

Abstract
The present study aimed at development of capsular dosage form of surface-adsorbed nanoemulsion (NE) of olmesartan medoxomil (OLM) so as to overcome the limitations associated with handling of liquid NEs without affecting their pharmaceutical efficacy. Selection of oil, surfactant, and cosurfactant for construction of pseudoternary phase diagrams was made on the basis of solubility of drug in these excipients. Rationally selected NE formulations were evaluated for percentage transmittance, viscosity, refractive index, globule size, zeta potential, and polydispersity index (PDI). Formulation (F3) comprising of Capmul MCM® (10% v/v), Tween 80® (11.25% v/v), polyethylene glycol 400 (3.75% v/v), and double-distilled water (75% v/v) displayed highest percentage cumulative drug release (%CDR; 96.69 ± 1.841), least globule size (17.51 ± 5.87 nm), low PDI (0.203 ± 0.032), high zeta potential (−58.93 ± 0.98 mV), and hence was selected as the optimized formulation. F3 was adsorbed over colloidal silicon dioxide (2 ml/400 mg) to produce free-flowing solid surface-adsorbed NE that presented a ready-to-fill capsule composition. Conversion of NE to surface-adsorbed NE and its reconstitution to NE did not affect the in vitro release profile of OLM as the similarity factor with respect to NE was found to be 66% and 73% respectively. The %CDR after 12 h for optimized NE, surface-adsorbed NE, and reconstituted NE was found to be 96.69 ± 0.54, 96.07 ± 1.76, and 94.78 ± 1.57, respectively (p > 0.05). The present study established capsulated surface-adsorbed NE as a viable delivery system with the potential to overcome the handling limitations of NE.
KEY WORDS: bioavailability, nanoemulsion, olmesartan medoxomil, oral
INTRODUCTION
Olmesartan medoxomil (OLM) is an extensively used antihypertensive drug (1). By the action of aryl esterases, situated in both intestine and plasma, it gets quickly de-esterified upon oral administration in to an active metabolite, i.e., olmesartan (2). Olmesartan is reported to be more helpful in patients with essential hypertension in comparison to other angiotensin II receptor blockers with respect to decrease in ambulatory blood pressure (3). Commercially available tablets of OLM exhibit reduced oral absorption leading to low oral bioavailability of 25.6% (4). OLM is highly lipophilic in nature (log p = 4.31) which attributes to its low aqueous solubility. Poor aqueous solubility and efflux of hydrophobic therapeutic agents by means of drug resistance pump in the gastrointestinal tract contribute to their low bioavailability (5,6). Oil to water nanoemulsion systems are reported to augment aqueous solubility of hydrophobic therapeutic agents by including them in the oil phase of the nanoemulsion (7–9). Non-ionic surfactants such as Tween 80 have been stated to be valuable pharmaceutical excipient in order to prevent the function of the drug-resistant P-glycoprotein (P-gp) efflux pump and thus augment the intestinal absorption of therapeutic agents susceptible to P-gp-mediated efflux in the intestine (10).
OLM has been formulated as a self-microemulsifying drug delivery system (SMEDDS) using capryol 90, tween 20 and tetraglycol (10:60:30, v/v/v) and Lee and his coworkers reported an improved relative bioavailability of 170% compared to the suspension in male Sprague–Dawley rats (11). SMEDDS as a delivery system have been reported to employ high concentration (30–60%) of surfactants that may lead to cellular toxicity (12). Furthermore, precipitation of the hydrophobic therapeutic agent formulated as SMEDDS and encapsulated in gelatin capsule may occur due to propensity of the volatile solvents used in formulation of SMEDDS to get transferred to the shell (13). One of the approaches to circumvent these limitations is to deliver hydrophobic therapeutic agents in the form of a nanoemulsion formulation that like SMEDDS is also isotropic, thermodynamically stable, transparent (or translucent) system of oil, water, and surfactants. Nanoemulsion (NE) possesses droplet size generally in the range of 10–100 nm but in contrast to SMEDDS, it employs a lesser amount (5–10%) of surfactant. Furthermore, NEs can be formulated with little energy input (heat or mixing), have a long shelf life, and is characterized with simplicity of scale up of the manufacturing process (14). Hence, it was hypothesized that developing a nanoemulsion-based drug delivery system utilizing a non-ionic surfactant as the emulsifier would help to improve solubility and prevent efflux of OLM out of the intestine eventually leading to enhanced oral bioavailability.
Although NE is one of the finest modes of delivery for hydrophobic therapeutic agents, but due to liquid nature of the dosage form, it would not be as accepted as the solid dosage form. Liquid dosage forms are normally associated with transportation issues, instability problems, and poor palatability due to the lipid content (in case of NE). Moreover, as potent drugs are incorporated in NE formulation, dose variability due to handling problems in case of select patient population may lead to toxicity. To manage these challenges, many attempts have been made to convert liquid formulations into the solid dosage forms like capsule (15–17) and tablet (18).
Based on these considerations, the current study was aimed at developing and characterizing NE of OLM so as to improve dissolution rate-limited absorption of the drug using a low surfactant concentration and to convert the developed liquid NE into a solid unit dosage form by adsorbing it over an inert, solid adsorbent so as to develop a patient friendly dosage form.
Materials
OLM was obtained as a gift sample from Sun Pharmaceutical Industries (Sikkim, India). Capmul MCM® (glycerol monodicaprylate) and Captex 100® were obtained as gift samples from Abitech Corporation Limited (Janesvile, WI, USA). Labrafil M 1944 CS® (oleoyl macrogoglyceride), Labrafil M 2125CS® (linoleoyl macrogolglycerides), Labrasol® (caprylo caproyl macrogol-8-glyceride), Peceol® (glyceryl oleate), Plurol oleique® (polyglycerol oleate), Lauroglucol 90® (propylene glycol monolaurate), and Transcutol® P (diethylene glycol monoethyl ether) were donated by Gattefosse (Saint Priest, Cedex, France). Tween 80® (polyoxyethylene sorbitan monooleate), Tween 20® (polyoxyethylene sorbitan monolaurate), ethanol, and polyethylene glycol 400 (PEG 400) were obtained from S.D. Fine-Chemicals Ltd. (Mumbai, India). Colloidal silicon dioxide and dialysis membrane (pore size of 25 Å) were obtained from Hi Media Laboratories Pvt. Ltd (Mumbai, India). All other chemicals and solvents were of analytical reagent grade and were used without further purification.
METHODS
Screening of Components
Solubility of OLM in various vehicles (oils, surfactants, and cosurfactants) was determined by shake flask method whereby an excess amount of drug was added to 2 ml of the selected vehicle and kept at 25 ± 1°C in an isothermal bath shaker (Hicon, New Delhi, India) for 72 h to reach equilibrium. The samples were centrifuged (Remi Pvt. Ltd., Vasai, India) at 3,000 rpm for 15 min. Supernatant was removed carefully and diluted suitably with ethanol (95%, v/v). The samples were analyzed for drug content at 257.8 nm using UV-visible spectrophotometer (Shimadzu, Pharmaspec1700, Kyoto, Japan). The solubility of drug in each component was calculated in triplicate and mean ± standard deviation (SD) was reported.
Construction of Pseudoternary Phase Diagram
On the basis of solubility study of drug, Capmul MCM was chosen as the oil phase, Tween 80 as the surfactant, and PEG 400 as the cosurfactant. Double-distilled water was used as the aqueous phase for the creation of pseudoternary phase diagram. Surfactant and cosurfactant were mixed (Smix) in different volume ratios (1:1, 1:2, 1:3, 2:1, 3:1, 4:1). For each phase diagram, oil and specific Smix ratio was mixed in different volume ratios from 1:9 to 9:1 so that maximum ratios were enclosed for the study in order to define the boundaries of phases formed in the phase diagram. Pseudoternary phase diagrams were developed by aqueous titration data using PCP Disso V2.08 software, Pune, India. Physical state of the blank NE was marked on a pseudo-three-component phase diagram in which one axis signified the aqueous phase, the second signified oil, and the third signified Smix. A total of 14 formulations were selected from the NE regions of the phase diagram.
Evaluation of Blank NEs
Thermodynamic Stress Stability Studies
Blank NE formulations were centrifuged at 3,500 rpm for 30 min and monitored for phase separation, creaming, or cracking. Those formulations that did not show any phase separation were subjected to heating–cooling cycle. Six cycles between refrigerator temperature (4°C) and 45°C with storage at each temperature for not less than 48 h were performed. The formulations that passed heating–cooling cycles were subjected to three freeze–thaw cycles at temperature between −21°C and +25°C with storage at each temperature for not less than 48 h. The formulations that passed the thermodynamic stress stability tests were further taken for dispersibility study in order to estimate the efficiency of emulsification.
Dispersibility Test
The efficiency of emulsification of oral NE was assessed using a standard USP XXII dissolution apparatus II. One milliliter of each formulation was added to 500 ml of double-distilled water at 37 ± 0.5°C. A standard stainless steel dissolution paddle rotating at 50 rpm presented gentle agitation. The in vitro behavior of the formulation was visually assessed using the grading system. Grade A was given to the formulations which exhibited clear or bluish appearance within 1 min, grade B included formulations with slightly less clear and bluish white appearance, grade C was given to the formulations with fine milky appearance within 2 min, grade D was assigned to the formulations with dull, grayish white having slightly oily appearance in more than 2 min, and graded E were the formulations with large oil globules present on the surface. Formulations, from each Smix ratio investigated, were selected on the basis of least Smix and suitable amount of oil (0.5 ml) that could completely dissolve the required amount (5 mg) of drug. Care was taken to select the formulations which passed the thermodynamic stress stability and dispersibility test in grades A and B.
Preparation of Drug-Loaded NE
Thermodynamically stable blank NEs were chosen for drug loading. Drug-loaded NEs were prepared by adding the calculated amount of drug (10.02 mg/ml of OLM) to the oil phase and stirring in isothermal bath shaker until whole of the drug was dissolved. Then, Smix in a fixed proportion was added to fixed volume of oil containing drug to produce a clear mixture. This was followed by adding definite proportion of water (drop wise) and shaking slowly until a clear NE was obtained.
Characterization of the NE
Globule Size, Zeta Potential, and Polydispersibility Index
Globule size of the NEs was determined by photon correlation spectroscopy that analyzes the fluctuation in light scattering due to Brownian motion of the globules, using a Zetasizer ver. 6.01 (Malvern Instrument Ltd., UK). The formulation was subjected to 500 times dilution with double-distilled water and light scattering was monitored at 25°C at a 90° angle. Zeta potential was also measured using the same instrument. The refractive index was kept at 1.33 and viscosity at 1.0 cps to mimic the values for pure water. Zeta potential values were determined from the electrophoretic mobility of oil droplets.
Percentage Transmittance, Refractive Index, and Viscosity
Percentage transmittance was determined spectrophotometrically. One milliliter of the formulation was diluted to 100 times with double-distilled water and percentage transmittance was measured against double-distilled water as blank at 630 nm. Abbe’s type refractrometer (Jindal Instruments, Ambala, India) was used to determine the refractive index. Few drops of NE were smeared on the lower prism surface. The eye piece cross-wire was adjusted so that a sharp demarcation line passes through the center having half light and half dark position. Then, readings were noted down from the scale. The viscosity of the formulations was determined without dilution using Brookfield viscometer DV-II+ Pro (Brookfield Engineering Laboratories, Inc, MA, USA) coupled with S-94 spindle at 100 rpm and 25 ± 2°C.
Drug Content
For the determination of drug content, 1 ml of the NE was taken in a 10 ml volumetric flask and shaken vigorously with ethanol (95%, v/v) for 10 min. Finally, the volume was made up to 10 ml with the same solvent and after proper dilution using ethanol, the drug content was analyzed at 257.8 nm by UV spectrophotometer (Shimadzu, Pharmaspec1700, Kyoto, Japan).
In Vitro Drug Release
In vitro drug release was measured by dialysis bag method using a pretreated dialysis membrane (MWCO 12–14 kD). Himedia dialysis membrane (Himedia Laboratories Pvt. Ltd., Mumbai, India) was kept in a normal saline solution for 2 h before study to ensure complete wetting of the membrane. Two milliliters of optimized formulations was placed in pretreated dialysis bag and drug release was studied using USP dissolution apparatus II (Hicon Enterprises, Delhi, India) containing 500 ml of phosphate buffer (pH 7.4) at 37 ± 0.5°C. The speed of the paddle was adjusted to 50 rpm. Two-milliliter sample was withdrawn at regular time intervals (0, 0.5, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, and 12 h) and the volume withdrawn was replaced with the fresh medium. The release of drug from the NE formulations was compared against the pure drug suspension. The samples were analyzed at 257.8 nm and the percentage cumulative drug release (%CDR) was calculated. The analysis of the samples was done in triplicate.
Transmission Electron Microscopy
Morphology of the oil droplets in the NE formulation was visualized using CM 10 transmission electron microscope (Mega View III FW, Philips, UK). NE formulation was diluted 100 times and a drop was applied to 300 mesh copper grid. The grid was inverted and a drop of phosphotungstic acid (PTA) was applied to the grid for 10 s. Excess of PTA was removed and grid was analyzed at 60–80 kV.
Selection of Optimized NE
Optimized formulation was selected on the basis of globule size, zeta potential, polydispersity index (PDI), percent drug content, and %CDR. The optimized NE was utilized for the preparation of surface-adsorbed NE by adsorption over a solid adsorbent.
Adsorption of NE Over a Solid Adsorbent
The optimized NE, F3 (5 ml) was placed in a glass mortar and colloidal silicon dioxide (400 mg) was added slowly and mixed gently to get the solid mass. The solid mass was passed through the sieve (22 mesh size) to get uniform free-flowing powder. The powder was stored over anhydrous calcium chloride in a dessicator until further evaluation.
Micromeritic and Rheological Characterization of Surface-Adsorbed NE
The flow properties of the surface-adsorbed NE were determined by the following tests: (1) Carr’s compressibility index, (2) Hausner’s ratio, and (3) angle of repose. The surface-adsorbed NE (5 g) was poured lightly into 50 ml measuring cylinder. The powder was subjected to tapping until no further change in volume was observed. Bulk density (Do) and tapped density (Df) of the powder was calculated by dividing the weight of the granules by its volume before and after tapping, respectively. Percentage compressibility was computed by subtracting Do from Df and divided by Df and multiplied by 100, Hausner’s ratio was computed as Df divided by Do. Angle of repose was measured by static funnel method. Angle of the heap of granules (5 g) formed by passing through a funnel placed at a height of 8 cm from the horizontal surface was measured using a protractor (17).
Drug Content Determination
Surface-adsorbed NE equivalent to 5 mg of OLM was dispersed in suitable quantity of ethanol (95%, v/v). The sample was mixed thoroughly to ensure complete dissolution of drug in ethanol. The sample was centrifuged using centrifuge (Remi Pvt Ltd., Vasai, India) at 3,500 rpm for 15 min to separate colloidal silicon dioxide particles. The supernatant was suitably diluted and analyzed spectrophotometrically at 257.8 nm. Drug content was computed from the validated calibration curve of drug in ethanol (95%, v/v).
Scanning Electron Microscopy
The morphological features of particles of colloidal silicon dioxide and surface-adsorbed NE were investigated by JEOL-5400 (Tokyo, Japan) scanning electron microscope. Gold sputter coating of all the samples was done to render the surface of particles electroconductive. The micrographs were viewed at ×100 and ×500 magnifications.
Assessment of Reconstituted NE
The surface-adsorbed NE powder (420 mg) was resuspended in 3.75 ml double-distilled water and shaken gently for 5 min. The reconstituted NE (RF3) was characterized for globule size and zeta potential using Zetasizer ver. 6.01 (Malvern Instrument Ltd., UK) as described earlier. The globule size was visualized by TEM as detailed previously.
In Vitro Drug Release
In vitro drug release study of surface-adsorbed NE was performed by introducing the powder equivalent to 5 mg drug into the release test media using USP dissolution apparatus II (Hicon Enterprises, Delhi, India) with paddle rotation of 50 rpm. The dissolution media consisted of 500 ml of phosphate buffer, pH 7.4 maintained at 37 ± 0.5°C and samples were withdrawn at predetermined time intervals. The amount of drug release was estimated by measuring absorbance of the samples at 257.8 nm and the release profiles of surface-adsorbed NE and RF3 were evaluated for similarity against the release profile of F3 (optimized NE).
Statistical Analysis
The results were expressed as mean ± SD and were analyzed statistically by one-way analysis of variance (ANOVA) using Graph Pad Prism V5.04 software (San Diego, CA, USA).
RESULTS AND DISCUSSION
Screening of Components
Solubility of the therapeutic agent in the oil phase of nanoemulsion governs the ability of the nanoemulsion formulation to preserve the drug in solubilised form during its shelf life and after oral administration. Hence, the solubility of the therapeutic agent in oils, surfactants, and cosurfactants was the most significant criterion for the screening of NE components. OLM is hydrophobic and less polar in nature that was confirmed by its poor solubility of 0.0071 mg/ml in water (polar solvent). However, the solubility of OLM was higher in nonpolar solvents. Consequently, the solubility of OLM was found to be maximum (10.023 ± 1.517 mg/ml) in Capmul MCM, a medium chain mono/diglyceride (Fig. 1). Higher solubility of drug in Capmul MCM may be attributed to the nonpolar nature of the poorly water-soluble drugs that supports their solubilization in oils like medium chain triglycerides or mono- or diglycerides (7). High solubility of drug in oil is particularly advantageous in NE formulation. The higher the solubility of the drug in oil phase, the lower will be the volume of oil required to dissolve the single dose of drug. As a result, less quantity of surfactant and cosurfactant may be required for NE formulation. In addition to oil, the solubility of drug in the surfactant is also important. The solubility of OLM was highest in Tween 80, among the examined surfactants and PEG 400 among the examined cosurfactants. Thus, for the formulation of NE, Capmul MCM, Tween 80, and PEG 400 were selected as oil phase, surfactant, and cosurfactant, respectively.
Fig. 1.
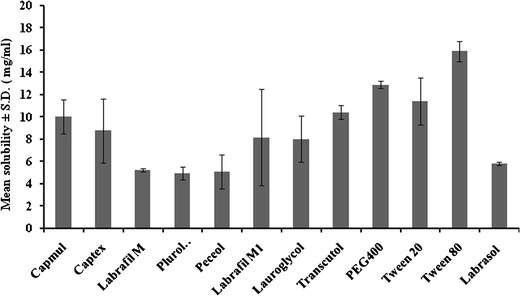
Saturation solubility bar chart for selection of excipients for nanoemulsion of OLM at 25 ± 1°C
Construction of Pseudoternary Phase Diagrams
The study of phase behavior helps to precisely characterize a phase boundary. Knowledge about the boundaries of the different phases as a function of composition variables can be attained by preparing phase diagrams. Selection of oil, surfactant, and the mixing ratio of oil to surfactant/cosurfactant mixture are vital for the NE formation (7). Low toxicity, resistance to pH, and ionic strength changes are some of the attributes that favor utilization of nonionic surfactants for the NE formulations. It was observed that when Tween80 (surfactant) was used alone, a significant zone of NE was obtained (Fig. 2a) but when PEG 400 (cosurfactant) was used along with the surfactant in a ratio of 1:1 (Fig. 2b), a tremendous decrease in the NE region was observed and 72.72% (v/v) of oil could be emulsified using 16.66% (v/v) of Smix. However, when the cosurfactant was utilized in 1:2 ratio (Fig. 2c), the amount of oil that could be emulsified was 52.17% (v/v) using 34.78% (v/v) of Smix. On further increasing the proportion of cosurfactant in the Smix to 1:3 (Fig. 2d), a decrease in NE region was observed with the maximum amount of oil that could be emulsified being reduced to 38.78% (v/v) using 52.17% (v/v) of Smix. With the Smix ratio of 1:4 (Fig. 2e), further decrease in the NE region was observed and the maximum amount of oil that could be emulsified was 36.36% (v/v).
Fig. 2.
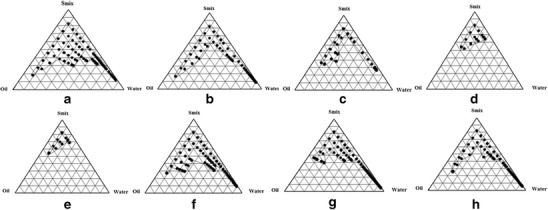
Pseudoternary phase diagrams involving Capmul MCM, Tween 80, and PEG 400 as the oil, surfactant, and cosurfactant, respectively. Ratio of surfactant to cosurfactant in a is 1:0, b 1:1, c 1:2, d 1:3, e 1:4, f 2:1, g 3:1, and h 4:1. Marked area oil in water NE region
In addition to varying cosurfactant in the Smix, the effect of varying the concentration of surfactant in the Smix from 1:1 to 2:1 (Fig. 2f) resulted in considerable increase in the NE region. On changing the concentration of surfactant in Smix from 2:1 to 3:1 (Fig. 2g) and then to 4:1 (Fig. 2h), it was observed that NE region did not change significantly. It was also concluded that 63.63% (v/v) of oil could be emulsified using 11.25% (v/v) of surfactant in comparison to emulsification of 10% (v/v) of oil by using 60% (v/v) of surfactant.
Selection of NEs from Phase Diagram
From the pseudoternary phase diagrams, 14 formulations were selected to fulfill the following criteria
The proportion of oil used should be able to solubilize the drug (single dose) completely. One milligram of OLM is dissolved easily in 0.1 ml of oil.
The minimum concentration of the Smix used for that amount of oil was taken.
The frequently used dose of the OLM is 5 mg (19). Therefore, 5 mg was selected as the dose for the development of NE formulation.
For convenience, 2 ml was selected as the volume of the NE formulation, so that it can be increased or decreased as per the requirement.
Thermodynamic Stress Stability
Thermodynamically stability of nanoemulsion formulations differentiates them from emulsions that have kinetic stability which, NEs by virtue of their thermodynamic stability do not exhibit phase separation, creaming, or cracking unlike emulsions that finally phase separate (20). Thus, the selected formulations were subjected to different thermodynamic stability tests such as centrifugation, heating–cooling cycles, and freeze–thaw cycles (Table I). The formulations that passed these tests were selected for the dispersibility study to evaluate the efficiency of emulsification.
Table I.
Selection of Final Test Nanoemulsion Formulations by Thermodynamic Stress Stability Studies and Dispersibilty Test
| Formulation code | S mix ratio | Percentage v/v of NE components | Observation | Results | Recoded formulations | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Oil | S mix | Water | Centrifugation | Heating-cooling cycle | Freeze-thaw cycle | Dispersibilty | ||||
| N1 | 1:0 | 10 | 40 | 50 | ✓ | ✓ | ✓ | Grade A | Passed | F1 |
| N2 | 1:0 | 10 | 38 | 52 | ✓ | ✓ | Х | Grade C | Failed | – |
| N3 | 1:0 | 10 | 36 | 54 | ✓ | ✓ | Х | Grade C | Failed | – |
| N4 | 2:1 | 10 | 15 | 75 | ✓ | ✓ | ✓ | Grade A | Passed | F2 |
| N5 | 2:1 | 10 | 42 | 48 | Х | – | – | Grade D | Failed | – |
| N6 | 2:1 | 10 | 40 | 50 | Х | – | – | Grade D | Failed | – |
| N7 | 2:1 | 10 | 38 | 52 | Х | – | – | Grade D | Failed | – |
| N8 | 2:1 | 10 | 37 | 53 | Х | – | – | Grade D | Failed | – |
| N9 | 3:1 | 10 | 15 | 75 | ✓ | ✓ | ✓ | Grade A | Passed | F3 |
| N10 | 3:1 | 10 | 43 | 47 | ✓ | Х | – | Grade C | Failed | – |
| N11 | 4:1 | 10 | 15 | 75 | ✓ | ✓ | ✓ | Grade B | Passed | F4 |
| N12 | 4:1 | 10 | 42 | 48 | Х | – | – | Grade D | Failed | – |
| N13 | 4:1 | 10 | 40 | 50 | Х | – | – | Grade D | Failed | – |
| N14 | 4:1 | 10 | 35 | 55 | ✓ | ✓ | ✓ | Grade B | Passed | F5 |
✓ = Pass, Х = fail
Dispersibilty
Gastrointestinal (GI) fluid is responsible for the dilution of oral NE and results in the gradual desorption of surfactant located at the oil–water interface. This process is thermodynamically governed by the tendency of the surfactant to preserve its concentration to its critical micelle concentration. In order to evaluate the emulsification efficiency of NE formulations upon infinite dilution in GI fluid, double-distilled water was used as a dispersion medium. This selection was based on the report corroborating insignificant difference in the behavior of NE, prepared using nonionic surfactants, dispersed in either water or simulated gastric or intestinal fluid (21,22). The formulations that passed the dispersibility test in double-distilled water in grade(s) A and B were selected for further study as these formulations were certain to remain as NE upon dispersion in the aqueous environment of the GI tract (Table I). Selected formulations were taken for globule size, zeta potential, PDI, refractive index, viscosity, percentage transmittance, drug content, and in vitro drug release determinations.
Characterization of NE
Globule Size, Zeta Potential, and Polydispersibility Index
Drug release and subsequent absorption are largely influenced by the globule size of NE (23). Rapid diffusion of drug from smaller droplets into aqueous phase favors drug dissolution. Globule size of the prepared NE was determined and results are shown in Table II. As observed, formulation F1 had the smallest globule size (15.08 ± 7.92 nm) followed by F3 (17.51 ± 5.87 nm). F1 formulation contained higher Smix ratio (40%) as compared to F3 (15%) that probably favored reduction in globule size. Diameter of the dispersed oil droplets of the NE was found to be much smaller than the diameter of smallest blood capillary (400 nm). This is advantageous as it avoids any probability of capillary blockage during transfer of the droplets in vivo. The small size of the globules also favors long circulation time (12,24)
Table II.
Mean (±SD, n = 3) Globule Size, Polydispersity Index (PDI), Zeta Potential, Drug Content, Refractive Index, Percentage Transmittance, and Viscosity of Final Test Formulations
| Formulation code | Mean globule size ± SD (nm) | Mean PDI ± SD | Mean zeta potential ± SD (mV) | Mean Drug content ± SD (%) | Mean refractive index ± SD | Mean percentage transmittance ± SD | Mean viscosity ± SD (cps) |
|---|---|---|---|---|---|---|---|
| F1 | 15.08 ± 7.92 | 0.150 ± 0.012 | −33.51 ± 0.21 | 85.66 ± 1.42 | 1.332 ± 0.004 | 88.67 ± 0.13 | 291 ± 0.78 |
| F2 | 79.09 ± 6.54 | 0.543 ± 0.021 | −48.72 ± 0.92 | 89.40 ± 2.04 | 1.389 ± 0.002 | 92.75 ± 0.64 | 286 ± 0.34 |
| F3 | 17.51 ± 5.87 | 0.203 ± 0.032 | −58.93 ± 0.98 | 95.60 ± 1.23 | 1.334 ± 0.003 | 99.78 ± 0.23 | 254 ± 0.35 |
| F4 | 29.21 ± 10.25 | 0.490 ± 0.041 | −58.32 ± 1.2 | 91.77 ± 3.21 | 1.374 ± 0.001 | 98.74 ± 0.08 | 293 ± 0.21 |
| F5 | 206.00 ± 8.76 | 0.480 ± 0.032 | −56.51 ± 1.09 | 86.24 ± 2.72 | 1.362 ± 0.013 | 96.88 ± 0.05 | 319 ± 1.04 |
Charge on the oil droplets in NE is another attribute that should be investigated while studying the absorption of nanoemulsion (25). Zeta potential also indicates NE stability with regards to degree of repulsion between adjacent, similarly charged globules in NE. Conventionally, zeta potential can be positive or negative in the range of −30 to +30 mV. The globules of OLM NEs displayed negative value of zeta potential (Table II) that could be due to the presence of negatively charged free fatty acids component of oil used in the formulation of nanoemulsion. Maximum value of zeta potential of −58.93 ± 0.98 mV was recorded for F3 closely followed by F4 and F5, and least zeta potential was documented for F1. Similar pattern was also observed for drug content determination and hence, it can be inferred that the amount of drug entrapped in the NE globule also influenced the magnitude of charge on the globules. The polydispersity index of F1 formulation was least (0.150 ± 0.012) followed by F3 formulation as 0.203 ± 0.032. Lower value of PDI is favorable, as it ensures uniformity in size of nanoemulsion globules.
Percentage Transmittance, Refractive Index, and Viscosity
The value of percentage transmittance of formulations F1 to F4 was closer to 100% indicating that the formulations were clear and transparent (Table II). Among all the formulations, F3 showed highest value of percentage transmittance (99.78 ± 0.23%) which was significantly (p < 0.001) higher in comparison to other formulations. The refractive index that also measures transparency was 1.334 ± 0.003 for F3 very close to that of water (1.334) and ensured its homogeneous character. The viscosity analysis revealed low viscosity for all formulations and F3 showed minimum viscosity of 254 ± 0.35 cps. Thus, F3 can be easily administered as a uniform dose.
In Vitro Drug Release
In vitro drug release profiles of OLM from NE formulations (F1–F5) and pure drug suspension (S) are shown in Fig. 3. The release of OLM from all the NE formulations was higher than the release profile of S (47.38% ± 0.352 in 12 h). Maximum %CDR of 96.69 ± 1.841 was displayed by formulation F3 followed by F4 (90.53% ± 3.311) in 12 h. The reason for the differences in percentage cumulative drug release of F3 and F4 can be correlated to the Smix composition and its quantity in NE. The Smix in F3 was in the ratio of 3:1whereas in F4 it was 4:1, both present in 15% by volume in respective NEs. As described in literature, an increase in surfactant concentration results in decrease in the droplet size, but this phenomenon levels off at a particular surfactant concentration whereby any further increase in surfactant concentration results in a raise in droplet size (26). Stabilization of oil droplets by virtue of decrease in droplet size can be attributed to the localization of surfactant monolayer at the oil–water interface (27). However, further rise in surfactant concentration leads to increased penetration of water into oil droplets causing breakdown of oil droplets and resulting in bigger droplets (28). Consequently, F4 had a larger globule size of 29.21 ± 10.25 nm in comparison to 17.51 ± 5.87 nm of F3 formulation that offered a larger surface area for the release of OLM. In F5, very high percentage of Smix (35%, v/v) resulted in large globules (206 ± 8.76 nm) that offered low surface area and consequently low %CDR of 72.16% in 12 h. In F1 and F2, low Smix ratios resulted in larger-sized globules and hence poor release than F3. Modeling of the in vitro release data indicated zero order as the best-fit model (Table III). On applying one-way ANOVA followed by Dunnett’s test, a significant difference was observed in the release profiles of F2, F4 (p < 0.05), and F3 (p < 0.01) in comparison to S (drug suspension).
Fig. 3.
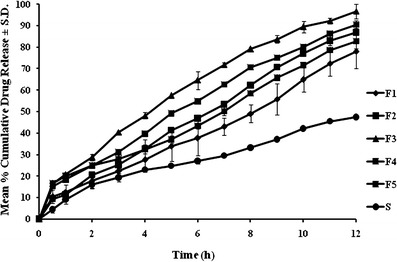
In vitro drug release study of NE formulations in phosphate buffer pH 7.4 at 37 ± 1 ±°C
Table III.
Interpretation of Drug Release Pattern of Nanoemulsion Formulations
| Formulation code | r 2 Value | Enhancement in % cumulative drug releasea | |||
|---|---|---|---|---|---|
| Zero order | First order | Higuchi | Peppas | ||
| F1 | 0.9723 | 0.9217 | 0.9143 | 0.8651 | 1.859 |
| F2 | 0.9876 | 0.8791 | 0.9163 | 0.8976 | 1.874 |
| F3 | 0.9854 | 0.8764 | 0.9747 | 0.9521 | 2.041 |
| F4 | 0.9882 | 0.8794 | 0.9643 | 0.9168 | 1.911 |
| F5 | 0.9458 | 0.9173 | 0.8976 | 0.9287 | 1.746 |
| S | 0.9256 | 0.8769 | 0.9043 | 0.8970 | – |
aEnhancement in %CDR after 12 h with respect to suspension
Selection and Development of Optimized Formulation
Formulation F3 with highest %CDR of 96.69 ± 1.841, least globule size (17.51 ± 5.87 nm), lower PDI value (0.203 ± 0.032), and high zeta potential (−58.93 ± 0.98 mV) was selected as the optimized formulation and was considered as stable and homogeneous formulation. Transmission electron microscopy of F3 revealed dark and spherical spots against a light background and the droplet size revealed by TEM was in conformity with the zeta sizing results. Although NE is one of the finest mode of delivery for hydrophobic therapeutic agent OLM, but due to liquid nature of the dosage form, it is normally associated with transportation issues, instability problems, and poor palatability due to the lipid content (in case of NE). Moreover, dose variability due to handling problems in case of select patient population may lead to toxicity. To manage these challenges, liquid NE can be developed as a solid dosage form by adsorbing on a highly porous solid with sufficient adsorbing capacity and convert it to a free-flowing powder. Colloidal silicon dioxide, a highly porous solid with a specific surface area of 200–400 m2/g was selected (29) for the development of capsular dosage form of surface-adsorbed NE.
Characterization of Surface-Adsorbed NE
Micromeritic and Rheological Characterization
The bulk density of surface-adsorbed NE was found to be 0.610 ± 0.103 g/cm3 and the tapped density was 0.7812 ± 0.052 g/cm3. Close values of bulk and tapped density will ensure uniform filling of capsule shell at industrial scale. The Hausner’s ratio derived from bulk and tapped density was found to be 1.287 ± 0.572. A Hausner’s ratio of less than 1.25 indicates good flow property of the granules (30) and slightly higher value of Hausner’s ratio can be improvised by use of flow activators. The angle of repose that was 31.79° ± 0.54, categorized the flow as passable (31) reconfirming the results of Hausner’s ratio. Carr’s compressibility index was found to be 28.073 ± 1.782% indicating compressible nature of the surface-adsorbed NE that presents an opportunity for tabletting of the surface-adsorbed NE. All these results proved that the surface-adsorbed NE powder can be aptly filled in the hard gelatin capsule as a solid unit dosage form.
Scanning Electron Microscopy
Colloidal silicon dioxide appeared to be spherical particles approximating 100 μm (Fig. 4a). The micrograph of surface-adsorbed NE was not different from colloidal silicon dioxide indicating uniform adsorption of NE (F3) over colloidal silicon dioxide (Fig. 4b). No unusual signs of precipitation/crystallization of drug/excipient on the surface of adsorbent were recognizable. This confirms that the NE was homogeneously adsorbed over colloidal silicon dioxide.
Fig. 4.

Scanning electron micrographs of a colloidal silicon dioxide and b surface-adsorbed NE
Reconstitution of Surface-Adsorbed NE and Evaluation
The particles of surface-adsorbed NE are expected to be released from the capsule and on contact with body fluids, these should instantaneously re-form into NE quite similar to the initial formulation. To assess the reconstitution ability, specified quantity of surface-adsorbed NE was shaken with specified quantity of water to get RF3 that was characterized for globule size, polydispersity index, zeta potential, and drug content. Results of the characterization are given in Table IV. Globule size, polydispersity index, zeta potential, and percentage drug content of formulation RF3 were found to be significantly different (p < 0.05) in comparison to formulation F3. The morphology of the reconstituted NE is shown in Fig. 5b. As observed, the globules were spherical but of slightly bigger in size than F3. The colloidal silicon dioxide particles might have interfered with the reconstitution of NE and consequently a higher globule size, PDI were observed. Lowering of drug content can be attributed to adsorption of OLM on colloidal silcon dioxide. The reconsitutional behavior of surface-adsorbed NE is expected to be different in vivo. The gastrointestinal fluid constituents containing biosurfactants in conjugation with the peristaltic movements will probably facilitate the reconstitution of NE favoring lower globule size, probably close to initial formulation F3.
Table IV.
Comparative Characterization of Reconstituted Nanoemulsion (RF3) and Nanoemulsion (F3)
| Formulation code | Mean globule size ± SD (nm) | Mean PDI ± SD | Mean zeta potential ± SD (mV) | Mean drug content ± SD (%) |
|---|---|---|---|---|
| RF3 | 28.98 ± 0.98 | 0.332 ± 1.35 | −35.90 ± 0.45 | 92.77 ± 2.16 |
| F3 | 17.51 ± 5.87 | 0.203 ± 0.03 | −58.93 ± 0.98 | 95.60 ± 1.23 |
Fig. 5.

Transmission electron micrographs of a optimized nanoemulsion (F3) and b reconstituted nanoemulsion (RF3)
Dosage Form Development of Surface-Adsorbed NE and its Evaluation
On the basis of bulk density of surface-adsorbed NE, bulk volume 0.61 cm3 was calculated for surface-adsorbed NE. This bulk volume can be suitably filled in size “0” capsule having body volume of 0.69 cm3 (32) for a dose size of 5 mg of OLM. The in vitro drug release profile (Fig. 6) of encapsulated surface-adsorbed NE showed lower drug release in the initial first hour in comparison to surface-adsorbed NE attributable to the additional disintegration step of the capsule shell. Later on, beyond 3 h, the release magnitude was similar to reconstituted NE(RF3) and NE(F3) till the 12th hour. The similarity factor (f2) of the release profiles was calculated using PCP Disso V2.08 software. The value of f2 between F3(reference) and surface-adsorbed NE (test) was found to be 66 and for F3(reference) and reconstituted NE it was found to be 73. A value of f2 factor between 50 and100 indicates similarity in given set of the reference and test samples. Hence, the in vitro drug releases of F3, surface-adsorbed NE and RF3 can be adjudged as similar. This study illustrates the potential of encapsulated surface-adsorbed NE as a suitable solid unit dosage form to circumvent the handling limitations associated with SMEDDS or SNEDDS or NE of water-insoluble therapeutic agents.
Fig. 6.
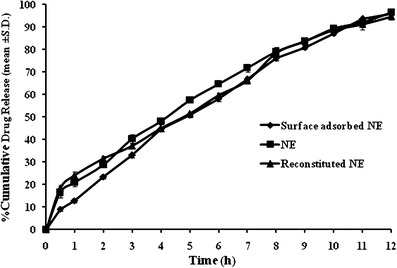
In vitro drug release profiles of F3, surface-adsorbed NE, and RF3 in phosphate buffer pH 7.4 at 37 ± 1 ±°C
CONCLUSION
OLM nanoemulsion was successfully prepared by the aqueous titration method. Formulation containing capmul MCM (10% v/v), tween 80 (11.25% v/v), PEG 400 (3.75% v/v), and double-distilled water (75% v/v) was optimized and was converted to solid powder by adsorbing onto colloidal silicon dioxide and encapsulated. The in vitro release profile of the encapsulated system was statistically similar to nanoemulsion proving the performance efficacy of the encapsulated system. However, the performance characteristics need in vivo evaluation. The developed system can be considered as a novel solid unit dosage form of OLM nanoemulsion that has the potential to circumvent the handling limitations associated with NE.
Acknowledgments
The authors are thankful to Sun Pharmaceutical Industries, Sikkim, India for providing the gift sample of olmesartan medoxomil.
References
- 1.Murakamia T, Konnoa H, Fukutsua N, Onoderaa M, Kawasakia T, Kusub F. Identification of a degradation product in stressed tablets of olmesartan medoxomil by the complementary use of HPLC hyphenated techniques. J Pharmaceut Biomed Anal. 2008;47:553–559. doi: 10.1016/j.jpba.2008.02.021. [DOI] [PubMed] [Google Scholar]
- 2.Park JH, Chang JS, El-Gamal MI, Choi WK, Lee WS, Chung HJ, et al. Novel amides and esters prodrugs of olmesartan: synthesis, bioconversion, and pharmacokinetic evaluation. Bioorg Med Chem Lett. 2010;20:5895–5899. doi: 10.1016/j.bmcl.2010.07.089. [DOI] [PubMed] [Google Scholar]
- 3.Klaus O, Stumpe MD. Olmesartan compared with other angiotensin II receptor antagonists: head to head trial. Clin Ther. 2004;26:A33–A37. doi: 10.1016/S0149-2918(04)90144-0. [DOI] [PubMed] [Google Scholar]
- 4.Wehling M. Can the pharmacokinetic characteristics of olmesartan medoxomil contribute to the improvement of blood pressure control? Clin Ther. 2004;26:A21–A27. doi: 10.1016/S0149-2918(04)90142-7. [DOI] [PubMed] [Google Scholar]
- 5.Nakagomi-Hagihara R, Nakai D, Kawai K, Yoshigae Y, Tokui T, Abe T. Oatp1b1, Oatp1b3 and Mrp2 are involved in hepatobiliary transport of olmesartan, a novel angiotensin II Blocker. Drug Metabol Dispos. 2006;34:862–869. doi: 10.1124/dmd.105.008888. [DOI] [PubMed] [Google Scholar]
- 6.Matsushima S, Maeda K, Kondo C, Hirano M, Sasaki M, Suzuki H, et al. Identification of the hepatic efflux transporters of organic anions using doubletransfected Madin-Darby canine kidney II cells expressing human organic anion-transporting polypeptide 1B1 (OATP1B1)/multidrug resistanceassociated protein 2, OATP1B1/multidrug resistance 1, and OATP1B1/breast. J Pharmacol Exp Ther. 2005;314:1059–1067. doi: 10.1124/jpet.105.085589. [DOI] [PubMed] [Google Scholar]
- 7.Lawrence MJ, Rees GD. Microemulsion-based media as novel drug delivery system. Adv Drug Deliv Rev. 2000;45:89–121. doi: 10.1016/S0169-409X(00)00103-4. [DOI] [PubMed] [Google Scholar]
- 8.Qian C, McClements DJ. Formation of nanoemulsions stabilized by model food-grade emulsifiers using high-pressure homogenization: factors affecting particle size. Food Hydrocolloids. 2011;25:1000–1008. doi: 10.1016/j.foodhyd.2010.09.017. [DOI] [Google Scholar]
- 9.Chen H, Khemtong C, Yang X, Chang X, Gao J. Nanonization strategies for poorly water soluble, drugs. Drug Discov Today. 2011;16:354–360. doi: 10.1016/j.drudis.2010.02.009. [DOI] [PubMed] [Google Scholar]
- 10.Shono Y, Nishihara H, Matsuda Y, Furukawa S, Okada N, Fujita T, et al. Modulation of intestinal P-glycoprotein function by cremophor EL and other surfactants by an in vitro diffusion chamber method using the isolated rat intestinal membranes. J Pharm Sci. 2004;93:877–885. doi: 10.1002/jps.20017. [DOI] [PubMed] [Google Scholar]
- 11.Lee BS, Kang MJ, Choi WS, Choi YB, Kim HS, Lee SK, et al. Solubilized formulation of olmesartan medoxomil for enhancing oral bioavailability. Arch Pharm Res. 2009;132:1629–1635. doi: 10.1007/s12272-009-2117-x. [DOI] [PubMed] [Google Scholar]
- 12.Bali V, Ali M, Ali J. Nanocarrier for the enhanced bioavailability of a cardiovascular agent: in vitro, pharmacodynamic, pharmacokinetic and stability assessment. Int J Pharm. 2011;403:46–56. doi: 10.1016/j.ijpharm.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 13.Kohli K, Chopra S, Dhar D, Arora S, Khar RK. Self-emulsifying drug delivery systems: an approach to enhance oral bioavailability. Drug Discov Today. 2010;15:958–965. doi: 10.1016/j.drudis.2010.08.007. [DOI] [PubMed] [Google Scholar]
- 14.Vior MCG, Monteagudo E, Dicelio LE, Awruch J. A comparative study of a novel lipophilic phthlocyanine incorporated into NE formulation: photophysics, size, solubility and thermodynamic stability. Dyes Pig. 2011;91:208–214. doi: 10.1016/j.dyepig.2011.03.011. [DOI] [Google Scholar]
- 15.Mandawgade SD, Sharma S, Pathak S, Patrawale VB. Development of SMEDDS using natural lipophile: application to artemether delivery. Int J Pharm. 2008;362:179–183. doi: 10.1016/j.ijpharm.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 16.Date AA, Nagarsenker MS. Design and evaluation of self nanoemulsifying drug delivery system (SNEDDS) for cefpodoxime proxetil. Int J Pharm. 2007;329:166–172. doi: 10.1016/j.ijpharm.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 17.Patel AR, Vavia PR. Preparation and in vivo evaluation of SMEDDS (self-microemulsifying drug delivery system) containing fenofibrate. AAPS J. 2007;9:E344–E352. doi: 10.1208/aapsj0903041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nazzal S, Khan MA. Controlled release of a self-emulsifying formulation from a tablet dosage form: stability assessment and optimization of some processing parameters. Int J Pharm. 2006;315:110–121. doi: 10.1016/j.ijpharm.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 19.Sinko PJ. Martin’s pharmacy and pharmaceutical sciences. 5th ed. Woltre Kulwer (India) Pvt Ltd.; 2008.
- 20.Laies P, Puchler K, Kirch W. The pharmacokinetics and metabolic profile of olmesartan medoxomil limits the risk of clinically relevant drug interaction. J Hypertens. 2001;19:S21–S32. doi: 10.1097/00004872-200101000-00003. [DOI] [PubMed] [Google Scholar]
- 21.Shinoda K, Kuneda H. The effect of salt concentration, temperature, and additives on the solvent property of aerosol OT solution. J Colloid Interface Sci. 1987;118:586–589. doi: 10.1016/0021-9797(87)90491-7. [DOI] [Google Scholar]
- 22.Shafiq S, Shakeel F, Talegaonkar S, Ahmad FJ, Khar RK, Ali M. Development and bioavailability assessment of ramipril nanoemulsion formulation. Eur J Pharm Biopharm. 2007;66:227–243. doi: 10.1016/j.ejpb.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 23.Khoo SM, Humberstone AJ, Porter CJH, Edwards GA, Charman WN. Formulation design and bioavailability assessment of lipidic self-emulsifying formulations of halofantrine. Int J Pharm. 1998;167:155–164. doi: 10.1016/S0378-5173(98)00054-4. [DOI] [Google Scholar]
- 24.Rajpoot P, Bali V, Pathak K. Anticancer efficacy, tissue distribution and blood pharmacokinetics of surface modified nanocarrier containing melphalan. Int J Pharm. 2012;426:219–230. doi: 10.1016/j.ijpharm.2012.01.027. [DOI] [PubMed] [Google Scholar]
- 25.Constanitinides PP, Scalart JP, Lancaster C, Marcello J, Marks G, Ellens H. Formulation and intestinal absorption enhancement evaluation of water-in-oil microemulsions incorporating medium-chain glycerides. Pharm Res. 1994;11:1385–1390. doi: 10.1023/A:1018927402875. [DOI] [PubMed] [Google Scholar]
- 26.Gershanik T, Benzeno S, Benita S. Interaction of the self-emulsifying lipid drug delivery system with mucosa of everted rat intestine as a function of surface charge and droplet size. Pharm Res. 1998;15:863–869. doi: 10.1023/A:1011968313933. [DOI] [PubMed] [Google Scholar]
- 27.Yoo JH, Shanmugam S, Thapa P, Lee ES, Balakrishnan P, Yoo SK. Novel self-nanoemulsifying drug delivery system for enhanced solubility and dissolution of lutein. Arch Pharm Res. 2010;33:417–426. doi: 10.1007/s12272-010-0311-5. [DOI] [PubMed] [Google Scholar]
- 28.Gursoy RN, Benita S. Self-emulsifying drug delivery system (SEDDS) for improved oral delivery of lipophilic drugs. Biomed Pharmacother. 2004;58:173–182. doi: 10.1016/j.biopha.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Pouton C. Formulation of self-emulsifying drug delivery systems. Adv Drug Deliv Rev. 1997;25:47–58. doi: 10.1016/S0169-409X(96)00490-5. [DOI] [Google Scholar]
- 30.Row CR, Sheskey PJ, Owe SC. Handbook of pharmaceutical excipients. 5th ed. Royal society of Great Britain; 2006.
- 31.Aulton ME. The science of dosage form design. 2. London: Churchill & Livingstone; 2002. [Google Scholar]
- 32.Gennaro AR. Remington: the science and practice of pharmacy. 20. Philadelphia: Lippincott Williams & Wilkins; 2004. [Google Scholar]