

Abstract
The specific effects of glucose deprivation on oxidative pentose phosphate cycle (OPPC) function, thiol homeostasis, protein function and cell survival remain unclear due to lack of a glucose-sensitive chemical probe. Using p53 wild type and mutant human colon cells, we determined the effects of hydroxyethyl disulfide (HEDS) on NADPH, GSH, GSSG, total glutathione, total non-protein and protein thiol levels, the function of the DNA repair protein Ku, and the susceptibility to radiation-induced free radicals under normal glucose or glucose-deprived conditions. HEDS is rapidly detoxified in normal glucose but triggered a p53-independent metabolic stress in glucose depleted state that caused loss of NADPH, protein and non protein thiol homeostasis and Ku function, and enhanced sensitivity of both p53 wild type and mutant cells to radiation induced oxidative stress. Additionally, high concentration of HEDS alone induced cell death in p53 wild type cells without significant effect on p53 mutant cells. HEDS offers a useful tool to gain insights into how glucose metabolism affects OPPC dependent stress-induced cellular functions and injury, including in tumor cells, where our findings imply a novel therapeutic approach to target glucose deprived tumor. Our work introduces a novel probe to address cancer metabolism and ischemic pathology.
Keywords: thiol homeostasis, glutathione, Ku function, glucose deprivation, ischemic pathology, oxidative stress
INTRODUCTION
Tissues are stressed by glucose deprivation during ischemia due to lack of blood flow (Russell et al., 2004; Martin et al., 2009; Romano et al., 1993; Brongholi et al., 2006; Goldberg et al., 1993; Milusheva et al., 1996). In solid tumors, the stress is constitutive, because overall steady-state levels of glucose are relatively low (Aronen et al., 2000; Rajendran et al., 2004; Izuishi et al., 2000). Ischemic/reperfusion models used to study such cellular stress (Ayene et al., 1992a; 1992b; 1993; Fisher et al., 1991; 1994; Zhao et al., 1997) suggest that it can be decreased by synthetic thiol compounds such as mercaptopropionylglycine (Ayene et al., 1993). Other evidence of a thiol depletion-dependent mechanism of tissue injury exists, based on documentation of decreased levels of intracellular glutathione after reperfusion of ischemic organs (Ceconi et al., 2000). While glucose deprivation and hypoxia are required to mimic ischemia, the relationship to OPPC, thiol homeostasis, protein function and cell survival during glucose deprivation-dependent tissue injury has yet to be established either in vitro or in vivo (Fang et al., 2009; Fern and Moller, 2000; Kintner et al., 2007; Wang et al., 2009; Zhou et al., 2009).
Cellular thiol homeostasis is tightly regulated by maintenance of the thiol status of certain protein and non-protein thiols. Homeostasis is crucial because oxidized and reduced forms of protein thiols determine the function of many cellular proteins. Thus, the operation of many metabolic pathways is tightly coordinated with cellular thiol homeostasis. A critical pathway regulating cellular thiol homeostasis during oxidative stress is the oxidative pentose phosphate cycle (OPPC), which delivers the major source of cellular NADPH that is critical to maintain reduced glutathione at levels needed for cellular thiol homeostasis and cell survival during oxidative stress (Ayene et al., 2000; 2002; 2008; Biaglow et al., 2000; Li et al., 2012; Pandolfi et al., 1995; Salvemini et al., 1999; Tuttle et al. 2007). Because of its reliance on glucose, the ability of OPPC to generate NADPH is likely to be attenuated by glucose deprivation that occurs commonly in ischemic tissues or solid tumors. In cancer cells, this question has intensified with direct evidence that steady-state concentrations of glucose are significantly reduced in solid tumors (Aronen et al., 2000; Izuishi et al., 2000; Rajendran et al., 2004). Unlike cells in normal tissue, where glucose deprivation is injurious, cancer cells in solid tumors can often adapt to glucose-deprivation induced oxidative stress by mechanisms that also permit them to acquire therapeutiuc resistance (Kato et al., 2002; Lee 2001; Li et al., 2009; Park et al., 2007; Wyld et al., 2002; Yun et al., 2005). While the effects of glucose deprivation have received significant attention, the specific role of glucose deprivation without the confounding effects of other nutrient, growth factors and oxygen depletion on the cellular control of thiol homeostasis and its subsequent effects on protein function, cell survival and response to free radicals induced oxidative stress have not been demonstrated in either cancer or normal cells.
To a large extent, this gap in knowledge is attributable to the lack of any chemical biological probes that can specifically assess thiol homeostasis in glucose-deprived cells. In earlier work, we discovered that a brief exposure of CHO cells, which lacks wild type p53, to hydroxyethyl disulfide, a thiol-specific oxidant, uniquely altered reduced glutathione levels and enhanced susceptibility to oxidative stress only in G6PD deficient rodent cells lacking OPPC activity but grown in normal growth medium (Ayene et al., 2000; 2002; 2008). Preliminary studies from our group have also demonstrated that CHO rodent cells incubated in phosphate buffered saline without glucose for 1 hour led to altered protein and non protein thiol homeostasis and radiation sensitization by 20 mM HEDS (Biaglow et al., 2003). These studies with CHO cells did not determine the specific role of glucose deprivation since these studies were carried out in phosphate buffered saline (PBS) that also lacked other growth factors and nutrirents that are normally present in tissue culture growth medium. The current studies will not only determine the use of HEDS as a bioactive probe for glucose regulated pathways against other type of oxidative stress in human cells but also will determine a comprehensive mechanism of HEDS effect as it relates to redox regulation of DNA repair protein Ku function, for the first time, in any type of glucose deprived cells.
MATERIALS AND METHODS
Cell Culture
HT29 and HCT116 human colon cancer cells obtained originally from the American Type Tumor Collection were a gift from Cameron Koch (University of Pennsylvania). These cells were selected on the basis of their known differential sensitivities to oxidative stress induced by hypoxia, radiation or chemotherapeutic agents that are derived in part from their differential p53 status (HCT116 cells are wild-type and HT29 cells are mutant), reflecting the most common alteration in human cancers generally. 8 × 105 cells cultured in DMEM containing 15% FCS and 25 mM HEPES were seeded in 60 mm Nunc dishes or six-well plates on the day before the experiment. Growth medium was removed and immediately replenished with glucose and HEPES containing DMEM medium and 2% dialyzed FCS. For glucose deprivation, the cells were replenished with glucose free DMEM, 2% dialyzed FCS and 25 mM HEPES. The cells in these plates under the conditions described above were incubated in 5% CO2 incubator at 37°C for 4 hrs before measuring glucose level, metabolic activity and radiation response in the presence and absence of HEDS.
HEDS treatment
A stock solution of 0.1 M HEDS (Sigma, USA) was prepared fresh in DMEM growth medium without glucose. After 4 hr glucose starvation cells were exposed to the indicated concentration of HEDS for 3 hr in 1 ml (six-well plate) or 1.5 ml (60 mm plate) of fresh DMEM growth medium with or without glucose, 2% dialyzed FCS and HEPES.
Extracellular Glucose Assay
Glucose levels in the medium before and after glucose starvation were quantified by following the reduction of NADP to NADPH in the reaction catalyzed by ATP, hexokinase (HK) and glucose 6- phosphate dehydrogenase (G6PD). Briefly, 25 μl of the sample is mixed with 175 μl of 0.1 M sodium acetate buffer (pH 4.6). Fifty μl of this sample mixture is mixed with 50 μl of ATP/NADP solution (455 mg ATP-Na2H2.3H2O/50 mg NADP-Na2H2 in 5 ml of Trizma (0.3 M), MgSO4 (0.3 M) buffer, pH 7.5), 20 μl of G6PD/HK (1:10 in 3.2 M ammonium sulfate) and 625 μl of 0.3 M Trizma/MgSO4 buffer. The O.D. was continuously measured at 340 nm every min for up to 10 min and the concentration was calculated using a glucose standard curve.
Quantification of HEDS detoxification/bioreduction
The amount of mercaptoethanol (ME) produced from HEDS by cells was estimated by either High Performance Liquid Chromatography and electrochemical detection (HPLC/EC), or 5, 5-dithiobis 2-nitrobenzoic acid (DTNB) assays (Ayene et al., 2000; 2002; 2008)). Cells treated with and without HEDS were cooled on ice. To quantify the bioreduction/ME production, 0.5 ml of extracellular medium was mixed with 0.5 ml of 100 mM sulfosalicyclic acid (SSA) lysis buffer in microfuge tubes and centrifuged in a microfuge. For HPLC/EC assay, the medium extract was analyzed using HPLC system consisting of a single pump, autosampler, guard cell, 5010 analytical cell and Colouchem III (ESA, USA). Diluted sample (10 μl) was loaded onto a C18 column and run in an isocratic mode using a mobile phase with 50 mM phosphate, pH 2.7, 0.05 mM octane sulfonic acid and 2.2% acetonitrile. For 5, 5-dithiobis 2-nitrobenzoic acid (DTNB) assay, 150 μl of the extract was mixed with 1200 μl of phosphate buffer and 150 μl of 10 mM DTNB. The optical density of this reaction mixture was measured at 412 nm and the concentration was calculated using an extinction coefficient of 1.36×104 for reduced DTNB.
Estimation of Intracellular Reductants NADPH by High Performance Liquid Chromatography (HPLC)
NADPH was quantified by a modified method as described previously (Noack et al. 1992) using an HPLC with fluorescence detection (Waters 474) at an excitation and emission wavelength of 340 and 455 nm, respectively. Pyridine nucleotides were extracted by 70% methanol directly added to the cells after two rinses with ice-cold PBS. NADPH was eluted with 0.1 M KH2PO4 (pH 6.1) using a step gradient of methanol; 5 min, 0% CH3OH; 6 min, 4% CH3OH; 5 min, 12% CH3OH; 14 min, 40% CH3OH. The data were collected and analyzed using Jasco Borwin software.
Quantification of intracellular non protein thiols (NPSH) and glutathione (GSH)
The intracellular non protein thiol (NPSH) was determined by DTNB assay as described previously (Li et al., 2009). Cells in the dish with the desired medium were rinsed twice and the attached cells were mixed with 1 ml of ice cold 50 mM SSA buffer. After 10 min on ice, cells were scraped with a teflon spatula and centrifuged in a microfuge. Three hundred microliter of the extract was mixed with 1050 μl of phosphate buffer and 150 μl of 10 mM DTNB, and the concentration was calculated as described above.
Reduced (GSH), oxidized (GSSG) and total glutathione (GSH+GSSG) were measured by a glutathione assay kit from Biovision, CA, USA. Cells were isolated and homogenized in 100 μl glutathione assay buffer. Sixty microliter of each homogenate mixed in 20 μl of perchloric acid (PCA) was vortexed, centrifuged and supernatant (40 μl) was mixed with 20 μl of ice cold 3N potassium hydroxide to precipitate PCA and neutralize the samples. The supernatant of these samples after centriguation was used for the assay as per the manufacturer’s instructions. Briefly, 10 μl of O-phthalaldehyde probe (OPA) was added to the standards and samples mixed with assay buffer with and without reducing agent mix or GSH quencher in a 96 well plate to measure GSH, total glutathione and GSSG. The glutathione concentration was calculated from the amount in samples determined using glutathione as a standard.
Quantification of intracellular protein thiols (PSH)
Cells attached to the dishes incubated with and without HEDS were washed three times with PBS and then treated with SSA as described above. This treatment precipitates cellular macromolecules in place without loss of protein. The acid was removed and dishes were washed two more times with 5 ml SSA. Under these experimental conditions, only the non protein thiols were washed off the cells. Acid-fixed cells were then incubated with 1 ml of 100 mM phosphate buffer, pH7.4, containing 1.5 mM dithiobis nitrobenzoic acid (DTNB) for 15 min at 37°C and the absorbancy read at 412 nm. The results were presented as percentage of untreated control.
Quantification of nuclear Ku function by Enzyme Linked Immunosorbent Assay (ELISA)
Cells grown in tissue culture dishes under different experimental conditions were cooled on ice and rinsed with 2.5 ml ice-cold phosphate buffered (0.01 M phosphate buffer, pH7.5, 0.15 M NaCl, 2.7 mM KCl) saline (PBS). These cells were isolated and centrifuged in ice cold PBS (1.5 ml) for 5 min at 500 rpm and cell pellet was used for the preparation of nuclear extract. Both nuclear extract preparation and ELISA analysis were carried out in the absence of DTT as described previously (Ayene et al. 2002). This modification of the Active Motif nuclear extract preparation is necessary to avoid the reversal of HEDS mediated oxidation of protein thiols including Ku protein thiols by DTT (Ayene et al. 2002). The nuclear proteins were prepared by membrane disruption using Active Motif hypotonic buffer, detergent and complete lysis buffer and centrifugation as per the manufacturer’s instructions (Li et al., 2009). The nuclear proteins were stored at −80°C. Protein concentration was quantified by Biorad Reagent.
The DNA binding efficiency of nuclear Ku was quantified by Ku DNA repair kits from Active Motif USA as described previously (Li et al., 2009). Briefly, nuclear protein (1 μg) mixed with AM6 binding buffer was incubated in an eight well strip provided by Active Motif for 1 hr at RT, washed and incubated with Ku70 antibody for 1 hr. After incubation, the wells were washed, incubated with developing solution for 4 min, mixed with stop solution and the O.D was read in a microplate reader at 450 nm with a reference wavelength of 655 nm. The results were presented as percentage of untreated control.
Cell Irradiation
Cells plated in 60 mm dishes with and without glucose were exposed to a single dose of 4 Gy of γ radiation (Dose rate-9.63Gy/min) at room temperature in air using a J.L. Shepherd Mark I 137Cs irradiator. This dose of radiation was most commonly used by us and others to determine the free radicals induced oxidative stress in mammalian cells. Cells were treated with HEDS as described above.
Determination of Cell Death by Clonogenic Assay
Mitotic cell death was quantified by clonogenic assay. Briefly, cells were harvested after treatment and plated at the required concentration in 100 mm dishes with no more than 250 colonies per dish; a viable colony was defined as having at least 50 cells. The surviving fraction of cells at a given dose of irradiation was calculated from the number of colonies formed, number of cells plated and plating efficiency of the control in glucose.
Statistical Analysis
Each data point presented was the mean + standard deviations (SD) of at least three to five experiments with SD as shown unless smaller than points plotted. The statistical significance of the differences between the groups was determined by Analysis of Variance (ANOVA) with the ‘p’ values presented in the legend of each figure.
RESULTS
Under the cell culture conditions employed, the accurate concentrations of glucose in glucose rich and glucose free media before and after a 4 hr incubation of HCT116 and HT29 cells was determined by enzymatic assay that uses hexokinase to oxidize glucose (Figure 1A,B). Glucose concentration measured in DMEM medium with glucose collected immediately after addition to the dish was 20 mM. The hexokinase assay demonstrated that the starting concentration of glucose in glucose free medium under these experimental conditions was approximately 0.3 mM. Our results showed no significant change in non-protein thiols (NPSH) by HEDS measured after a 3 hr incubation in the presence of glucose, suggesting that the NPSH level is maintained by OPPC (Figure 1C,D). However, depletion of glucose in the medium significantly decreased the NPSH level in both cell lines, at all concentrations of HEDS examined, suggesting a direct correlation between the lack of glucose and altered thiol homeostasis during oxidative stress induced by HEDS as a bioactive thiol-specific oxidant (Figure 1C,D). Although the intracellular NPSH showed a HEDS concentration-dependent decrease in the absence of glucose, we used 0, 0.7 and 3.3 mM HEDS for our studies since these two concentrations showed a marked difference in NPSH.
Figure 1. HEDS decreases non-protein thiols (NPSH) in glucose-deprived human colon cancer cells independent of p53 status.
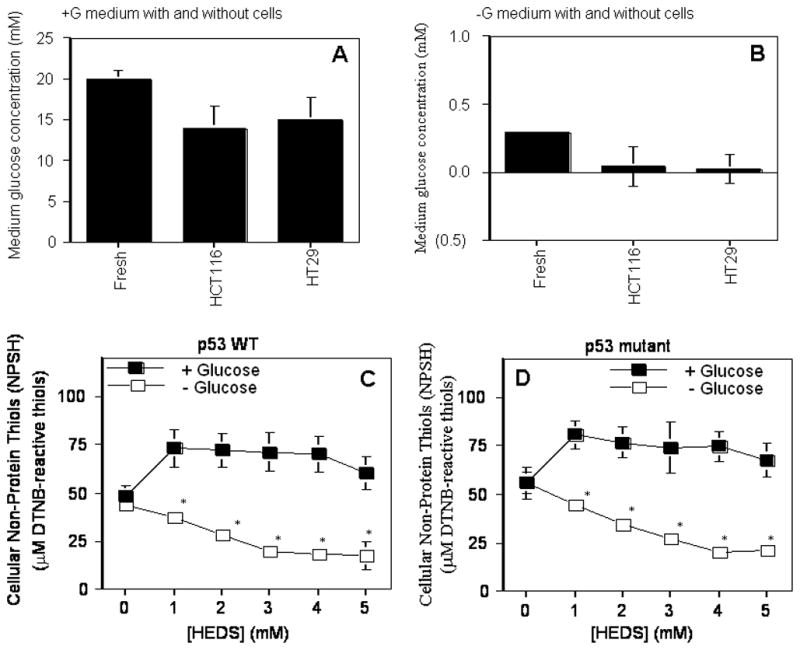
(A, B). Glucose level in DMEM medium with (A) or without (B) glucose plus 2% dialyzed FBS. Glucose concentration measured in DMEM medium with glucose was 20 mM as compared to 0.3 mM in glucose free medium. (C, D). Quantification of intracellular non protein thiols. The effect of glucose deprivation on intracellular non-protein thiol was determined in the absence or presence of HEDS at the indicated concentration in cells that are radiation-sensitive and p53 wild-type HCT116 (C) or radiation-resistant and p53 mutant HT29 (D). Mean for three to five independent experiments are shown with SD. A statistically significant reduction in NPSH levels in cells cultured in glucose-deprived media occurred after HEDS treatment (*P<0.01).
We quantified the effect of HEDS (0, 0.7 and 3.3 mM) on glutathione (GSH) that plays a major role in cellular thiol redox homeostasis (Figure 2A – F). Glutathione was not affected by HEDS measured after three hours incubation in the presence of glucose consistent with its effect on total non protein thiols (Figure 1) that includes glutathione. Depletion of glucose in both these cells led to a decrease in glutathione (GSH) with concomitant increase in oxidized glutathione (GSSG) but with no significant decrease in total glutathione (GSH+GSSG).
Figure 2. HEDS decreases glutathione (GSH) with concomitant increase in oxidized glutathione (GSSG) in glucose-deprived human colon cancer cells independent of p53 status.
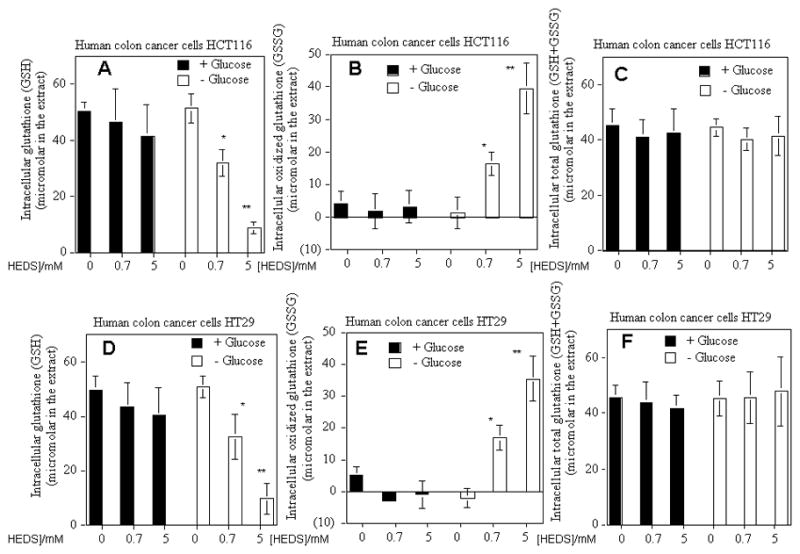
The effect of glucose deprivation on intracellular glutathione levels (GSH (A,D), GSSG (B,C) and total GSH+GSSG (C,F) was determined in the absence or presence of HEDS at the indicated concentration in cells that are radiation-sensitive and p53 wild-type HCT116 (A,B,C) or radiation-resistant and p53 mutant HT29 (D,E,F). Mean for three to five independent experiments are shown with SD. A statistically significant reduction in GSH with concomitant increase in GSSG but without any change in total glutathione in cells cultured in glucose-deprived media occurred after HEDS treatment (*P<0.01; **P<0.001).
We next measured protein thiol (PSH) levels to determine whether HEDS can also deplete PSH in cells cultured in low glucose medium but not normal glucose medium (Figure 3). The results showed only a small change in PSH levels in HCT116 and HT29 cells measured after incubation with HEDS, in the presence of glucose, suggesting that the overall PSH level is maintained fairly well in the cells by the presumptive OPPC-dependent reductants (Figure 3). However, depletion of glucose in the medium significantly decreased the PSH levels by about 15% in cells treated with low (0.7 mM) concentration of HEDS (Figure 3). At high (3.3 mM) concentration of HEDS, the PSH level was decreased by about 36% in glucose deprived cells (Figure 3).
Figure 3. HEDS decreases levels of protein thiols (PSH) in glucose-deprived human colon cancer cells independent of p53 status.
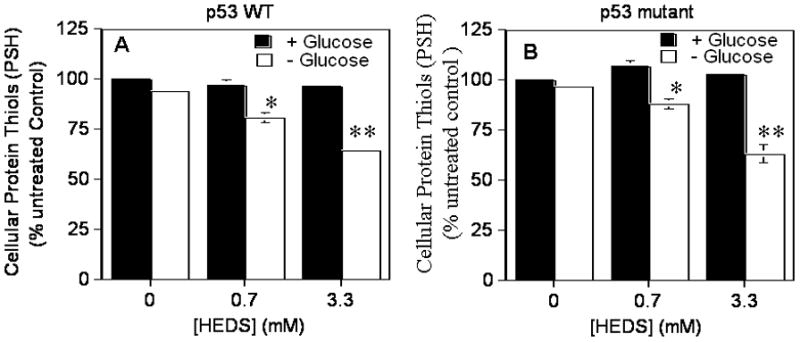
The effect of glucose deprivation on intracellular protein thiol levels was determined in the absence or presence of HEDS at the indicated concentration in cells that are radiation-sensitive and p53 wild-type HCT116 (A) or radiation-resistant and p53 mutant HT29 (B). Mean for three to five independent experiments are shown with SD. A statistically significant reduction in PSH levels in cells cultured in glucose-deprived media occurred after HEDS treatment (0.7 mM, *P<0.01; 3.3 mM, **P<0.001).
Although many proteins are likely affected by HEDS-mediated changes in NPSH and PSH in glucose-deprived cells, we examined effects on the DNA binding protein Ku to assess functional impact, since Ku is centrally involved in DNA repair, DNA recombination, telomere maintenance, radiation resistance and anti-apoptosis in cancer, ischemia, and other disorders. We observed that DNA binding by Ku protein was higher in both HCT116 and HT29 cells after glucose deprivation. However, depletion of glucose in the medium has significantly decreased the DNA binding activity of Ku by about 15% in cells treated with low concentration of HEDS (Figure 4). At high concentration of HEDS, the DNA binding activity of Ku was decreased by about 40% in glucose-deprived cells (Figure 4). In contrast, HEDS did not have significant effects on the DNA binding activity of Ku in cells measured after incubation with HEDS (0.7 and 3.3 mM) in the presence of glucose (Figure 4).
Figure 4. HEDS inhibits the function of redox-dependent DNA binding by Ku protein in glucose deprived cells.
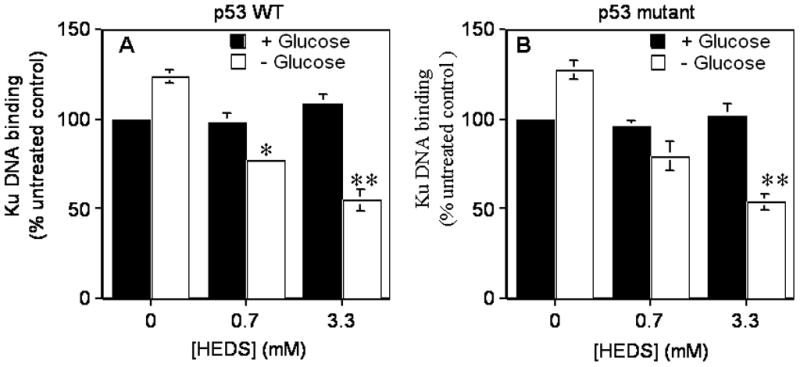
Ku binding activity was compared after HEDS treatment of p53 wild type HCT116 (A) and p53 mutant HT29 (B) cells cultured in normal or glucose-deprived media. Mean for three to five independent experiments are shown with SD. A statistically significant reduction in Ku function in cells cultured in glucose-deprived media occurred after HEDS treatment (0.7 mM, *P<0.01; 3.3 mM, **P<0.001).
We next determined whether the changes in thiol homeostasis affected the survival of these cells. In the presence of glucose, HEDS alone had no significant effect on the survival of either HCT116 or HT29 cells (Figures 5A–D). Short-term glucose deprivation (7 hr) without HEDS did not affect the survival of these cells measured 7 days later after restoring cells to normal glucose medium. However, the addition of a high concentration of HEDS (3.3 mM) incubated for 3 hrs during the short-term glucose deprivation decreased the survival of HCT116 cells by 80% but with no significant effect on HT29 cells (Figures 5A–D). The observed differences between these two cell lines to HEDS induced oxidative stress in low glucose medium are consistent with other evidence of their differences in responding to oxidative stress induced by radiation, hypoxia and chemotherapeutic agents (Blaszyk et al., 2000; Yao et al., 2005).
Figure 5. HEDS reduces the survival of wild-type p53 human colon cancer cells under glucose-deprived conditions.
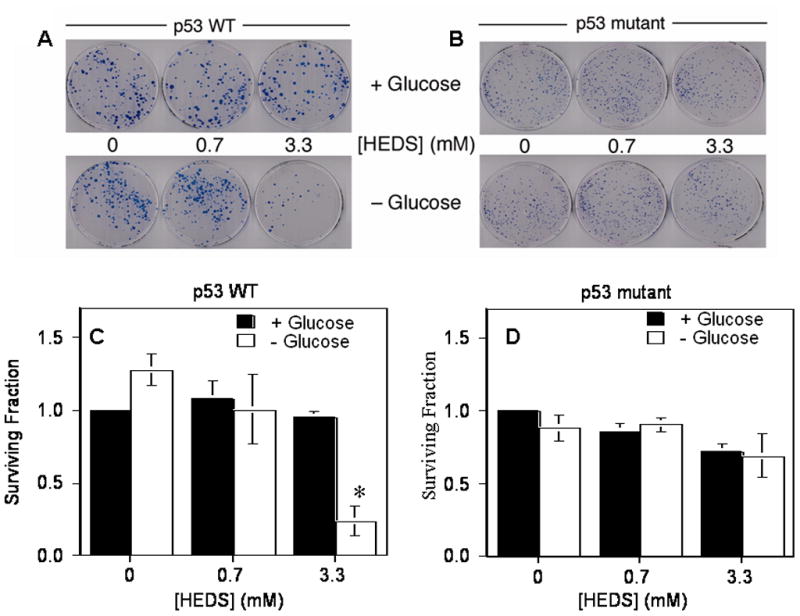
(A,B). Representative plates stained with methylene blue after clonogenic colony formation assay of HCT116 (A) and HT29 (B) cells exposed to HEDS in the presence or absence of glucose. (C,D). Quantification of reduced survival fractions in the assay. Cytotoxic effects of HEDS under low glucose conditions were apparent only in HCT116 (C) cells harboring wild-type p53. In contrast, HT29 (D) cells harboring mutant p53 were not responsive to HEDS under low glucose conditions. Mean for three to five independent experiments are shown with SD. A statistically significant reduction in the surviving fraction of cells in the clonogenic assay occurred only in HCT116 cells exposed to 3.3 mM HEDS (*P<0.001).
Studies from our lab and others have shown that cells deficient in Ku protein induced by mutagenesis or Ku siRNA are sensitive to radiation-induced cell death (Ayene et al., 2005; Critchlow et al., 1998). Therefore, we used radiation as a tool to determine whether the loss of Ku function in glucose-deprived cells after HEDS treatment affects the cellular ability to overcome free radical induced oxidative stress. Previous reports have demonstrated that glucose deprivation increased the resistance of cancer cells to other types of oxidative stress induced cell death (Li et al., 2009; Park et al., 2007; Wyld et al., 2002). Consistent with these reports, our current results also showed that both HCT116 and HT29 cells have resistance to oxidative stress induced by radiation during glucose deprivation (Figures 6A–D). However, both HCT116 and HT29 human cells exhibited greater susceptibility to radiation in the absence of glucose after 3 hr incubation with HEDS at either low (0.7 mM) or high (3.3 mM) concentrations (Figures 6A–D). However, HEDS did not affect the susceptibility of these cells to cell death under the same conditions in the presence of glucose (Figures 6A–D). These results suggested that glucose-mediated regulation of thiol homeostasis is required to inhibit the HEDS-mediated increase in susceptibility of these cells to free radical-induced cell death.
Figure 6. HEDS accentuates the human colon cancer cell cytotoxicity of ionizing radiation under glucose-deprived conditions.
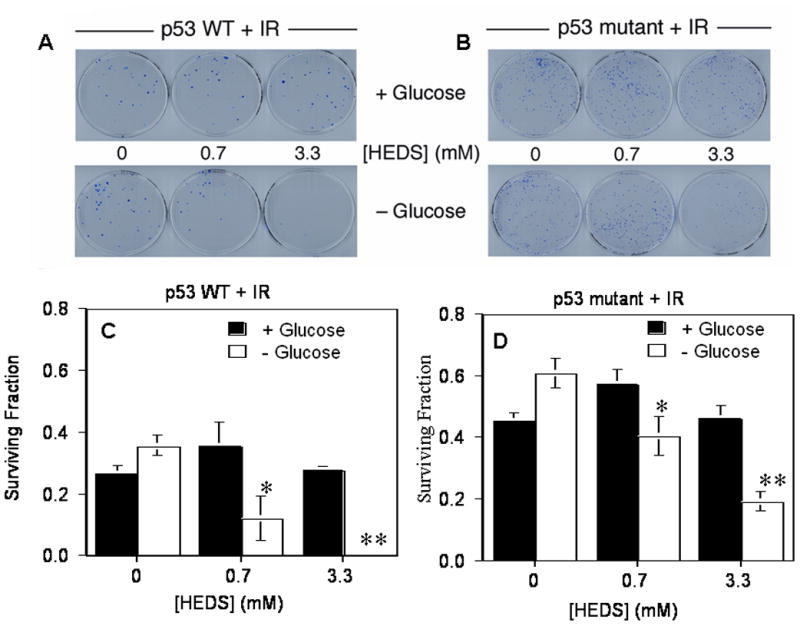
(A,B). Representative plates stained with methylene blue after clonogenic colony formation assay of HCT116 (A) and HT29 (B) cells exposed to HEDS in the presence or absence of glucose before exposure to 4 Gy ionizing irradiation (IR). (C,D). Quantification of reduced survival fractions in the assay. Increased cytotoxic effects of radiation under low glucose conditions were apparent after HEDS treatment in both p53 wild-type HCT116 (C) cells and p53 mutant HT29 (D) cells, though more accentuated in HCT116 cells. Mean for three to five independent experiments are shown with SD. In glucose-deprived media, HEDS induced a statistically significant reduction in the surviving fraction of cells in the radiation response assay, when added at 0.7 mM (*P<0.01) or 3.3 mM (**P<0.001) in both cell lines.
We determined whether the changes in thiol homeostasis and survival is due to its effect on NADPH since it is produced by OPPC and used by various enzymes to regulate thiol homeostasis in cells. In the presence of glucose, low concentration of HEDS (0.7 mM) alone had no significant effect on NADPH of either HCT116 or HT29 cells (Figures 7A, B). However, higher concentraton of HEDS (3.3 mM) decreased NADPH more in the absence of glucose than in the presence of glucose suggesting that NADPH is depleted to detoxify HEDS but replenished better in the presence of glucose. Taken together, our results defined HEDS as a glucose-sensitive chemical probe for OPPC-dependent cellular thiol homeostasis in human cells and defined the basis for its selective cytotoxic properties.
Figure 7. Glucose deprivation limits the NADPH recycling.
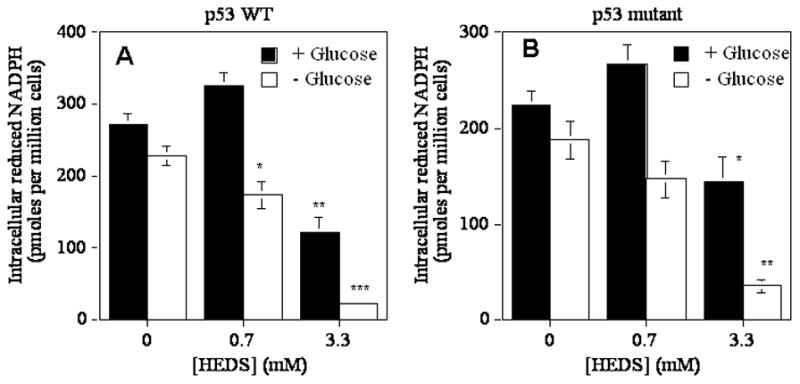
Glucose-dependent regulation of NADPH in p53 wild type HCT116 (A) and HT29 (B) human colon cancer cells treated with HEDS. Mean levels quantified by HPLC/Fluorescence are presented with standard deviation in the data. A statistically significant decrease in cells with normal glucose at 3.3 mM HEDS (**P<0.001) and a higher decrease in NADPH occurred in glucose-deprived cells as compared to normal glucose independent of p53 status in the presence of 0.7 mM (*P<0.01) or 3.3 mM (**P<0.001) HEDS. A st
In order to determine whether the lack of detoxification of HEDS is responsible for the loss of NPSH and PSH homeostasis and radiation sensitization in human cells, we have measured the cellular detoxification/bioreduction of HEDS. We observed a concentration-dependent conversion of HEDS into mercaptoethanol (ME) up to 3.0 mM in three hours by both HCT116 and HT29 cells in the presence of glucose (Figure 8). This bioreductive capacity suggested a fully operational OPPC in these cells (Figure 8). Surprisingly, both HCT116 and HT29 even with a higher concentration of NADPH compared to CHO failed to detoxify HEDS when incubated for 3 hr after a short period (4 hr) of glucose deprivation (Figure 8). These results demonstrated, for the first time, that glucose-deprived human cells with different p53 status and higher concentrations of DNA repair proteins and NADPH than CHO cells phenocopy G6PD/OPPC deficiency, which is necessary for the detoxification of HEDS.
Figure 8. Glucose deprivation limits the rapid detoxification of HEDS by cellular bioreduction to mercaptoethanol (ME).
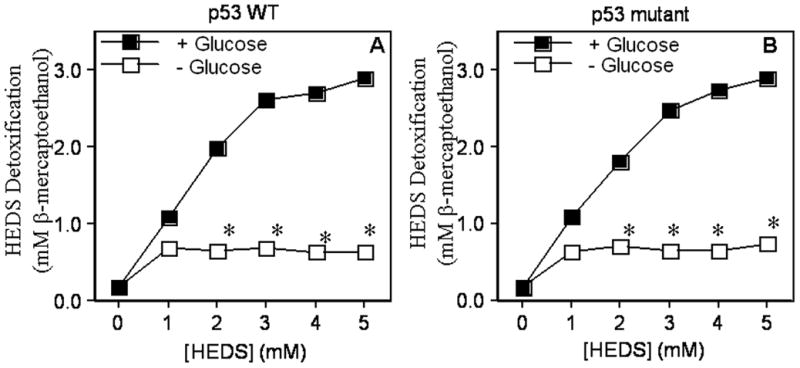
Culture media was assayed for ME by DTNB assay following HEDS exposure of p53 wild type HCT116 (A) and p53 mutant HT29 (B) cells cultured in normal or glucose-deprived media. Decreased HEDS conversion to ME occurred preferentially in glucose-deprived cells. Mean for three to five independent experiments are shown with SD. A statistically significant decrease in extracellular ME levels occurred independent of p53 status in glucose deprived cells (*P<0.01).
Although the DTNB assay employed demonstrated efficient bioreduction of different concentrations of HEDS (Figure 8), we confirmed the observations obtained from the DTNB assay using an HPLC/electrochemical (EC) assay to further document HEDS detoxification. Unlike the DTNB assay, which measures all DTNB reactive thiols in the medium, HPLC/EC can specifically quantify ME without interference from other types of non-protein thiols in the cells and medium ( Ayene et al., 2000). HPLC tracings highlighting the ME peak extracted from the culture medium of cells treated with 0.7 and 3.3 mM HEDS are shown in Figure 9A,B. In the presence or absence of glucose, there was no peak at 3.8 min elution time (results not shown). However, the medium from cells treated with HEDS (0.7 and 3.3 mM) for 3 hr in the presence of glucose showed a ME peak at 3.8 min elution time. We did not detect other thiols in a similar concentration range in the extracellular medium (data not shown). Further, the bioreduction/detoxification data quantified from the HPLC peaks showed a glucose-dependent detoxification of HEDS (0.7 and 3.3 mM) similar to that observed for the DTNB assay (Figure 9C,D). These results suggested that the detoxification of 0.7 and 3.3 mM HEDS, which were used for studies to determine its effect on PSH, Ku protein function and survival of mammalian cells, was dependent on the glucose concentration in the medium.
Figure 9. Quantification of HEDS bioreduction by HPLC/EC detection.
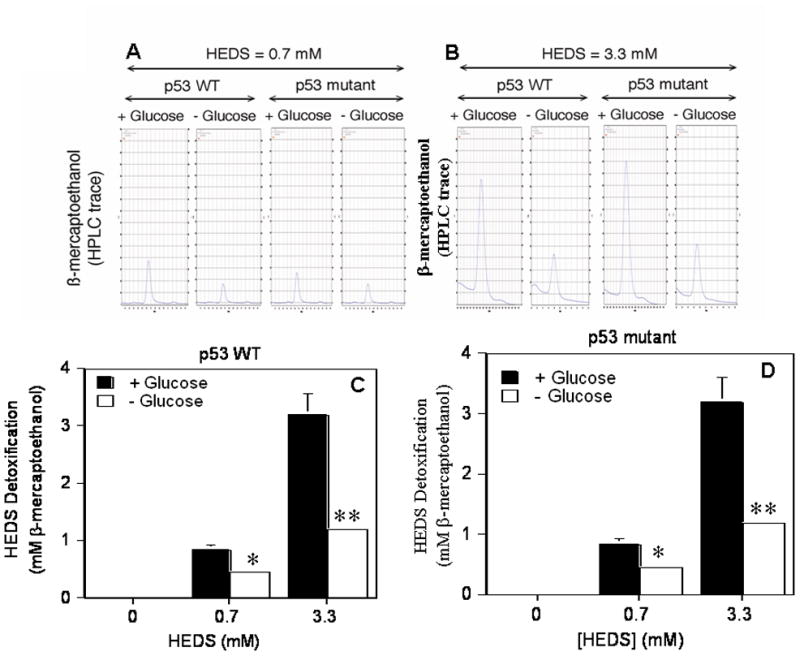
(A,B). Representative HPLC/EC tracings of mercaptoethanol production illustrating the rapid glucose-dependent detoxification of 0.7 mM (A) and 3.3 mM (B) HEDS in human colon cancer cells which occurs independent of p53 status. (C,D). Quantification of mercaptoethanol levels produced by HEDS bioreduction in p53 wild type HCT116 (A) and p53 mutant HT29 (B) cells. Mean levels quantified by HPLC/EC are presented with standard deviations in the data. A statistically significant decrease in HEDS bioreduction to mercaptoethanal occurred in glucose-deprived cells independent of p53 status in the presence of 0.7 mM (*P<0.01) or 3.3 mM (**P<0.001) HEDS.
DISCUSSION
Our results identify HEDS as a specific chemical probe of cellular thiol homeostasis and survival even in human colon cancer cells. The identification of this probe offers an important advance to studies of how glucose deprivation affects cellular function and survival, because unlike other chemical probes the activity of HEDS is selective for the glucose-deprived state rather than the associated confounding non specific effects produced by free radicals in an ischemic state. The comparison of current results with our previously published data showed that human cells have higher level of NADPH as compared to CHO cells (Ayene et al., 2002). We found that human cancer cells were not susceptible to HEDS-mediated loss of NPSH and PSH in the presence of glucose. However, these cells were highly susceptible to HEDS mediated loss of NPSH and PSH in the absence of glucose. These results demonstrate that glucose deficiency which occurs in ischemic tissues and tumors directly impairs the cellular control of thiol redox homeostasis even in human cells independent of their p53 status. To our knowledge a similar specificity for cellular stress and injury in glucose- deprived cells has yet to be demonstrated for other oxidants. Most chemical oxidants, such as menadione, diamide, hydrogen peroxide and tert-butylhydroperoxide, are quite toxic due to general oxidation of lipids, proteins and DNA and are not specific for thiols. Further, these other oxidants deplete NPSH even in the presence of glucose. Hence, they are not suitable to specifically identify the impact of glucose deprivation on NPSH and PSH thiol redox homeostasis, protein function and cell survival after free radical induced oxidative stress.
Protein thiols have been shown to affect the function of many proteins, but there has not been any demonstration of the impact of glucose deprivation on protein function in intact cells during oxidative stress, mediated via effects on non-protein thiol homeostasis, because of the lack of a specific thiol oxidant that only affects glucose-deprived cells. Many proteins containing cysteine thiols may be affected by low glucose, and our focus here on Ku protein provided an illustration of how protein function is specifically altered by glucose deprivation through this mechanism. Ku is highly sensitive to redox modification and has multifunctional roles in V(D)J recombination, antiapoptotic activity and telomere lengthening in mammalian cells. Most importantly, it also plays a major role in DNA double strand break repair (DSB) through non-homologous DNA end joining (NHEJ), the major pathway of DSB repair in mammalian cells (Critchlow et al., 1998; Mahaney et al., 2009; Scott & Pandita 2006). The efficiency of this complex mechanism of DSB repair is dependent on the DNA-binding activity of the Ku protein, which requires reduced sulfhydryl groups and may be regulated by oxidation-reduction of its cysteine residues (50). Ku is an ideal candidate to evaluate the functional impact of glucose deprivation mediated by altered thiol redox homeostasis, since its Ku70 subunit must be in a reduced form to bind DNA (Zhang & Yaneva 1993). The ability of HEDS to attenuate Ku function offers the first direct evidence that redox-sensitive proteins require glucose to maintain their function during stress that affect thiol redox status. These findings establish the critical requirement for OPPC activity in maintaining cellular protein function in cells subjected to glucose starvation during tissue ischemia.
Our study determined the impact of glucose deprivation not only on cells with wild type p53 (HCT116), which is present in normal mammalian cells, but also on cells with mutant p53 (HT29), which is mostly found in cancer cells. Consistent with previous reports, we found that HT29 cells were less responsive to radiation than the HCT116 cells in complete growth medium. Irrespective of the different p53 status between these cells, both these cells were able to regulate glucose-dependent thiol homeostasis during HEDS mediated oxidative stress. HEDS alone caused cell death only in p53 wild-type HCT116 cells with very little effect on the survival of radiation resistant HT29 cells in the absence of glucose. These results raised the possibility that glucose deprivation mediated loss of control of thiol homeostasis, which occurs in ischemic tissues, will be detrimental to normal cells since all normal human cells have wild type p53. It also raises the possibility for the use of this approach for elucidating the mechanisms of the resistance of p53 mutant cells to oxidative stress. However, HEDS increased the susceptibility of glucose-deprived p53 wild type and mutant cell lines to free radicals induced by radiation. The response of glucose-deprived radiation resistant and sensitive cells to HEDS demonstrated an additional advantage of such disulfides in targeting cancer cells without affecting normal tissues since the steady state concentration of glucose is much lower in solid tumor than the normal tissues (Aronen et al., 2000; Rajendran et al., 2004; Izuishi et al., 2000). This can also be construed to be clinically relevant since glucose-deprived cancer cells have better survival mechanisms and are less responsive to radiation and photodynamic therapy than glucose-rich cells (Li et al., 2009; Park et al., 2007; Wyld et al., 2002). Additionally, it also raises the possibility that hypoxic cancer cells, which are resistant to radiation, can also be targeted by HEDS since hypoxia is associated with low glucose (Rajendran et al., 2004). Although the radiation sensitization by HEDS in glucose deprived cells is due to a combination of cell death induced at high concentration of HEDS, the radiation sensitization at low concentration of HEDS with out HEDS toxicity (cell death) and the synergestic effect at high concentration of HEDS suggest that molecular changes such as loss of Ku protein function may be responsible for the radiation sensitizing effect of HEDS. This is further confirmed by the data in p53 mutant cells that is not susceptible to HEDS induced cell death but to radiation sensitization by HEDS.
While many biochemical pathways are affected by glucose deprivation during oxidative stress, our findings establish for the first time a direct link between glucose deprivation, NADPH depletion, loss of control of cellular thiol homeostasis and protein function, and cell death during oxidative stress. Unlike many oxidants, HEDS at low concentrations is not inherently toxic, as it does not affect either non-protein thiols (NPSH) or protein thiols (PSH) (Ayene et al., 2000; 2002; 2008). The major component of NPSH in mammalian cells is glutathione, which helps maintain intracellular redox status either by scavenging reactive oxygen species (ROS) directly or as a substrate for other enzymes that reduce oxidatively modified proteins including glutathionylated proteins (Ayene et al., 2000; 2002; 2008; Ellison et al., 2012; Franco and Cidlowski 2009; Tsou et al., 2009; Yang et al., 2006 ). Low molecular mass disulfides are oxidizing agents that are primarily limited in their chemical reactivities to chemical or enzyme mediated thiol-disulfide exchange reactions. The cysteine residue of glutathione may be oxidized to inter molecular disulfides through addition of such compounds. However, HEDS altered the intracellular glutathione only in glucose deprived cells due to decreased OPPC associated with low intracellular glucose. Several of the OPPC dependent biochemical pathways may also become ineffective in repairing the HEDS mediated redox modification of GSH and PSH in glucose deprived cells (see Figure 10).
Figure 10. Models of HEDS cellular reactions.
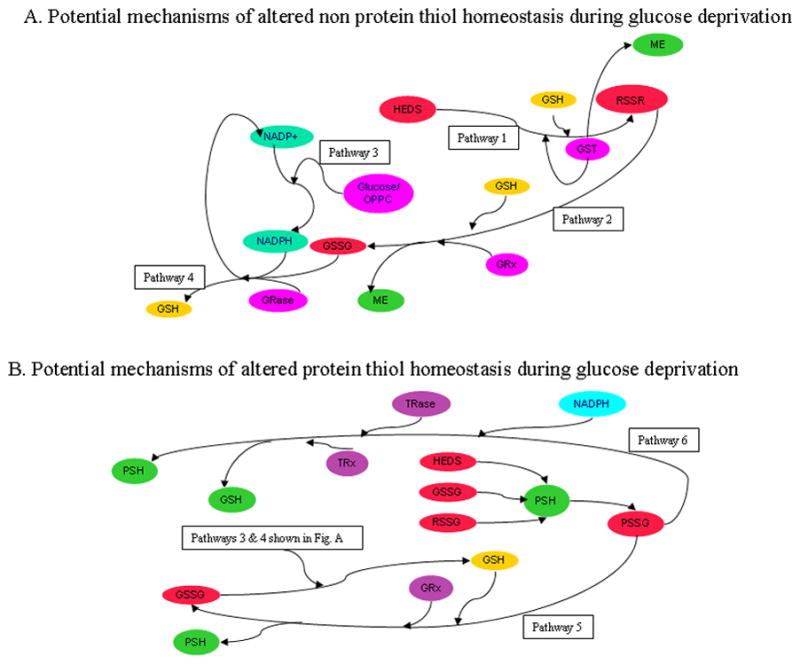
The ability of HEDS to interfere with thiol redox homeostasis and elicit loss of protein function and cell death occurs in glucose-deprived cells where HEDS detoxification is inefficient (Pathways 1–6). (A.) HEDS effect on non-protein thiols (NPSH). Bioreductive/detoxification pathways are similarly active in cells receiving adequate glucose independent of p53 status. HEDS may react spontaneously with GSH or in a reaction catalyzed by glutathione-S-transferase (GST) may produce non protein disulfides (RSSR including GSSG and/or mixed disulfide (MESSG) of glutathione (GSH)) and mercaptoethanol (ME) (Pathway 1). MESSG, if produced, may also react with GSH to produce ME and oxidized GSH (GSSG) through the catalytic action of glutaredoxin (GRX) (Pathway 2). In contrast, glucose-deprived cells convert HEDS into ME with poor efficiency, demonstrating that reductants needed to detoxify HEDS require intracellular glucose (Pathway 3). GSH loss in glucose-depleted cells is likely caused by reduced activity of the oxidative pentose phosphate cycle, a crucial factor in GSH homeostasis by recycling oxidized GSSG back to GSH via glutathione reductase (GRase) and NADPH (Pathway 4). (B). HEDS effect on protein thiols. HEDS, GSSG and/or HEDS-glutathione mixed disulfide (MESSG) produced by HEDS can react with protein thiols (PSH). Oxidation of PSH and loss of Ku function in glucose-depleted cells argues that oxidative pentose phosphate cycle (Pathways 3 and 4) is a crucial pathway for repair of oxidized protein thiols, either through the action of thioredoxin reductase (TRase)/thioredoxin (TRx)/NADPH (Pathway 5) or glutaredoxin (GRx) and GSH (Pathway 6).
The recycling of disulfides of non protein thiols (GSSG) to GSH is generally associated with OPPC activity that is dependent on glucose substrate (Fig. 10). The HEDS dependent depletion of NPSH/GSH in glucose deprived cells is consistent with the lack of reduction of NPSSPN/GSSG or NPS/GS- adducts back to NPSH/GSH presumably due to the decreased generation of NADPH from low activity of OPPC resulting from glucose deprivation (Fig. 10). Addititonally, the altered thiol homeostasis is also dependent on how effectively HEDS is reduced to mercaptoethanol. The lack of altered thiol homeostasis in glucose rich cells suggested a rapid reduction of HEDS by intracellular non protein thiols or direct reduction by enzymes (Fig. 10). Depletion of PSH by HEDS in glucose deprived cells may involve direct modification by HEDS and/or NPSSPN/GSSG (Fig. 10). The maintenance of protein thiols (PSH) in cells with normal glucose may involve enzyme linked- and/or direct NADPH- reduction of PSSR to PSH. Considering that the major non protein thiols in most mammalian cells is GSH, glutaredoxin (Grx) and thioredoxin (Trx) may also play a significant role in PSH homeostasis (Fig. 10). The relative importance of these pathways may depend on the level of NPSH/GSH Vs NPSSPN/GSSG, as well as enzymes, which may catalyze protein mixed disulfide removal (e.g. thioredoxin reductase, glutaredoxin). Nevertheless, glucose deprivation dependent depletion of PSH in both these cells indicated that glucose associated oxidative pentose cycle activity consistent with these pathways is essential for protein thiol redox homeostasis of important proteins such as Ku.
In conclusion, our results demonstrated that the loss of NADPH, GSH and protein thiols and the inhibition of DNA repair protein function via oxidation of sulfhydryl groups by HEDS rely on the level of glucose availability and the associated activity of the oxidative pentose phosphate cycle in human cells. This is an important finding since it demonstrated, for the first time, glucose deprivation mediated loss of control of cellular thiol redox homeostasis can cause loss of function of important proteins and enhanced susceptibility to oxidative stress during which glucose is depleted. Specifically, it reports the discovery of a specific and essential role for the oxidative pentose phosphate cycle (OPPC) in maintaining the survival of glucose-deprived cells generally found in ischemic regions of damaged or malignant tissues. Additionally, it also suggests that such a probe may be useful in other fields of biological and medical research where the metabolic pathophysiology of tissue ischemia is relevant. Specifically, it can be used to identify the specific and essential role for the OPPC in maintaining the survival of cells in stroke, heart failure, and other common diseases where cellular oxidative stress engendered by tissue ischemia and glucose deprivation play a major role. Additionally, this approach also raises the potential use of HEDS or HEDS-like compounds to target most solid tumors that are likely to have low glucose due to high metabolic activity and low glucose supply due to disorganized vasculature without any detrimental effect on normal tissues. Our approach is clinically relevant for understanding the mechanisms of normal cell injury during ischemia and targeting glucose deprived cancer cells considering that GSH, which requires OPPC for recycling, is a major protector of oxidative damage caused by oxidative stress, certain chemotherapeutic agents and radiation.
Hydroxyethyldisulfide a bioactive probe for cancer metabolism/ischemic pathology
Pentose cycle disruption by hydroxyethyldisulfide causes cell death in colon cells
Hydroxyethyldisulfide improves the response of colon cancer cells to radiation
Hydroxyethyldisulfide alters NADPH and thiol redox leading to cell death
Hydroxyethyldisulfide alters thiol redox dependent DNA repair protein Ku function
Acknowledgments
This work was supported by grants from the National Institute of Health National Cancer Institute (CA109604) and The Pennsylvania Department of Health to I.S. Ayene. K. M. Ward is a recipient of graduate research assistantship from the Brook J. Lenfest Foundation.
Abbreviations
- G6PD
glucose-6-phosphate dehydrogenase
- HEDS
hydroxyethyldisulfide
- OPPC
oxidative pentose phosphate cycle
- ME
mercaptoethanol
- GSH
glutathione
- GSSG
oxidized glutathione
- NPSH
non protein thiol
- PSH
protein thiol
- DTNB
dithiobisnitrobenzoic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aronen HJ, Pardo FS, Kennedy DN, Belliveau JW, Packard SD, Hsu DW, Hochberg FH, Fischman AJ, Rosen BR. High microvascular blood volume is associated with high glucose uptake and tumor angiogenesis in human gliomas. Clin Cancer Res. 2000;6:2189–2200. [PubMed] [Google Scholar]
- Ayene IS, Dodia C, Fisher AB. Role of oxygen in oxidation of lipid and protein during ischemia/reperfusion in isolated perfused rat lung. Arch Biochem Biophys. 1992a;296:183–189. doi: 10.1016/0003-9861(92)90561-a. [DOI] [PubMed] [Google Scholar]
- Ayene IS, Dodia C, Fisher AB. Oxygen-dependent oxidative damage during lung ischemia and reperfusion. In: Yagi i, Kondo m, Niki E, Yoshikawa T., editors. V International Congress of Oxygen Radicals. 1992b. pp. 457–460. [Google Scholar]
- Ayene IS, Almehdi AB, Fisher AB. Inhibition of Lung Tissue Oxidation during Ischemia/Reperfusion by 2-Mercaptopropionylglycine. Arch Biochem Biophys. 1993;303:307–312. doi: 10.1006/abbi.1993.1288. [DOI] [PubMed] [Google Scholar]
- Ayene IS, Koch CJ, Tuttle SW, Stamato TD, Perez ML, Biaglow JE. Oxidation of cellular thiols by hydroxyethyldisulphide inhibits DNA double-strand-break rejoining in G6PD deficient mammalian cells. Int J Radiat Biol. 2000;76:1523–1531. doi: 10.1080/09553000050176289. [DOI] [PubMed] [Google Scholar]
- Ayene IS, Stamato TD, Mauldin SK, Biaglow JE, Tuttle SW, Jenkins SF, Koch CJ. Mutation in the glucose-6-phosphate dehydrogenase gene leads to inactivation of Ku DNA end binding during oxidative stress. J Biol Chem. 2002;277:9929–9935. doi: 10.1074/jbc.M111366200. [DOI] [PubMed] [Google Scholar]
- Ayene IS, Ford LP, Koch CJ. Ku protein targeting by Ku70 small interfering RNA enhances human cancer cell response to topoisomerase II inhibitor and gamma radiation. Mol Cancer Ther. 2005;4:529–536. doi: 10.1158/1535-7163.MCT-04-0130. [DOI] [PubMed] [Google Scholar]
- Ayene IS, Biaglow JE, Kachur AV, Stamato TD, Koch CJ. Mutation in G6PD gene leads to loss of cellular control of protein glutathionylation: mechanism and implication. J Cell Biochem. 2008;103:123–135. doi: 10.1002/jcb.21394. [DOI] [PubMed] [Google Scholar]
- Biaglow JE, Ayene IS, Koch CJ, Donahue J, Stamato TD, Tuttle SW. G6PD deficient cells and the bioreduction of disulfides: effects of DHEA, GSH depletion and phenylarsine oxide. Biochem Biophys Res Commun. 2000;273:846–852. doi: 10.1006/bbrc.2000.3024. [DOI] [PubMed] [Google Scholar]
- Biaglow JE, Ayene IS, Koch CJ, Donahue J, Stamato TD, Mieyal JJ, Tuttle SW. Radiation response of cells during altered protein thiol redox. Radiat Res. 2003;159:484–494. doi: 10.1667/0033-7587(2003)159[0484:rrocda]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Blaszyk H, Hartmann A, Cunningham JM, Schaid D, Wold LE, Kovach JS, Sommer SS. A prospective trial of midwest breast cancer patients: a p53 gene mutation is the most important predictor of adverse outcome. Int J Cancer. 2000;89:32–8. doi: 10.1002/(sici)1097-0215(20000120)89:1<32::aid-ijc6>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Brongholi K, Souza DG, Bainy AC, Dafre AL, Tasca CI. Oxygen-glucose deprivation decreases glutathione levels and glutamate uptake in rat hippocampal slices. Brain Res. 2006;14:211–218. doi: 10.1016/j.brainres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Ceconi C, Bernocchi P, Boraso A, Cargnoni A, Pepi P, Curello S, Ferrari R. New insights on myocardial pyridine nucleotides and thiol redox state in ischemia and reperfusion damage. Cardiovasc Res. 2000;47:586–594. doi: 10.1016/s0008-6363(00)00104-8. [DOI] [PubMed] [Google Scholar]
- Critchlow SE, Jackson SP. DNA end-joining: from yeast to man. Trends Biochem Sci. 1998;23:394–398. doi: 10.1016/s0968-0004(98)01284-5. [DOI] [PubMed] [Google Scholar]
- Ellison I, Richie JP. Mechanisms of glutathione disulfide efflux from erythrocytes. Biochem Pharmacol. 2012;83:164–169. doi: 10.1016/j.bcp.2011.09.016. [DOI] [PubMed] [Google Scholar]
- Fang SH, Yuan YM, Peng F, Li CT, Zhang LH, Lu YB, Zhang WP, Wei EQ. Pranlukast Attenuates Ischemia-like Injury in Endothelial Cells Via Inhibiting Reactive Oxygen Species Production and Nuclear Factor-[kappa]B Activation. J Cardiovas Pharm. 2009;53:77–85. doi: 10.1097/FJC.0b013e318196736c. [DOI] [PubMed] [Google Scholar]
- Fern R, Moller T. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci. 2000;20:34–42. doi: 10.1523/JNEUROSCI.20-01-00034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher AB, Dodia C, Ayene I, al-Mehdi A. Ischemia-reperfusion injury to the lung. Ann N Y Acad Sci. 1994;723:197–207. [PubMed] [Google Scholar]
- Fisher AB, Dodia C, Tan ZT, Ayene I, Eckenhoff RG. Oxygen-dependent lipid peroxidation during lung ischemia. J Clin Invest. 1991;88:674–679. doi: 10.1172/JCI115352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco R, Cidlowski JA. Apoptosis and glutathione: beyond an antioxidant. Cell Death Diff. 2009;16:1303–1314. doi: 10.1038/cdd.2009.107. [DOI] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–3524. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izuishi K, Kato K, Ogura T, Kinoshita T, Esumi H. Remarkable tolerance of tumor cells to nutrient deprivation: possible new biochemical target for cancer therapy. Cancer Res. 2000;60:6201–6207. [PubMed] [Google Scholar]
- Kato K, Ogura T, Kishimoto A, Minegishi Y, Nakajima N, Miyazaki M, Esumi H. Critical roles of AMP-activated protein kinase in constitutive tolerance of cancer cells to nutrient deprivation and tumor formation. Oncogene. 2002;21:6082–6090. doi: 10.1038/sj.onc.1205737. [DOI] [PubMed] [Google Scholar]
- Kintner DB, Luo J, Gerdts J, Ballard AJ, Shull GE, Sun D. Role of Na+-K+-Cl- cotransport and Na+/Ca2+ exchange in mitochondrial dysfunction in astrocytes following in vitro ischemia. Am J Physiol Cell Physiol. 2007;292:11. doi: 10.1152/ajpcell.00412.2006. [DOI] [PubMed] [Google Scholar]
- Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001;26:504–510. doi: 10.1016/s0968-0004(01)01908-9. [DOI] [PubMed] [Google Scholar]
- Li J, Ayene R, Ward KM, Dayanandam E, Ayene IS. Glucose deprivation increases nuclear DNA repair protein Ku and resistance to radiation induced oxidative stress in human cancer cells. Cell Biochem Funct. 2009;27:93–101. doi: 10.1002/cbf.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang D, Ward KM, Prendergast GC, Ayene IS. Hydroxyethyl disulfide as an efficient metabolic assay for cell viability in vitro. Toxicol In Vitro. 2012;26:603–612. doi: 10.1016/j.tiv.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahaney B, Meek K, Lees-Miller S. Repair of ionizing radiation-induced DNA double-strand breaks by non-homologous end-joining. Biochem J. 2009;417:639–645. doi: 10.1042/BJ20080413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Rojas S, Pareto D, Santalucia T, Millan O, Abasolo I, Gomez V, Llop J, Gispert JD, Falcon C, Bargallo N, Planas AM. Depressed glucose consumption at reperfusion following brain ischemia does not correlate with mitochondrial dysfunction and development of infarction: an in vivo positron emission tomography study. Curr Neurovasc Res. 2009;6:82–88. doi: 10.2174/156720209788185650. [DOI] [PubMed] [Google Scholar]
- Milusheva EA, Doda M, Baranyi M, Vizi ES. Effect of hypoxia and glucose deprivation on ATP level, adenylate energy charge and [Ca2+]o-dependent and independent release of [3H]dopamine in rat striatal slices. Neurochem Int. 1996;28:501–507. doi: 10.1016/0197-0186(95)00129-8. [DOI] [PubMed] [Google Scholar]
- Noack H, Kunz WS, Augustin W. Evaluation of a procedure for the simultaneous determination of oxidized and reduced pyridine nucleotides and adenylates in organic phenol extracts from mitochondria. Anal Biochem. 1992;202:162–165. doi: 10.1016/0003-2697(92)90222-s. [DOI] [PubMed] [Google Scholar]
- Pandolfi PP, Sonati F, Rivi R, Mason P, Grosveld F, Luzzatto L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J. 1995;14:5209–5215. doi: 10.1002/j.1460-2075.1995.tb00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HR, Ryoo IJ, Choo SJ, Hwang JH, Kim JY, Cha MR, Shin-Ya K, Yoo ID. Glucose-deprived HT-29 human colon carcinoma cells are sensitive to verrucosidin as a GRP78 down-regulator. Toxicology. 2007;229:253–261. doi: 10.1016/j.tox.2006.11.049. [DOI] [PubMed] [Google Scholar]
- Rajendran JG, Mankoff DA, O’Sullivan F, Peterson LM, Schwartz DL, Conrad EU, Spence AM, Muzi M, Farwell DG, Krohn KA. Hypoxia and glucose metabolism in malignant tumors: evaluation by [18F]fluoromisonidazole and [18F]fluorodeoxyglucose positron emission tomography imaging. Clin Cancer Res. 2004;10:2245–2252. doi: 10.1158/1078-0432.ccr-0688-3. [DOI] [PubMed] [Google Scholar]
- Romano C, Price M, Bai HY, Olney JW. Neuroprotectants in Honghua: glucose attenuates retinal ischemic damage. Invest Ophthalmol Vis Sci. 1993;34:72–80. [PubMed] [Google Scholar]
- Russell RR, 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvemini F, Franze A, Iervolino A, Filosa S, Salzano S, Ursini MV. Enhanced Glutathione Levels and Oxidoresistance Mediated by Increased Glucose-6-phosphate Dehydrogenase Expression. J Biol Chem. 1999;274:2750–2757. doi: 10.1074/jbc.274.5.2750. [DOI] [PubMed] [Google Scholar]
- Scott S, Pandita T. The cellular control of DNA double-strand breaks. J Cell Biochem. 2006;99:1463–1475. doi: 10.1002/jcb.21067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou PS, Addanki V, Haas JA, Page NA, Fung HL. Role of glutaredoxin-mediated protein S-glutathionylation in cellular nitroglycerin tolerance. J Pharmacol Exp Therap. 2009;329:649–56. doi: 10.1124/jpet.108.149997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle SW, Maity A, Oprysko PR, Kachur AV, Ayene IS, Biaglow JE, Koch CJ. Detection of reactive oxygen species via endogenous oxidative pentose phosphate cycle activity in response to oxygen concentration: implications for the mechanism of HIF-1alpha stabilization under moderate hypoxia. J Biol Chem. 2007;282:36790–36796. doi: 10.1074/jbc.M700327200. [DOI] [PubMed] [Google Scholar]
- Wang X, Figueroa BE, Stavrovskaya IG, Zhang Y, Sirianni AC, Zhu S, Day AL, Kristal BS, Friedlander RM. Methazolamide and melatonin inhibit mitochondrial cytochrome C release and are neuroprotective in experimental models of ischemic injury. Stroke. 2009;40:1877–1885. doi: 10.1161/STROKEAHA.108.540765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyld L, Tomlinson M, Reed MW, Brown NJ. Aminolaevulinic acid-induced photodynamic therapy: cellular responses to glucose starvation. Br J Cancer. 2002;86:1343–1347. doi: 10.1038/sj.bjc.6600234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Ebbert JO, Sun Z, Weinshilboum RM. Role of the Glutathione Metabolic Pathway in Lung Cancer Treatment and Prognosis: A Review. J Clin Onc. 2006;24:1761–1769. doi: 10.1200/JCO.2005.02.7110. [DOI] [PubMed] [Google Scholar]
- Yao K, Shida S, Selvakumaran M, Zimmerman R, Simon E, Schick J, Haas N, Balke M, Ross H, Johnson S. Macrophage migration inhibitory factor is a determinant of hypoxia-induced apoptosis in colon cancer cell lines. Clin Cancer Res. 2005;11:7264–7271. doi: 10.1158/1078-0432.CCR-05-0135. [DOI] [PubMed] [Google Scholar]
- Yun H, Lee M, Kim SS, Ha J. Glucose Deprivation Increases mRNA Stability of Vascular Endothelial Growth Factor through Activation of AMP-activated Protein Kinase in DU145 Prostate Carcinoma. J Biol Chem. 2005;280:9963–9972. doi: 10.1074/jbc.M412994200. [DOI] [PubMed] [Google Scholar]
- Zhang WW, Yaneva M. Reduced sulphydryl groups are required for DNA binding of Ku protein. Biochem J. 1993;293:769–774. doi: 10.1042/bj2930769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Ayene IS, Fisher AB. Role of iron in ischemia-reperfusion oxidative injury of rat lungs. Am J Respir Cell Mol Biol. 1997;16:293–299. doi: 10.1165/ajrcmb.16.3.9070614. [DOI] [PubMed] [Google Scholar]
- Zhou M, Xu W, Liao G, Bi X, Baudry M. Neuroprotection against neonatal hypoxia/ischemia-induced cerebral cell death by prevention of calpain-mediated mGluR1alpha truncation. Exp Neurol. 2009;218:75–82. doi: 10.1016/j.expneurol.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]