

Abstract
Using a neuronal model of serum starved SK-N-SH neuroblastoma cells, we showed previously that the phosphorylation of Akt and the mTOR substrates S6K and S6 through the vascular endothelial growth factor receptor VEGFR2 was enhanced by treatments with the phosphatase PP2A inhibitor okadaic acid (OA). These findings suggested that PP2A inhibition uncouples the regulation of Akt signaling by mTOR and affects cell survival. We therefore examined the effects of mTOR inhibition on Akt phosphorylation at sites threonine 308 (T308) and serine 473 (S473) and survival in OA treated cells. OA induced a loss in cell viability, the accumulation of hyperactivated Akt as monomeric and ubiquitinated forms and an increase in the total levels of ubiquitinated proteins. These events were exacerbated by treatments with an allosteric (rapamycin) but not an active-site inhibitor (PP242) of mTOR. Notably, rapamycin augmented the OA-induced hyperphosphorylation of Akt by suppressing a negative feedback loop of Akt activation through VEGFR2 and its downstream target phosphatidylinositol 3-kinase (PI3K). Treatments with the antioxidant N-acetlycysteine but not the pan caspase inhibitor Z-VAD-FMK promoted survival. Unlike reports that rapamycin promotes survival through increased Akt activation, these findings show that rapamycin-induced hyperphosphorylation of Akt fails to rescue our neuronal model from an oxidative stress-induced and caspase-independent cell death mediated by PP2A inhibition. Moreover, the exacerbation of OA-induced events by rapamycin suggests that mTOR and PP2A work in concert to regulate cell survival, activated Akt and the levels of ubiquitinated proteins.
Introduction
The target of rapamycin (mTOR) is a serine/threonine protein kinase that modulates protein synthesis and survival in response to various stimuli [25]. mTOR exists as two functionally distinct complexes mTORC1 and mTORC2. mTORC1 is activated by the serine/threonine protein kinase B or Akt to signal cap dependent protein translation through phosphorylation of the 40S ribosomal protein S6 kinase 1 (S6K1), the ribosomal protein S6 and the eukaryotic initiation factor 4E (eIF-4E)-binding protein 1 (4E-BP1). 4E-BP1 negatively regulates translation by sequestering the initiation factor eIF4E whereupon 4E-BP1 phosphorylation by S6K1 releases eIF4E to allow protein synthesis [7]. Growth factors activate Akt through a phosphatidylinositol 3-kinase (PI3K)-dependent mechanism involving phosphorylation within the activation loop at Threonine 308 (T308) by phosphoinositide-dependent protein kinase 1 (PDK1) and a second phosphorylation within the hydrophobic C-terminal domain at Serine 473 (S473) by the mTORC2 complex [9]. Increasing evidence shows that the mTOR pathway is functionally linked to aberrant Akt activation in cancer and neurodegeneration [1, 25]. For example, insulin signaling through the downstream adaptor protein, insulin receptor substrate-1 (IRS1) is negatively regulated by a feedback loop from activated S6K1 that diminishes Akt phosphorylation through PI3K [7]. Inhibition of mTOR by rapamycin disrupts this feedback mechanism, leading to Akt hyperactivation that promotes survival [1, 25].
Akt phosphorylation at T308 and S473 is negatively regulated through dephosphorylation (inactivation) by the serine/threonine phosphatase protein phosphatase 2A (PP2A) [10, 12]. This is substantiated by demonstrations that the inhibition of PP2A with okadaic acid (OA), a neutralizing antibody or a knock down of catalytic or regulatory subunits elevate Akt phosphorylation [12]. Furthermore, PP2A interacts directly with Akt [12], controls Akt-mediated survival [14] and is negatively regulated by mTOR [7]. The ubiquitin proteasome pathway also regulates activation or degradation through attachment of polyubiquitin chains with lysine-63 (K63) or lysine-48 (K48) linkages, respectively [18, 21]. Since phosphorylated Akt is more resistant to degradation by the proteasome [14], PP2A inhibition would dramatically influence Akt turnover and cell survival.
In endothelial cells the vascular endothelial growth factor (VEGF) signals Akt stability and survival through mTOR since treatments with rapamycin lead to apoptosis, caspase activation and Akt degradation [16]. Using a neuronal model of serum starved SK-N-SH neuroblastoma cells, we showed previously that the signaling of Akt, S6K1 and S6 activation by VEGF through its receptor VEGFR2 is enhanced by OA, suggesting that that PP2A inhibition uncouples the regulation of Akt activation by mTOR [3]. We therefore examined the interplay among mTOR substrate activation, Akt phosphorylation at T308 and S473 and survival when serum deprived SK-N-SH cells were treated with OA.
Materials and Methods
Materials
Recombinant human VEGF 165 was obtained from PeproTech. Inhibitors for VEGFR2 (SU1498), PI3K (LY294002), mTOR (rapamycin) and PP2A (OA) were obtained from LC Labs. The mTOR inhibitor PP242 was from Chemdea and the pan caspase inhibitor carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone (Z-VAD-FMK) was from Axxora. N-acetylcysteine (Nac) and epoxomicin were obtained from Sigma.
Cell Culture and Inhibitor Treatment
Human neuroblastoma SK-N-SH cells were maintained and serum starved as described previously [3]. For inhibitor studies, cells were pretreated with and without predetermined concentrations of rapamycin (48 hr), SU1498, LY294002, Z-VAD-FMK, Nac, PP242, and epoxomicin (2 hr) while OA (400 nM) was added during the last hr of incubation.
Protein Extraction and Immunoblotting
Cells were harvested in lysis buffer and quantified for protein content as described previously [3]. Primary and secondary antibodies were from Cell Signaling Technology except for those against actin (Sigma) and ubiquitinated protein conjugates (Dako). Immunoblots were visualized with the SuperSignal West Pico chemiluminescent substrate (Pierce) and quantified, where indicated, using ImageJ (NIH, http://rsb.info.nih.gov/ij/). Data were normalized to their respective total protein levels and expressed as the fold difference of the vehicle control.
Protein Immunoprecipitation
Akt was immunoprecipitated from equal concentrations (400 μg) of total cell lysates using an anti-Akt antibody (Cell Signaling Technology) and incubated overnight at 4°C. Antibody-antigen complexes were selectively removed by binding to protein G magnetic beads according to the manufacturer’s directions (Millipore). Immunoprecipitated protein was eluted from the beads in SDS loading buffer and analyzed by immunoblotting.
Cell viability
Cells were plated in 96-well plates at a concentration of 5 × 104 cells/well and treated as indicated. Viability was determined using a colorimetric MTS based CellTiter Aqueous One Solution Cell Proliferation Assay (Promega) according to manufacturer’s instruction. Survival measurements are expressed as the percent of the vehicle control.
Caspase 3/7 activity
Caspase activity was measured in a 96-well format using the fluorescence cell-based Apo-ONE Homogeneous Caspase 3/7 Assay (Promega) according to manufacturer’s directions and quantified using the Molecular Dynamics Typhoon 9410 Imaging System (Amersham). Data were normalized as fluorescent units/μg protein.
Statistical analyses
Data are expressed as the mean ± SEM of cell treatments from at least three independent experiments. Statistical significance was determined with GraphPad Prism Software using a one-way analysis of variance (ANOVA) with Bonferroni’s posttest and values of P <0.05 as significant.
Results
Rapamycin enhances Akt phosphorylation through VEGFR2
Based on previous findings [3], we addressed whether prolonged (48 hr) treatments with rapamycin influenced the OA-induced increase in Akt phosphorylation through VEGFR2 in serum starved SK-N-SH cells. Western blots show that rapamycin augmented an OA-induced phosphorylation of monomeric (60 kDa) and a HMW form (135 kDa) of Akt at T308 and S473 that decreased total Akt (Fig. 1A, lanes 2 and 3). To address VEGFR2 participation, cells were incubated under the same conditions without and with VEGF alone or in the presence of the VEGFR2 inhibitor SU1498 (Fig. 1B). VEGFR2 inhibition reduced the monomeric and HMW forms of phosphorylated Akt at T308 and S473 (short and long exposures) in the absence (Fig. 1B, lanes 3 and 4) and presence of VEGF (lanes 5 and 6) while total levels increased, suggesting that autocrine and paracrine VEGF signaling promotes Akt activation through VEGFR2 [3]. PP2A levels were unchanged. Rapamycin inhibited S6K1 but not ERK1/2 phosphorylation while SU1498 blocked the activation of both kinases (Fig. 1C, compare lanes 2 with 4 and 5) as expected [3]. The sustained Akt activation at S473 suggests that prolonged rapamycin treatments did not affect mTORC2 function [17].
Fig. 1. Rapamycin enhances Akt phosphorylation induced by OA through the VEGF receptor VEGFR2.
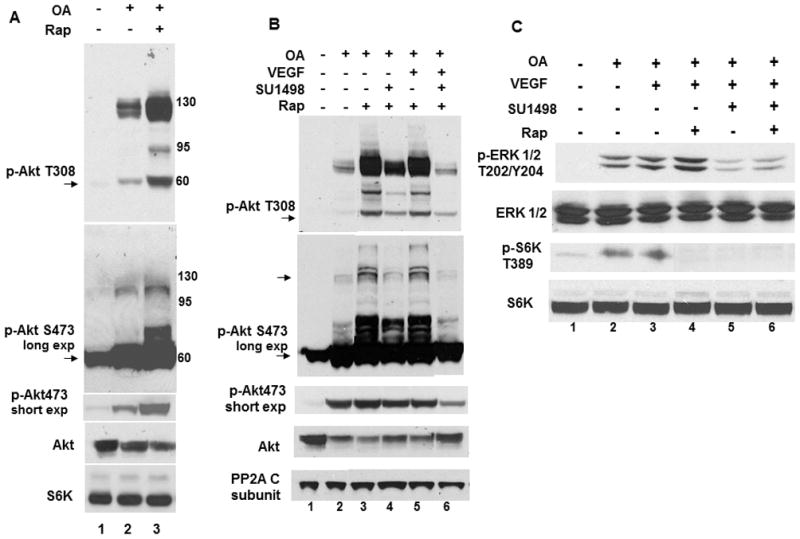
(A) Cells were pretreated with 1 μM rapamycin (Rap; 48 hr) and 400 nM OA was added during the last 1 hr where indicated. Immunoblots were probed for monomeric (60 kDa) and HMW (135 kDa) forms of Akt phosphorylation at T308 and S473 using short and long exposures. Blots were also probed for total Akt (monomeric) with S6K as a loading control. (B) Cells were treated as in A with 10 μM SU1498 and VEGF (10 ng/ml) added during the last 2 hr and 15 minutes, respectively. Immunoblots were probed Akt phosphorylation at T308, S473 or total Akt with the PP2A catalytic subunit C as a loading control. (C) Immunoblots of the same treatments in B were probed for the phosphorylation and total levels of ERK1/2 or S6K. Arrows designate monomeric forms of phosphorylated Akt. Data are typical of 3 independent experiments
Inhibitors of mTOR differentially affect Akt phosphorylation through PI3K
Treatments with a predetermined concentration of PP242, an active-site inhibitor of mTORC1/mTORC2 [4], blocked OA-induced S6K1 and S6 phosphorylation (Fig. 2A, lane 2) and prevented Akt phosphorylation at T308 and S473 alone (Fig. 2B, lanes 2 and 4) or in combination with rapamycin at 48 (lanes 3 and 5) or 2 hr (lanes 6 and 7). A failure by rapamycin to inhibit 4EBP1 phosphorylation at 2 and 48 hr (lanes 3 and 6) was attenuated by PP242 (lanes 4, 5 and 7).
Fig. 2. mTOR inhibitors elicit differential effects in OA treated cells.
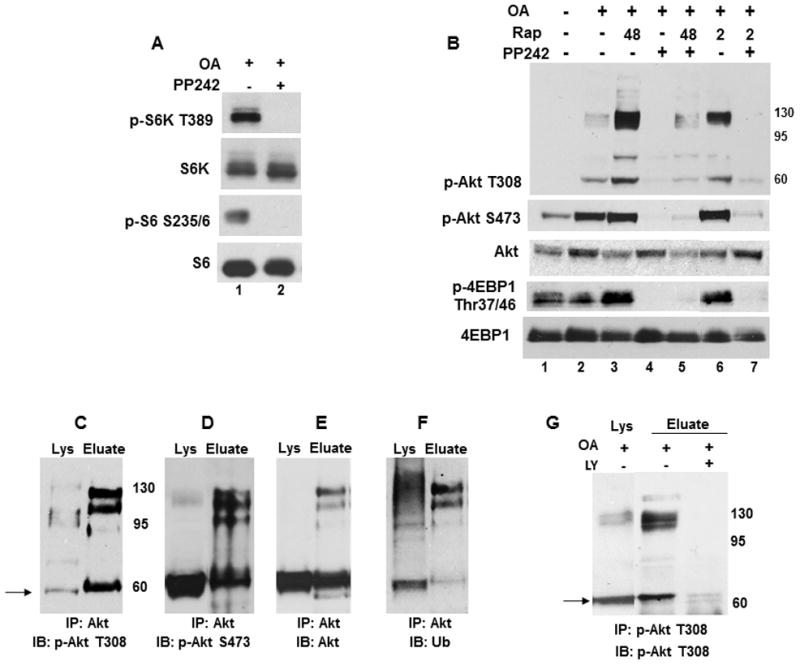
(A) Cells were treated with OA without and with 2.5 μM PP242 for 2 hr and analyzed for phosphorylated and total S6K and S6. (B) OA treated cells were incubated without or with 1 μM rapamycin (2 or 48 hr) or 2.5 μM PP242 (2 hr) alone or in combination and analyzed for Akt phosphorylation at T308 and S473, total Akt and phosphorylated and total 4EBP1. (C–G) Hyperphosphorylated Akt accumulates as PI3K-dependent ubiquitinated forms. Akt was immunoprecipitated from lysates of OA treated cells as described in the Material and Methods. Blots of immunopreciptated protein (Eluate) and total lysates (Lys) were probed for (C) Akt T308, (D) Akt S473, (E) total Akt or (F) ubiquitin conjugates. (G) Akt was immunoprecipitated from cells treated with OA without or with 20 μM LY294002 (LY) for 2 hr and analyzed for Akt phosphorylation at T308 (Eluate).
A confirmation of Akt specificity by immunoprecipitation showed that Akt phosphorylation at T308 (Fig. 2C), S473 (Fig. 2D) and total Akt (Fig. 2E) were detected as monomeric and HMW forms that are polyubiquitined (Fig. 2F). A probing of duplicate blots from several experiments with an antibody that only detects K63-linked polyubiquitin chains (data not shown) showed no reactivity with the HMW forms of Akt, suggesting that the ubiquitin tag designates K48 linkages for degradation. Incubations with the PI3K inhibitor LY294002 (LY) or Wortmannin (data not shown) prevented Akt phosphorylation at T308 (Fig. 2G), suggesting that PI3K mediates Akt hyperphosphorylation.
OA induces a caspase independent cell death that is blocked by N-acetylcysteine and potentiated by rapamycin
Since OA induces oxidative stress in neuronal cells [22], cell viability and caspase activation were measured alone or following pretreatments with rapamycin or PP242 with and without the antioxidant Nac (Fig. 3). OA induced a loss in viability with caspase-3/7 activation (Figs. 3A and 3B, lanes 5, 6 and 7) that was attenuated with Nac (lanes 8, 9 and 10). Notably, rapamycin but not PP242, significantly exacerbated the cell death induced by OA (Fig. 3A, lane 6). Measurements of viability and caspase-3/7 activity with the pan caspase inhibitor Z-VAD-FMK substituted for Nac showed an attenuation of caspase-3/7 activity that failed to rescue cells from the oxidative stress elicited by OA (Figs. 3C and 3D, lanes 4, 5 and 6) and the significantly greater loss in cell viability induced by rapamycin (lane 5).
Fig. 3. OA induces caspase activation and an oxidative stress-induced cell death.
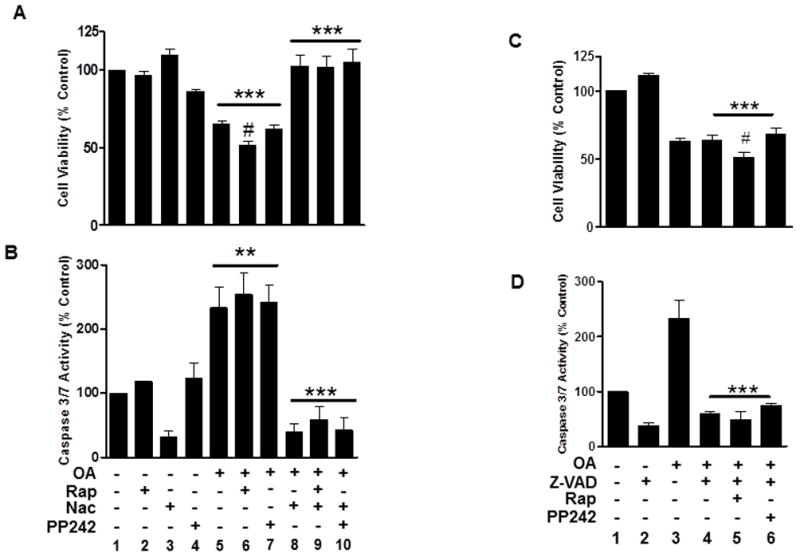
(A, B) Cells were treated with OA without or with rapamycin (48 hr), 5 mM Nac or PP242 (2 hr) as indicated. (C, D), Cells were treated as in A with 40 μM Z-VAD-FMK substituted for Nac. Cells were then assayed for viability (A, C) or (B, D) caspase-3/7 activation as described in the Material and Methods. Survival represents the percent viability relative to the vehicle-treated control (100%) ±S.E.M from at least three independent experiments. Caspase measurements (units/μg protein) were normalized as the percent activity relative to control ±SEM. *** (P<0.001) Indicates a significant difference between OA, OA/Rap or OA/PP242 versus control or versus the same treatments with Nac in A or in B and between OA/Z-VAD, OA/Rap/Z-VAD or OA/PP242/Z-VAD versus control or Z-VAD alone in C or versus OA alone in D; ** (P<0.01) Indicates a significant difference between OA, OA/Rap or OA/PP242 versus control in B; # (P<0.05) indicates a significant difference between OA or OA/PP242 versus OA/Rap in A and between OA/Z-VAD or OA/PP242/Z-VAD versus OA/Rap/Z-VAD in C.
Proteasome inhibition differentially modulates activated Akt and ubiquitinated protein levels
To address Akt degradation, the effects of the proteasome inhibitor epoxomicin were examined on the monomeric form of phosphorylated Akt in OA treated cells (Fig. 4A). Since caspases cleave Akt [14] and are induced by oxidative stress, these experiments included incubations with Z-VAD-FMK alone or in combination with Nac. Epoxomicin increased the levels of phosphorylated Akt at T308 by 4.0-fold in untreated control cells (Fig. 4A, lanes 1 with 8), 1.6-fold with OA alone (lanes 2 and 5), 1.8-fold with Z-VAD-FMK alone (lanes 3 and 6) and 2.4 with co-incubations of Z-VAD-FMK/Nac (lanes 4 and 7). These results suggest that caspase activation and oxidative stress influence Akt removal.
Fig. 4. Proteasome inhibition increases the levels of phosphorylated Akt while rapamycin augments an OA-induced increase in ubiquitinated proteins.
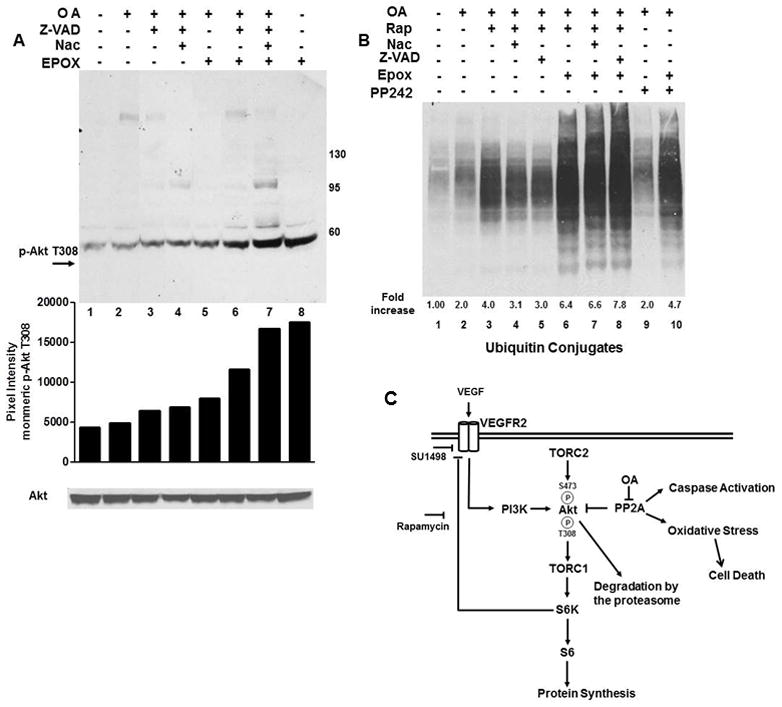
(A) OA treated cells were incubated with Nac, Z-VAD-FMK or 100 nM epoxomicin for 2 hr as indicated. Immunoblots (light exposure) were probed for phosphorylation of monomeric Akt at T308 or total Akt. (B) Cells were treated with OA and with rapamycin, Nac, PP242 or Z-VAD-FMK alone or in combination with epoxomicin and analyzed for ubiquitinated conjugates. Data in A and B were quantified using ImageJ and expressed as relative pixel intensity in A and the fold increase relative to control in B. (C) Model of mTOR, PP2A and VEGFR2 crosstalk in serum starved SK-N-SH cells. OA exacerbates the VEGF/VEGFR2-directed phosphorylation of S6K1 and S6 and Akt phosphorylation at T308 and S473 and induces an oxidative stress-induced cell death. PP2A and mTOR negatively control Akt phosphorylation through dephosphorylation and a S6K1 directed negative feedback of VEGFR2 signaling through PI3K, respectively.
Since oxidative stress [11] or rapamycin [6] increase ubiquitinated protein levels, ubiquitin protein conjugate levels were measured in OA/rapamycin treated cells alone or with Nac or Z-VAD-FMK without and with epoxomicin (Fig. 4B) and compared to OA/PP242 treated cells incubated alone or with epoxomicin. When compared to the untreated control, OA increased ubiquitinated proteins 2-fold (lane 1). OA/PP242 induced a 2-fold increase in ubiquitinated protein levels (lanes 2 and 9) that increased nearly 5-fold with epoxomicin (lane 10) while rapamycin induced a 4-fold (lane 3) or 3-fold increase with Nac (lane 4) or Z-VAD-FMK (lane 5) that were augmented to nearly 7-fold with epoxomicin (lanes 6, 7 and 8).
Discussion
Using a neuronal model of serum starved SK-N-SH neuroblastoma cells, we show that OA induces Akt hyperphosphorylation, increased levels of ubiquitinated proteins and an oxidative stress-induced cell death in serum starved SK-N-SH cells that are augmented by mTOR inhibition with rapamycin but not PP242. While these results are consistent with evidence that OA induces oxidative stress leading to caspase activation and cell death in neurons [22], they are in stark contrast to reports that hyperactivated Akt is sufficient for protecting neurons from harmful stimuli [1]. Moreover, rapamycin is a promising therapeutic for neurodegenerative disorders that promotes survival through Akt [1]. The failure of rapamycin to rescue SK-N-SH cells from the oxidative insult induced by OA, suggests that mTOR must cooperate with PP2A to promote survival. Consistent with this notion, PP2A activity prevented apoptosis induced by oxidative stress and reduced neurotoxicity in a mouse model of Parkinson’s disease [13]. The lack of survival with Z-VAD-FMK suggests that cell death is caspase-independent. Autophagy may mediate this event since it is associated with caspase-independent death, induced by OA [20, 23] and underlies rapamycin-enhanced neuronal cell death [2]. Rapamycin also sensitizes cells to oxidative insults through an Akt-dependent signaling of reactive oxygen species [15]. Therefore, rapamycin may increase the susceptibility of serum starved SK-N-SH cells to the pro-oxidant cell death induced by OA via enhanced Akt phosphorylation. These findings also suggest that rapamycin as a therapeutic has the risk of sensitizing neurons to the oxidative stress associated with neurodegenerative disorders.
Rapamycin enhances Akt activation by relieving a negative feedback loop in which S6K1 targets IRS-1 [9] or growth factor receptors [8] to downregulate Akt signaling through PI3K. Our finding that VEGFR2 inhibition attenuates rapamycin-mediated Akt hyperphosphorylation suggests that rapamycin counteracts a mTORC1/S6K1-mediated feedback inhibition of Akt activation through the VEGF/VEGFR2 pathway. These findings reveal a novel role for VEGFR2. S6K1 may function in this feedback by targeting VEGFR2 directly as shown for the tyrosine kinase receptors for PDGF [24] and insulin [8] or through IRS-1 since VEGFR2 colocalizes with IRS-1 to stimulate PI3K activation in VEGF treated endothelial cells [5]. We attribute the OA-induced phosphorylation of Akt at T308 and S473 in SK-N-SH cells to regulation by mTORC1 and mTORC2, respectively, since both phosphorylations are augmented by rapamycin and abrogated by the mTORC1/mTORC2 inhibitor PP242. Presumably, rapamycin enhances the recruitment of Akt to the plasma membrane for T308 and S473 phosphorylation by PDK1 and mTORC2, respectively [9]. In contrast, PP242 may block this feedback stimulation by destabilizing Akt activation at T308 through its suppression of mTORC2-mediated Akt phosphorylation at S473 [4].
The OA-induced accumulation of ubiquitinated forms of activated Akt and their enhancement by rapamycin is consistent with evidence that dephosphorylation by PP2A regulates Akt removal by proteasomal-mediated degradation [14, 16]. This is further supported by the epoxomicin-induced increase in phosphorylated Akt and evidence that Akt S473 hyperphosphorylation promotes K48-linked polyubiquitination [18, 19] while mTOR inhibition destabilizes Akt activated by VEGFR2 [16]. The accumulation of total ubiquitinated proteins with and without rapamycin and their further enhancement by epoxomicin suggests that PP2A exerts a global effect on protein turnover that is further modulated by mTOR. This accumulation could result from Akt-dependent mechanisms in that hyperphosphorylated Akt would generate either high levels of activated substrates or oxidatively damaged proteins for proteasomal removal [15] or increase the levels of E3 ligases for protein ubiquitination. Alternatively, the failure of rapamycin to block 4E-BP1 phosphorylation [4] would increase translation, leading to an elevated pool of ubiquitinated proteins.
Conclusions
In summary, PP2A inhibition in serum starved SK-N-SH cells leads to Akt hyperphosphorylation, increased ubiquitinated protein levels and a oxidative stress-induced cell death that is exacerbated by mTOR inhibition with rapamycin (Figure 4C). Notably, rapamycin augments Akt phosphorylation by counteracting a negative feedback loop of VEGFR2-directed Akt phosphorylation through PI3K. Therefore, PP2A and mTOR and VEGFR2 act in concert to regulate Akt activation, survival and protein degradation in response to serum starvation.
Highlights.
The effects of the PP2A inhibitor okadaic acid (OA) are examined in a neuronal model of SK-N-SH cells
OA induces increased Akt phosphorylation, cell death and ubiquitinated protein levels
Rapamycin exacerbates OA-induced Akt phosphorylation, death and ubiquitinated protein levels
Rapamycin augments Akt phosphorylation by suppressing a negative feedback loop through VEGFR2
mTOR and PP2A cooperate to regulate Akt activation, survival and ubiquitinated protein levels
Acknowledgments
This investigation was funded by a grant from the National Center for Research Resources [RR003037], a component of the National Institutes of Health (NIH) and a NINDS/NIH grant [SC1NS066033]. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011;12:437–452. doi: 10.1038/nrn3068. [DOI] [PubMed] [Google Scholar]
- 2.Choi KC, Kim SH, Ha JY, Kim ST, Son JH. A novel mTOR activating protein protects dopamine neurons against oxidative stress by repressing autophagy related cell death. J Neurochem. 2010;112:366–376. doi: 10.1111/j.1471-4159.2009.06463.x. [DOI] [PubMed] [Google Scholar]
- 3.Edelstein J, Hao T, Cao Q, Morales L, Rockwell P. Crosstalk between VEGFR2 and muscarinic receptors regulates the mTOR pathway in serum starved SK-N-SH human neuroblastoma cells. Cell Signal. 2011;23:239–248. doi: 10.1016/j.cellsig.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Feliers D, Chen X, Akis N, Choudhury GG, Madaio M, Kasinath BS. VEGF regulation of endothelial nitric oxide synthase in glomerular endothelial cells. Kidney Int. 2005;68:1648–1659. doi: 10.1111/j.1523-1755.2005.00575.x. [DOI] [PubMed] [Google Scholar]
- 6.Harston RK, McKillop JC, Moschella PC, Van Laer A, Quinones LS, Baicu CF, Balasubramanian S, Zile MR, Kuppuswamy D. Rapamycin treatment augments both protein ubiquitination and Akt activation in pressure-overloaded rat myocardium. Am J Physiol Heart Circ Physiol. 2011;300:H1696–1706. doi: 10.1152/ajpheart.00545.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 8.Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA, Sabatini DM. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332:1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–222. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kondo M, Oya-Ito T, Kumagai T, Osawa T, Uchida K. Cyclopentenone prostaglandins as potential inducers of intracellular oxidative stress. J Biol Chem. 2001;276:12076–12083. doi: 10.1074/jbc.M009630200. [DOI] [PubMed] [Google Scholar]
- 12.Kuo YC, Huang KY, Yang CH, Yang YS, Lee WY, Chiang CW. Regulation of phosphorylation of Thr-308 of Akt, cell proliferation, and survival by the B55alpha regulatory subunit targeting of the protein phosphatase 2A holoenzyme to Akt. J Biol Chem. 2008;283:1882–1892. doi: 10.1074/jbc.M709585200. [DOI] [PubMed] [Google Scholar]
- 13.Lee KW, Chen W, Junn E, Im JY, Grosso H, Sonsalla PK, Feng X, Ray N, Fernandez JR, Chao Y, Masliah E, Voronkov M, Braithwaite SP, Stock JB, Mouradian MM. Enhanced phosphatase activity attenuates alpha-Synucleinopathy in a mouse model. J Neurosci. 2011;31:6963–6971. doi: 10.1523/JNEUROSCI.6513-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao Y, Hung MC. Physiological regulation of Akt activity and stability. Am J Transl Res. 2010;2:19–42. [PMC free article] [PubMed] [Google Scholar]
- 15.Nogueira V, Park Y, Chen CC, Xu PZ, Chen ML, Tonic I, Unterman T, Hay N. Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis. Cancer Cell. 2008;14:458–470. doi: 10.1016/j.ccr.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riesterer O, Zingg D, Hummerjohann J, Bodis S, Pruschy M. Degradation of PKB/Akt protein by inhibition of the VEGF receptor/mTOR pathway in endothelial cells. Oncogene. 2004;23:4624–4635. doi: 10.1038/sj.onc.1207596. [DOI] [PubMed] [Google Scholar]
- 17.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 18.Suizu F, Hiramuki Y, Okumura F, Matsuda M, Okumura AJ, Hirata N, Narita M, Kohno T, Yokota J, Bohgaki M, Obuse C, Hatakeyama S, Obata T, Noguchi M. The E3 ligase TTC3 facilitates ubiquitination and degradation of phosphorylated Akt. Dev Cell. 2009;17:800–810. doi: 10.1016/j.devcel.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 19.Wu YT, Ouyang W, Lazorchak AS, Liu D, Shen HM, Su B. mTOR complex 2 targets Akt for proteasomal degradation via phosphorylation at the hydrophobic motif. J Biol Chem. 2011;286:14190–14198. doi: 10.1074/jbc.M111.219923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Kim SO, Li Y, Han J. Autophagy contributes to caspase-independent macrophage cell death. J Biol Chem. 2006;281:19179–19187. doi: 10.1074/jbc.M513377200. [DOI] [PubMed] [Google Scholar]
- 21.Yang WL, Zhang X, Lin HK. Emerging role of Lys-63 ubiquitination in protein kinase and phosphatase activation and cancer development. Oncogene. 2010;29:4493–4503. doi: 10.1038/onc.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yi KD, Covey DF, Simpkins JW. Mechanism of okadaic acid-induced neuronal death and the effect of estrogens. J Neurochem. 2009;108:732–740. doi: 10.1111/j.1471-4159.2008.05805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoon SY, Choi JE, Kweon HS, Choe H, Kim SW, Hwang O, Lee H, Lee JY, Kim DH. Okadaic acid increases autophagosomes in rat neurons: implications for Alzheimer’s disease. J Neurosci Res. 2008;86:3230–3239. doi: 10.1002/jnr.21760. [DOI] [PubMed] [Google Scholar]
- 24.Zhang H, Bajraszewski N, Wu E, Wang H, Moseman AP, Dabora SL, Griffin JD, Kwiatkowski DJ. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J Clin Invest. 2007;117:730–738. doi: 10.1172/JCI28984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]