpharm.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 44.Berg OA, Hurler J, Skalko-Basnet N. Advanced delivery system for skin and burns therapy : mupirocin as an antibacterial model drug. Eur J Pharm Sci. 2011;44:46–47. [Google Scholar]
- 45.Brittain HG. Profiles of drug substances, excipients and related methodology. 1. Oxford: Elsevier; 2007. [DOI] [PubMed] [Google Scholar]
- 46.PhysProp database. http://www.vcclab.org/lab/alogps/. Accessed at 22 March 2012.
- 47.Zaki NM, Artursson P, Bergström CA. A modified physiological BCS for prediction of intestinal absorption in drug discovery. Mol Pharm. 2010;7:1478–1487. doi: 10.1021/mp100124f. [DOI] [PubMed] [Google Scholar]
- 48.Kim JS, Mirchell S, Kijek P, Tsume Y, Hilfinger J, Amidon GL. The suitability of an in situ perfusion model for permeability determinations: utility for BCS class I biowaiver requests. Mol Pharm. 2006;65:681–694. doi: 10.1021/mp060042f. [DOI] [PubMed] [Google Scholar]
- 49.Avdeef A. Absorption and drug development—solubility, permeability, and charge state. New Jersey: Wiley; 2003. [Google Scholar]