

Wnt-11/frizzled-7 reduces the lateral clustering of C-cadherin by capturing the protocadherin PAPC and C-cadherin into distinct adhesion-modulating complexes.
Abstract
Wnt-11/planar cell polarity signaling polarizes mesodermal cells undergoing convergent extension during Xenopus laevis gastrulation. These shape changes associated with lateral intercalation behavior require a dynamic modulation of cell adhesion. In this paper, we report that Wnt-11/frizzled-7 (Fz7) controls cell adhesion by forming separate adhesion-modulating complexes (AMCs) with the paraxial protocadherin (PAPC; denoted as AMCP) and C-cadherin (denoted as AMCC) via distinct Fz7 interaction domains. When PAPC was part of a Wnt-11–Fz7 complex, its Dynamin1- and clathrin-dependent internalization was blocked. This membrane stabilization of AMCP (Fz7/PAPC) by Wnt-11 prevented C-cadherin clustering, resulting in reduced cell adhesion and modified cell sorting activity. Importantly, Wnt-11 did not influence C-cadherin internalization; instead, it promoted the formation of AMCC (Fz7/Cadherin), which competed with cis-dimerization of C-cadherin. Because PAPC and C-cadherin did not directly interact and did not form a joint complex with Fz7, we suggest that Wnt-11 triggers the formation of two distinct complexes, AMCC and AMCP, that act in parallel to reduce cell adhesion by hampering lateral clustering of C-cadherin.
Introduction
Morphogenetic movements in gastrulation, organ formation, or tissue remodeling are composed of different cell behaviors, e.g., modulation of cell adhesion, cell polarity formation, and cytoskeleton rearrangements. Common molecular principles regulating cadherin-mediated cell adhesion or tissue polarity by the Wnt/planar cell polarity (PCP) signaling have been described for invertebrates and vertebrates (Wang and Nathans, 2007; Hammerschmidt and Wedlich, 2008; Goodrich and Strutt, 2011; Niessen et al., 2011). But how cell adhesion gets adjusted to tissue remodeling processes controlled by Wnt/PCP signaling is less understood.
In Xenopus laevis gastrulation, β-catenin–independent Wnt signaling and the paraxial protocadherin (PAPC) orchestrate multiple cellular properties to coordinate the movements of different tissue layers. The classical type I C-cadherin (cdhcA; former term, EP-Cadherin) is the primary mediator of cell adhesion in the early Xenopus embryo and ubiquitously distributed in all tissue layers (Müller et al., 1994; Lee and Gumbiner, 1995). This raises the question how C-cadherin adhesion is modulated in gastrulation.
PAPC and the noncanonical Wnt ligands, Wnt-11 and Wnt-5a, are strongly interconnected in the Wnt signaling network. Wnt/PCP signaling is mediated by the frizzled-7 (Fz7) receptor, which binds to PAPC, and both are required to keep the involuting mesoderm separated from the overlying ectoderm (Winklbauer et al., 2001; Medina et al., 2004). Upon Wnt/PCP ligand binding to Fz7, disheveled (dsh/dvl) shifts to the membrane and activates RhoA, Rac1, and JNK (Habas et al., 2001, 2003). PAPC, which is a target gene of the noncanonical Wnt-5a–Ror2 pathway (Schambony and Wedlich, 2007), activates RhoA and JNK but inhibits Rac1 (Medina et al., 2004; Unterseher et al., 2004; Schambony and Wedlich, 2007). ANR5 and Sprouty interact with the cytoplasmic domain of PAPC, mediate RhoA activation, and enhance PCP signaling (Chung et al., 2007; Wang et al., 2008). In contrast to the signaling function of PAPC, its role in cell adhesion is still unresolved. Based on dissociation and reaggregation assays of animal caps injected with Xenopus PAPC RNA, cell adhesion activity was explained with homophilic binding of PAPC (Kim et al., 1998; Yamamoto et al., 1998). Calcium-dependent homophilic binding was indeed reported for arcadlin, the PAPC homologue in rat (Yamagata et al., 1999). Strong concerns against an intrinsic adhesion activity of PAPC are given by the finding that PAPC alters C-cadherin–mediated adhesion (Chen and Gumbiner, 2006). As possible mechanisms, oligomerization of PAPC via Cys residues (Chen et al., 2007) or complex formation of PAPC and FLRT3 (fibronectin leucine-rich domain transmembrane protein-3) with C-cadherin (Chen et al., 2009) is in discussion.
In contrast to the ubiquitous presence of C-cadherin, PAPC expression is restricted to the mesoderm at the blastopore lip, where morphogenetic movements start (Kim et al., 1998). Thus, a spatially restricted slight decrease of C-cadherin adhesion through PAPC in the involuting mesoderm is an attractive model (Chen and Gumbiner, 2006).
Here, we report that Wnt-11 diminishes cis-dimerization of C-cadherin by capturing PAPC and C-cadherin into two distinct Wnt-11/Fz7 adhesion-modulating complexes (AMCs). Both complexes inhibit lateral clustering of C-cadherin and thereby weaken cadherin-mediated adhesion. Our experiments reveal that, via this novel mechanism, adjustment of cell adhesion and polarity formation is achieved by the same signal, Wnt-11.
Results
PAPC cell membrane localization is regulated by Wnt-11
Although the interconnection of Wnt/PCP and PAPC signaling is well known, its physiological role remains obscure. Therefore, we examined the influence of the PCP ligand Wnt-11 on the subcellular localization of PAPC. For this purpose, fluorescent-labeled PAPC (PAPC-mCherry) was injected into the dorsal marginal zone (DMZ) of 16-cell–stage Xenopus embryos together with membrane-anchored (growth-associated protein 43 [GAP43]) GFP. The latter served to label cell membranes. PAPC-mCherry localization was monitored in DMZ explants by in vivo time-lapse microscopy (Fig. 1 A). Membrane versus vesicle localization of PAPC was evaluated by double-blind scoring, and DMZ explants were classified according to five categories, explained in Fig. S1 A. At the onset of gastrulation, when mesodermal cells align along the mediolateral axis, PAPC was both localized at the cell membrane and in cytoplasmic vesicles (Fig. 1, B and H). However, when Wnt-11 was coinjected, enhanced PAPC membrane localization was observed (Fig. 1, C and H). Depletion of Wnt-11, instead, resulted in loss of PAPC at cell membranes and in increased numbers of vesicles (Fig. 1, D and H). These observations indicate that PAPC subcellular localization is controlled by Wnt-11.
Figure 1.

Wnt-11 regulates PAPC cell membrane localization through Fz7 during convergent extension. (A) Scheme showing time-lapse xPAPC localization analyses in DMZ explants. The two dorsal blastomeres of 16-cell–stage embryos were injected with 500 pg xPAPC-mCherry RNA alone or in combination with 20 pg xWnt-11 RNA, 1 pmol xWnt-11 MO, 500 pg xFz7 RNA, 1.6 pmol xFz7 MO, and/or 500 pg dnDyn1 RNA. 50 pg GAP43-GFP RNA was injected in all samples as a cell membrane tracer. At stage 10.25, DMZ explants were cut, and subcellular localization of xPAPC-mCherry was analyzed. (B–G) Representative images of time-lapse videos are shown. xWnt-11 stabilized xPAPC at the cell membrane, and this activity required xFz7. Bars, 60 µm. (H) Counting of DMZ explants showing xPAPC subcellular localization according the observed phenotypes showed in Fig. S1 A. n, number of DMZ explants. # exp., number of independent experiments. (I) Western blot (WB) analysis of total xPAPC-myc (gray arrowhead) protein amount performed with cell lysates of stage-11 embryos. xWnt-11 did not influence total xPAPC-myc protein amount, whereas xFz7 depletion reduced the overall protein level of xPAPC-myc, even in the presence of xWnt-11. GAP43-GFP (black arrowhead) served as an injection control, α-tubulin (black star) as a loading control, and PonceauS as a transfer control. Injection amount was as follows: 1 ng xPAPC-myc RNA, 40 pg xWnt-11 RNA, 1 pmol xWnt-11 MO, 500 pg xFz7 RNA, 1.6 pmol xFz7 MO, and 100 pg GAP43-GFP RNA. (J) Relative xPAPC-myc signal intensity in dependency of xWnt-11 and xFz7. For more details, see Materials and methods. Error bars show SEM. Student’s t test was performed (*, P < 0.05; **, P < 0.005).
Next, we investigated whether Wnt-11–induced membrane stabilization of PAPC also requires the Wnt-11 receptor Fz7 and performed Fz7 gain- and loss-of-function experiments in Wnt-11–expressing embryos. Fz7 coexpression led to a slight increase in the membrane signal (Fig. 1, E and H). In Fz7 morphants, however, PAPC was strongly reduced at cell membranes, but, in contrast to Wnt-11 morphants also, fewer intracellular PAPC vesicles were observed (Fig. 1, F and H). As Wnt-11/Fz7 does not regulate PAPC gene expression during gastrulation (Schambony and Wedlich, 2007), these observations indicate that Fz7 is required for PAPC protein stabilization in general, whereas Wnt-11 together with Fz7 influences PAPC subcellular localization. To corroborate this hypothesis, we performed Western blot analysis with DMZ explants expressing PAPC-myc in dependency of Wnt-11/Fz7 (Fig. 1, I and J). The overall level of PAPC-myc protein remained unchanged in the presence or absence of Wnt-11. In the absence of Fz7, however, the total level of PAPC-myc dropped down, even when Wnt-11 was coexpressed, whereas in the presence of both, an increase in PAPC protein was observed (Fig. 1, I and J). These Western blot data confirm our experiments in which fluorescent PAPC protein was analyzed microscopically (Fig. 1, B–F).
The observed changes in subcellular distribution of PAPC were Wnt-11 specific, as Wnt-5a gain- or loss-of-function experiments had no influence on PAPC localization (Fig. S1 B). Additionally, we investigated the influence of Wnt-11 and Fz7 on the cellular distribution of C-cadherin, the prominent cell adhesion mediator in the early Xenopus embryo. Strikingly, neither Wnt-11 and Fz7 overexpression nor depletion altered the subcellular localization of C-cadherin (Fig. 2, A–E). These results show that Wnt-11 specifically influences the subcellular localization of PAPC.
Figure 2.
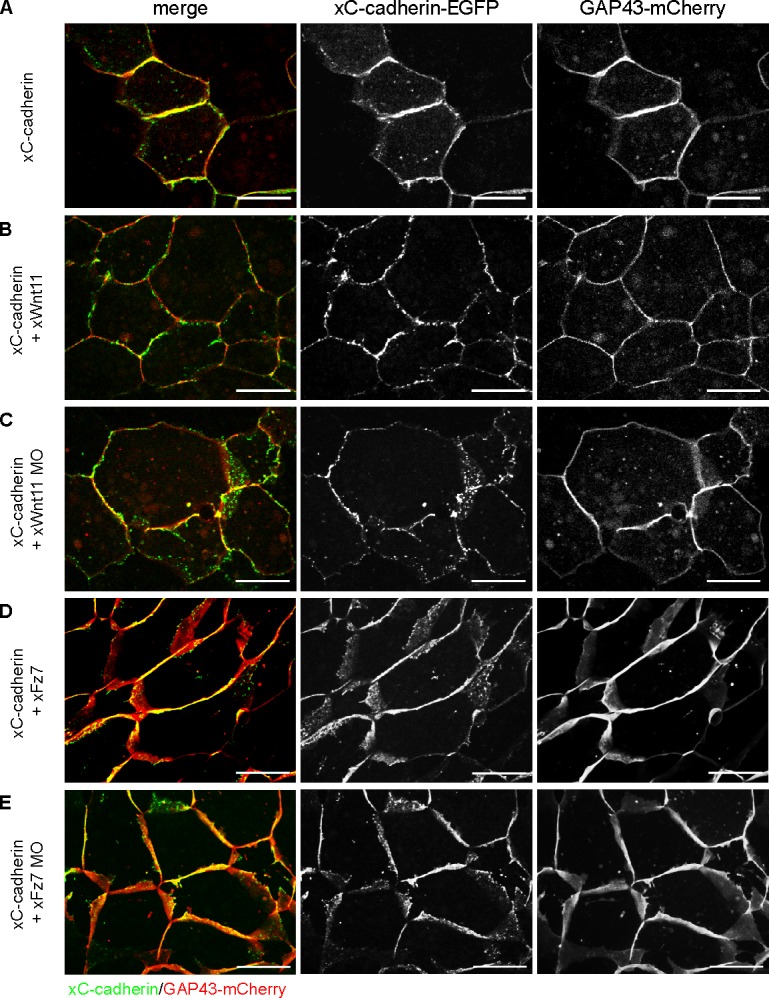
C-cadherin surface expression is not affected by Wnt-11 and Fz7. Time-lapse microscopic analysis of xC-cadherin subcellular localization during convergent extension. 16-cell–stage embryos were injected into the DMZ region with 500 pg xC-cadherin–EGPF RNA alone or in combination with 20 pg xWnt-11 RNA, 1 pmol xWnt-11 MO, 500 pg xFz7 RNA, or 1.6 pmol xFz7 MO. 200 pg GAP43-mCherry RNA was coinjected in all samples as a cell membrane tracer. At stage 10.25, DMZ explants were cut, and subcellular localization of xC-cadherin–EGFP was analyzed. (A–E) Representative images of confocal time-lapse videos are shown. Neither overexpression nor depletion of xWnt-11 or xFz7 had an effect on xC-cadherin–EGFP subcellular localization. Bars, 20 µm.
Wnt-11 stabilizes PAPC at cell membranes by blocking its internalization
Dynamin1 is a GTPase required in endocytosis (Mettlen et al., 2009). Mutants with defects in the GTP-binding site behave as dominant-negative proteins inhibiting the first step of endocytosis by blocking endocytotic vesicle formation at the plasma membrane (Herskovits et al., 1993; Damke et al., 1994). To investigate whether Wnt-11 depletion leads to PAPC endocytosis, we injected dominant-negative Dynamin1 (dnDyn1) RNA into Wnt-11 morphants. As seen in Fig. 1 G, dnDyn1 prevented the formation of intracellular PAPC-positive vesicles, indicating that Wnt-11 controls internalization of PAPC, thereby regulating its cell membrane localization. To further confirm these observations, we determined and compared the cell surface protein level of PAPC in Wnt-11–overexpressing and Wnt-11–depleted DMZ explants by surface biotinylation assay (Fig. 3 A). Quantification of three independent experiments revealed that the cell surface level of PAPC protein decreased in Wnt-11 morphants (Fig. 3 B), pointing to an increased endocytosis of PAPC. Interestingly, PAPC expression as well as Wnt-11 did not influence the surface protein level of C-cadherin. In summary, these results confirmed our in vivo time-lapse microscopy data of DMZ explants expressing PAPC-mCherry or C-cadherin–EGFP (Figs. 1, 2, and S2). However, as we did not detect a significant increase of PAPC in the membrane fraction when Wnt-11 was overexpressed (Fig. 3, A and B) compared with our time-lapse data (Fig. 1 C), we performed biotin pulse-chase experiments. By labeling the cell surface protein fraction with Sulfo-NHS-SS-biotin followed by endocytosis induction via a temperature switch, we could directly analyze whether Wnt-11 stabilized PAPC at the cell membrane by blocking or retarding its internalization. At the same time, we investigated whether C-cadherin surface expression remained unaffected in Wnt-11–overexpressing and Wnt-11–depleted DMZ explants. After endocytosis induction, we detected the residual biotin-labeled PAPC and C-cadherin fractions at certain time points (Fig. 3 C). The amount of biotinylated PAPC continuously decreased in untreated DMZ explants, whereas a reduction of biotinylated C-cadherin was first observed after 45 min. In the presence of Wnt-11, however, biotinylated PAPC was stabilized at the membrane, and a decrease was not seen until 45 min, similar to C-cadherin. As expected, in the absence of Wnt-11, a dramatic drop of biotinylated PAPC was measured already after 10 min, whereas biotinylated C-cadherin was found to be more stable, similar to C-cadherin in untreated DMZ explants (Fig. 3 C). These results underline the specific influence of Wnt-11 on the subcellular distribution of PAPC: while it stabilizes PAPC at cell membranes, the cell surface expression of C-cadherin remained unaffected.
Figure 3.

Wnt-11 stabilized cell surface PAPC, whereas C-cadherin remained unaffected. (A) Cell surface biotinylation assay performed with DMZ explants. Cell surface and total protein amount of xPAPC-myc (9E10) and endogenous xC-cadherin (6B6) were detected by Western blotting. xWnt-11 depletion reduced cell surface amount of xPAPC-myc, whereas cell surface level of endogenous xC-cadherin was not effected. neg. KO, negative control. (B) Relative cell surface amounts of xPAPC-myc and endogenous xC-cadherin in dependency of xWnt-11, calculated as described in Materials and methods. Error bars show SEM. Student’s t test was performed (*, P < 0.05). # exp., number of independent experiments. (C) Biotin pulse-chase experiment performed with DMZ explants. At the indicated induction times, the residual biotin-labeled (surface) and total protein amount of xPAPC-myc (9E10) and endogenous xC-cadherin (α-Ccad) were detected by Western blotting. Relative surface amounts of xPAPC-myc and endogenous xC-cadherin were plotted over endocytosis induction time. Cell surface xPAPC was stabilized in xWnt-11–overexpressing DMZ explants, whereas xWnt-11 depletion led to a faster degradation of xPAPC membrane fraction. xWnt-11 did not affect the cell surface expression of xC-cadherin. Injection amount was as follows: 1 ng xPAPC-myc RNA, 40 pg xWnt-11 RNA, and 1 pmol xWnt-11 MO. pos. KO, positive control. n, number of independent experiments.
Next, we examined whether PAPC endocytosis requires clathrin or caveolin1. Treatment of DMZ explants with specific inhibitors for clathrin- or caveolin1-dependent internalization and analysis of the membrane versus cytoplasmic pool of PAPC using surface biotinylation assay revealed that PAPC is internalized in vivo via the clathrin-mediated endocytosis during Xenopus gastrulation (Fig. 4 A). Both clathrin-specific inhibitors, chlorpromazine and monodansylcadaverine, increased the cell surface protein level of PAPC, whereas caveolin-specific inhibitors (filipin and genistein) did not change the cell surface amount of PAPC (see ratio surface/cytoplasmic PAPC pool; Fig. 4 A).
Figure 4.

Wnt-11 controls PAPC membrane localization by blocking its clathrin-mediated internalization. (A) Cell surface biotinylation assay performed with DMZ explants, injected with 1 ng xPAPC-myc RNA, and treated with clathrin- and caveolin1-specific inhibitors. Cell surface, cytoplasmic (cyto), and total protein amounts of xPAPC-myc were detected by Western blotting using specific myc tag antibody (9E10). The relative xPAPC-myc signal intensity (IN) of cell surface and cytoplasmic fractions were measured as described in Materials and methods. Finally, the ratio surface versus cytoplasmic fraction was calculated. The cell surface amount of xPAPC increased in the presence of clathrin-specific inhibitors. Used clathrin-specific inhibitors were 60 µM chlorpromazine (CPZ) and 300 µM monodansylcadaverine (MDC). Used caveolin1-specific inhibitors were 5 µg/ml filipin and 200 µM genistein. neg. KO, negative control. (B) Confocal microscopic analysis of DMZ explants, cryosectioned, and immunostained for clathrinHC and xPAPC-EGFP. Nuclei were stained with DAPI. xWnt-11 expression resulted in clathrin-xPAPC accumulation at cell membranes. In xWnt-11 morphants, membrane localization of clathrin and PAPC was strongly reduced but increased in intracellular vesicles. Injection amount was as follows: 500 pg xPAPC-EGFP RNA, 20 pg xWnt-11 RNA, and 1 pmol xWnt-11 MO. Bars, 20 µm. (C) Ratio number of membrane versus cytoplasmic colocalization of xPAPC (xPAPC-clathrin-CL) and clathrinHC in dependency of xWnt-11 (for more details, see Materials and methods). Error bars show SEM. Student’s t test was performed (**, P < 0.005). n, number of cells.
Finally, immunofluorescence analysis showed that clathrin-mediated internalization of PAPC is controlled by Wnt-11 (Fig. 4 B). In the presence of Wnt-11, clathrin accumulated at the cell membrane and colocalized with membrane-stabilized PAPC. In Wnt-11 morphants, however, membrane localization of both PAPC and clathrin decreased, and more intracellular double-positive vesicles for PAPC and clathrin were observed (Fig. 4 B; also see ratio of membrane/cytoplasmic colocalization of PAPC and clathrin in Fig. 4 C). In summary, these experiments showed that Wnt-11 specifically stabilizes PAPC at the cell membrane by blocking its clathrin- and Dynamin1-mediated internalization (Figs. 1 G, 3, and 4).
Cell sorting and cell adhesion are influenced by PAPC, Fz7, and Wnt-11
The Wnt-11–dependent PAPC stabilization at the cell membrane prompted us to test whether this could decrease C-cadherin–mediated cell adhesion. It was demonstrated in detail that PAPC shows no intrinsic adhesion property but instead induces cell sorting by lowering C-cadherin adhesion (Chen and Gumbiner, 2006). Therefore, we used an in vivo cell dispersion assay, which directly monitors cell sorting activities (Kim et al., 1998). Synthetic GFP RNA was injected into a single animal blastomere (A-tier) of 32-cell–stage embryos (Fig. 5 A). These animal blastomeres express a low level of Fz7 but no PAPC or Wnt-11 (Abu-Elmagd et al., 2006; Schambony and Wedlich, 2007). At late-gastrula stage 12.5 (Nieuwkoop and Faber, 1967), GFP-expressing cells were found extensively interspersed with unlabeled cells, which was not changed upon Wnt-11 injection (Fig. 5 B). In contrast, cells derived from blastomeres injected with Fz7 or coinjected with Fz7 and Wnt-11 formed tight patches and showed sharp borders to the unlabeled surrounding cells, indicating that cadherin-mediated adhesion was reduced (Fig. 5 B; Chen and Gumbiner, 2006). When PAPC was injected, cell sorting was observed, which increased when Fz7 and Wnt-11 were coinjected (Fig. 5 B). We analyzed three different egg batches according to the criteria of Chen and Gumbiner (2006) for statistical evaluation (Fig. 5 C). In addition, we quantified the results of the cell dispersal assay by measuring the spot size and fluorescence intensity and monitored the cell sorting effect as intensity/size ratio (Fig. 5 D; for method description, see Fig. S3). Thereby, we confirmed that Fz7 by itself induced cell sorting but slightly less than PAPC. Cell sorting through PAPC was not significantly increased in the presence of Wnt-11. However, when all three components were coinjected, a robust increase in cell sorting was measured.
Figure 5.

Wnt-11/Fz7 and PAPC cooperate in decreasing C-cadherin–mediated cell adhesion. (A) Scheme of cell dispersal assay. One animal blastomere of a 32-cell–stage embryo was injected with synthetic mRNAs together with the lineage tracer GAP43-GFP. At stage 12.5, intermingling of GFP-expressing cells was monitored. The patch morphology reflects changes in cell sorting activity as a result of changes in cell adhesion: tight patches with sharp borders and high fluorescence intensity indicate strong cell sorting properties. (B) Cell dispersal assay. Representative images of stage-12.5 embryos. Dashed lines reflect embryo size. Bar, 900 µm. Corresponding pseudocolor images show fluorescence intensity. Relative intensity of fluorescence is indicated by the color bar, where blue and violet represent the lowest and the highest level of detectable fluorescence, respectively. (C) Evaluation of the increasing changes in cell sorting activity. n, number of embryos; # exp., number of experiments. (D) Quantitative evaluation of cell dispersal assay. The diagram displays the cell sorting activity expressed as the relative ratio between intensity and area (for more details, see Fig. S3 and Materials and methods). Error bars indicate SEM. Student’s t test was performed (**, P < 0.005). (E) Scheme of blastomere adhesion assay. Synthetic mRNAs were injected in the animal hemisphere of 4-cell–stage embryos together with the lineage tracers GAP43-GFP and H2B-mCherry. At stage 8.5, animal caps were dissected. The blastomeres of the inner layer were dissociated, spotted on 1 µg/ml CEC1-5–coated substrates, and allowed to adhere. The adhesion strength of blastomeres was measured by the ratio of the number of blastomeres remaining attached after (Nt) versus the number before (N0) flipover of the Petri dish. (F) Representative images of dissociated blastomeres expressing the indicated constructs attached to the CEC1-5 substrate before and after the flipover. Bars, 100 µm. (G) Quantification of the blastomere adhesion assay. The diagram displays the xC-cadherin adhesion index as the relative ratio between blastomeres attached to the CEC1-5 substrate after and before flipover. Injection amount was as follows: 500 pg xPAPC RNA, 500 pg xFz7 RNA, 20 pg xWnt-11 RNA, 100 pg GAP43-GFP RNA, and 250 pg H2B-mCherry RNA. Error bars indicate SEM. Student’s t test was performed (*, P < 0.05; **, P < 0.005). n, number of counted blastomeres.
To further confirm that the cell sorting effects result from an altered C-cadherin–mediated adhesion, blastomere adhesion assays were performed using purified C-cadherin extracellular cadherin repeat (CEC) 1-5 as substrate (Niessen and Gumbiner, 2002). Blastomeres obtained from dissociated animal caps (stage 8.5) were allowed to adhere to CEC1-5 for 30 min. Then, the dish was flipped, which results in the detachment of cells that cannot adhere to the cadherin-coated surface (Fig. 5 E). When cells were counted before and after flipping, a decrease of ∼50% in adhesion to CEC1-5 was observed in the presence of PAPC and ∼70% in the presence of Fz7. However, the strongest decrease in C-cadherin adhesion was measured when PAPC, Wnt-11, and Fz7 were coexpressed (∼85%; Fig. 5, F and G).
The cell sorting and C-cadherin adhesion effects of PAPC and of Fz7 observed here are in line with previous reports (Medina et al., 2000; Chen and Gumbiner, 2006; Chen et al., 2007, 2009). However, the cooperative effect of Wnt-11/Fz7/PAPC has not been investigated so far. We assume that the latter is in consequence of PAPC membrane stabilization by Wnt-11/Fz7 (see Figs. 1, 3, and 4).
The molecular mechanism of how Fz7 alone influences the adhesion activity of C-cadherin is not yet understood. Therefore, we analyzed which part of the Fz7 receptor is required for cell sorting in the absence of PAPC. Using cell dispersal assay, we investigated the cell sorting capacity of several Fz7 deletion mutants, depicted in Fig. S4 A. Only two deletion mutants consisting of the cytoplasmic tail alone (CxFz7) or the cytoplasmic tail with the seven transmembrane regions (ΔNxFz7) of Fz7 led to cell sorting comparable with the full-length Fz7 receptor (Fig. S4, B and C). Strikingly, the robust increase in cell sorting when Fz7, Wnt-11, and PAPC were coexpressed seems to be mediated by another Fz7-interacting domain. The cell dispersal assay revealed that the Fz7 deletion mutant consisting of the extracellular domain and the first transmembrane unit (xFz7-TM1; Fig. S4, A, D, and E) exhibited quite similar sorting activity as the full-length Fz7 receptor, whereas the effect of the cytoplasmic domain (CxFz7) was moderate. Thus, cell sorting in the presence of PAPC requires the extracellular domain of Fz7, which represents the PAPC-binding site (Medina et al., 2004). For Fz7-induced cell sorting independently of PAPC, however, the cytoplasmic domain of Fz7 seems to be essential.
Fz7 physically interacts with C-cadherin and PAPC
Based on our cell dispersal results with the different Fz7 deletion mutants, we investigated complex formation between cadherins and Fz7 by coimmunoprecipitations. As shown in Fig. 6 A, Fz7 coprecipitates C-cadherin and PAPC. We also expanded these experiments by performing coimmunoprecipitation in the presence and absence of Wnt-11. However, we could not detect significant changes depending on Wnt-11 (Fig. 6, B and C). Next, we asked whether a trimeric complex is formed between Fz7, C-cadherin, and PAPC. This we can exclude because C-cadherin coprecipitates with Fz7 to a much lesser extent when PAPC is overexpressed (Fig. 6 D). This effect was also observed when coprecipitation of endogenous C-cadherin with Fz7 was analyzed (Fig. 6 E). Thus, Fz7 forms distinct complexes either with C-cadherin or PAPC, which we termed AMCC and AMCP for AMCs containing either C-cadherin or PAPC. Thus, PAPC competes with C-cadherin for Fz7 binding, and the AMCP seems to be preferentially formed.
Figure 6.

Fz7 complex formation with C-cadherin and PAPC shown by coimmunoprecipitations. For coimmunoprecipitation (co-IP) assays, the indicated constructs were injected into the DMZ of 8-cell–stage embryos. At stage 10.5, DMZ explants were dissected and lysed. In all cases, xFz7 was precipitated with myc antibody (9E10), and complex formation was detected by Western blotting (WB) for the proteins shown (arrows). (A) xPAPC and xC-cadherin coimmunoprecipitations with xFz7. xFz7 interacted with both xC-cadherin and xPAPC. (B and C) xWnt-11–dependent coimmunoprecipitations of xFz7 with xC-cadherin (B) or xPAPC (C). No significant changes could be detected. (D and E) xC-cadherin coimmunoprecipitations with xFz7 in the presence of xPAPC. xPAPC competed with both exogenous (D) and endogenous (E) xC-cadherin for xFz7 complex formation with preference for xPAPC/xFz7 complex. Injection amount was as follows: 1 ng xC-cadherin–EGFP RNA, 1 ng xPAPC-HA RNA, 500 pg xFz7-myc RNA, 40 pg xWnt-11 RNA, and 1 pmol xWnt-11 MO.
To monitor complex formation in vivo, we used the bimolecular fluorescence complementation (BiFC) assay, which is based on two nonfluorescence fragments of a fluorescent protein (here, for example, YFP). When they are brought in proximity to each other by direct interaction between proteins fused to the fragments, the fluorophore is activated, and the YFP signal appears (Kerppola, 2008). For this purpose, we cloned different split YFP constructs: PAPC and C-cadherin both C-terminally fused either to the N-terminal (YN) or the C-terminal (YC) half of YFP, full-length Fz7 C-terminally fused to YN, and a short form of Fz7 (Fz7-TM1) fused to YC (Fig. 7 A). The Fz7-TM1 construct consists of the extracellular part and the first transmembrane domain of Fz7. All constructs also contained a myc tag, which served to control their successful expression (Fig. 7 A). We expressed different combinations of these constructs in DMZ explants to monitor complex formation during convergent extension. C-cadherin–YC and Fz7-YN formed complexes at cell membranes displayed by the YFP signal. No YFP signal was observed when Fz7-TM1-YC was combined with C-cadherin–YN (Fig. 7 B). Vice versa results were obtained for PAPC. Fz7-TM1-YC and PAPC-YN led to a positive YFP signal in the cell membrane, whereas no signal was observed when the full-length Fz7 was combined with PAPC (Fig. 7 B). The latter could be explained by sterical hindrance of split YFP reconstitution as a result of the distance given by seven transmembrane domains. From these results, we conclude that C-cadherin most likely interacts with the cytoplasmic part of Fz7, whereas PAPC binds to its extracellular part, as reported by Medina et al. (2004). Importantly, we did not observe a direct interaction of PAPC and C-cadherin in the BiFC assay (Fig. 7 B).
Figure 7.

Fz7 interacts with C-cadherin and PAPC at the cell membrane in vivo, shown by BiFC. (A) Primary structure of the different split YFP constructs used in the BiFC assay. The putative interacting partners were either fused to YFP fragment consisting of the first N-terminal 155 amino acids (YN) or the last C-terminal 83 amino acids (YC) of YFP. All constructs contained a myc tag that served as a control for successful expression. β, β-catenin binding site; CRD, cysteine-rich domain; cyto, cytoplasmic region; EC, extracellular cadherin repeat; p120, p120 binding site; SS, signal peptide sequence; TM, transmembrane domain. (B) Expression of different combinations of the split YFP constructs in DMZ explants to monitor complex formation in vivo during convergent extension. DMZ explants were either coinjected with GAP43-mCherry (GAP43) to label cell membranes or fixed and fluorescently immunostained for split YFP constructs using myc antibody (9E10) to control their successful expression. Nuclei were stained with DAPI. Protein–protein interaction was displayed by the YFP fluorescence (BiFC). Full-length xFz7 interacted with xC-cadherin, whereas xFz7-TM1 interacted with xPAPC at the cell membranes. No direct interaction between xC-cadherin and xPAPC was observed. (C–G) In vivo xFz7–xC-cadherin complex formation in the presence or absence of xPAPC or xWnt-11. Cell membranes were labeled with GAP43-mCherry, and protein–protein interaction was displayed by YFP fluorescence (BiFC). Depletion of xPAPC promoted the interaction between xFz7 and xC-cadherin, whereas in Wnt11 morphants, xFz7–xC-cadherin complex formation was abolished. Injection amount was as follows: 1 ng xPAPC-YN/YC RNA, 1 ng xC-cadherin-YC/YN RNA, 500 pg xFz7-YN RNA, 500 pg xFz7-TM1-YC, 20 pg xWnt-11 RNA, 1 pmol xWnt-11 MO, 1 ng xPAPC RNA, 1.6 pmol xPAPC MO, and 200 pg GAP43-mCherry RNA. Bars, 20 µm.
Although C-cadherin and PAPC bind to different parts of Fz7, coimmunoprecipitation assays (Fig. 6, D and E) revealed that both are not part of the same Fz7 complex. This we confirmed by monitoring in vivo Fz7/C–cadherin complex formation in the presence or absence of PAPC. The YFP signal of Fz7-YN/C-cadherin–YC was reduced when PAPC was coexpressed (Fig. 7 D), whereas the signal was found increased in PAPC morphants (Fig. 7 E). We also could demonstrate the Wnt-11–dependent formation of the Fz7/C–cadherin complex in vivo using the BiFC assay. In Wnt-11 morphants, the YFP signal for the Fz7/C–cadherin complex was lost (Fig. 7 G). Instead, a clustering effect was observed when Wnt-11 was coexpressed (Fig. 7 F). Thus, our in vivo complex formation results (Fig. 7) coincide with our coimmunoprecipitation data (Fig. 6).
Fz7–cadherin complexes prevent cis-dimerization of C-cadherin
As we observed cell sorting by Fz7 or PAPC supported by Wnt-11 (Fig. 5) but no degradation or internalization of C-cadherin (Figs. 2, 3, and S2), we speculated that the hindrance of lateral clustering or cis-dimerization of C-cadherin might cause reduction in cell adhesion. Enforcement in cell adhesion by cis-dimerization and/or lateral clustering of cadherins has been shown by biochemical, biophysical, and theoretical approaches (Brieher et al., 1996; Yap et al., 1997; Zhang et al., 2009; Wu et al., 2010). A physiological role for dimerization in morphogenetic processes instead failed so far. As dimerization/lateral clustering is closely connected with the association of cadherins to the actin cytoskeleton network (Yap et al., 1997), we supposed that reduction of cis-dimerization or lateral clustering of C-cadherin upon PAPC or Fz7 expression should be seen by a decrease of endogenous C-cadherin or β-catenin membrane staining in fixed and Triton X-100–permeabilized DMZ explants. Under these conditions, the cytoskeletal-associated cadherin–catenin adhesion complexes are labeled, whereas the unbound cadherins are mainly removed by Triton X-100 treatment. Indeed, a reduced C-cadherin signal was observed upon PAPC expression (Fig. 8 A), which was also seen for β-catenin (Fig. 8 B). This is not in contradiction to our time-lapse imaging data shown in Fig. S2 because, in these experiments, C-cadherin–EGFP was overexpressed. To quantify the reduction in C-cadherin and β-catenin seen by immunostainings of fixed and permeabilized DMZ explant, we calculated the mean gray values. PAPC expression resulted in nearly threefold reduction of C-cadherin staining (Fig. 8 A, bottom) and a twofold decrease of the β-catenin signal (Fig. 8 B, bottom). Comparable effects were observed when the complex-forming BiFC constructs were expressed (Fig. 8, A and B). As Wnt-11 promotes membrane localization of PAPC by preventing its internalization (Figs. 1, 3, and 4), we also investigated the influence of Wnt-11. As shown in Fig. 8 C, the reduction of β-catenin signal in the presence of PAPC was further enforced by Wnt-11. In Wnt-11 morphants, β-catenin staining was partly restored.
Figure 8.

PAPC–Fz7 and C-cadherin–Fz7 interactions reduce cadherin–catenin complexes at cell membranes. DMZ explants were injected with the indicated constructs, fixed, and fluorescently immunostained using either specific xC-cadherin (6B6) or xβ-catenin antibody (PGDS 7D12). (A, top) Fluorescent immunostainings for endogenous xC-cadherin. Endogenous xC-cadherin membrane staining was reduced upon xPAPC-GFP expression and upon formation of the xPAPC–xFz7–TM1 complex (BiFC). (bottom) Quantitative evaluation of endogenous xC-cadherin staining. The mean grayscale value (intensity) of xC-cadherin staining was measured and plotted into box and whisker diagrams (box plots). For more details, see Materials and methods. (B and C) Fluorescent immunostainings for endogenous xβ-catenin. Endogenous xβ-catenin membrane staining was reduced upon xPAPC-GFP expression and upon formation of the xPAPC–xFz7–TM1 and xC-cadherin–xFz7 complexes. xPAPC-induced reduction of xβ-catenin could be restored by Wnt-11 depletion. The graphs show quantitative evaluation of endogenous xβ-catenin staining. The mean grayscale value (intensity) of xβ-catenin staining was measured and plotted into box and whisker diagrams (box plots). Injection amount was as follows: 1 ng xPAPC-GFP RNA, 1 ng xPAPC-YN RNA, 1 ng xC-cadherin–YC RNA, 500 pg xFz7-YN RNA, 500 pg xFz7-TM1-YC, 20 pg xWnt-11 RNA, and 1 pmol xWnt-11 MO. Bars, 20 µm. In the box plots, each box represents the values between the 25th (dark gray) and 75th quartiles (light gray). The line within the boxes indicates the median values, and the blue rhombi show the mean values. Error bars indicate SEM. Student’s t test was performed (*, p < 0.05 to wild type; **, p < 0.05 to xPAPC). # exp., number of experiments.
To monitor the cis-dimerization of C-cadherin in vivo, we expressed split YFP C-cadherin constructs (Fig. 9). In this BiFC assay, the YFP signal occurs when two cadherins with different YFP halves directly interact, which points to lateral cis-dimerization. When we traced the C-cadherin YFP signal, we called it dimerization, although these dimers can form larger clusters. In DMZ explants, C-cadherin cis-dimers were recognized in cell membranes by the YFP signal. Nearly no YFP membrane signal was detected when PAPC, Wnt-11, and Fz7 were coexpressed, whereas single injection of PAPC or Wnt-11/Fz7 coinjection did not prevent cis-dimerization (Fig. 9 A). As these molecules in the DMZ are endogenously present, we used the animal cap to confirm that PAPC, Fz7, and Wnt-11 counteract C-cadherin dimerization. In the animal cap, C-cadherin and low amounts of Fz7 are present, whereas PAPC and Wnt-11 are undetectable (Abu-Elmagd et al., 2006; Schambony and Wedlich, 2007). C-cadherin dimerization was displayed by the YFP signal in animal cap cells (Fig. 9 B). Again, only when PAPC, Fz7, and Wnt-11 were coexpressed, the YFP signal diminished. Thus, PAPC, Fz7, and Wnt-11 counteract C-cadherin cis-dimerization.
Figure 9.

Lateral cis-dimerization of C-cadherin is reduced by coexpression of Wnt-11, Fz7, and PAPC in animal caps and DMZ explants. (A and B) In vivo monitoring of lateral clustering of xC-cadherin by expression of the split YFP xC-cadherin constructs in DMZ explants (A) and in animal caps (B). Cell membranes were labeled in all samples with GAP43-mCherry. Whereas xWnt-11/xFz7 and xPAPC alone had no apparent effect, the combination of all three reduced lateral clustering of xC-cadherin. Injection amount was as follows: 1 ng xC-cadherin–YC/YN RNA, 500 pg xFz7 RNA, 500 pg xPAPC RNA, or 20 pg xWnt-11 RNA. Bars, 20 µm.
Discussion
Wnt-11/Fz7 triggers the formation of two distinct AMCs
Gastrulation, predominantly convergent extension movement of a tissue sheet, requires fine-tuned, spatially regulated cell adhesion. Here, we report that Wnt-11 reduces cell adhesion in Xenopus through the formation of two distinct AMCs. One complex includes Wnt-11, Fz7, and C-cadherin (AMCC), and, in the second complex (AMCP), Wnt-11 and Fz7 associate with PAPC that is stabilized in the cell membrane via this mechanism. PAPC has been shown to reduce C-cadherin–mediated adhesion (Chen and Gumbiner, 2006) and to bind the extracellular domain of Fz7 (Medina et al., 2004). With our novel findings, we link both processes. We show complex formation of PAPC and Fz7 in vivo during convergent extension and demonstrate that Wnt-11 and Fz7 are essential to maintain PAPC at the membrane. Together, they cooperate in cell sorting by reducing cell adhesion. Thus, PAPC-mediated reduction of C-cadherin adhesion requires Wnt-11 and Fz7 because, without both, PAPC is internalized. In agreement with Chen and Gumbiner (2006) and Chen et al. (2009), we do not observe an internalization of C-cadherin dependent on Wnt-11, Fz7, or PAPC (Figs. 2 and S2) in vivo. Instead, we explain the reduced adhesion by a loss of lateral clustering in C-cadherin–β-catenin adhesion complexes. We could not detect any changes in C-cadherin membrane expression by surface biotinylation assays or biotin pulse-chase experiments (Fig. 3). However, in immunostainings of fixed and permeabilized DMZ explants detecting only cadherins associated to actin cytoskeleton, we observed a reduction in endogenous C-cadherin and β-catenin (Fig. 8). In addition, we detected no YFP signal in our BiFC experiments with the C-cadherin split YFP constructs in the presence of Wnt-11, PAPC, and Fz7 (Fig. 9). Thus, reduction of C-cadherin and β-catenin seen in immunostainings might be a result of the lack of C-cadherin–lateral cis-dimerization and clustering.
We also observed a cell sorting effect of Fz7 independently of PAPC (Fig. 5), already mentioned in Medina et al. (2000). This can be explained by complex formation of C-cadherin with Fz7, which is augmented by Wnt-11 (Figs. 6 and 7). Chen and Gumbiner (2006) excluded a separate influence of Fz7 on C-cadherin adhesion and cell sorting based on their Fz7 depletion data. As Fz7 expression in the animal cap is very low and Wnt-11 is absent, an expected increase in C-cadherin adhesion might be at the detection limit. Instead of the full-length Fz7, they expressed the cytoplasmic truncated Fz7 (dnFz7), which cannot interfere with C-cadherin adhesion because dnFz7 lacks the binding site for this cadherin (Fig. 7). We showed that, in the absence of PAPC, the cytoplasmic domain of Fz7 is required to induce cell sorting (Fig. S4).
PAPC and C-cadherin bind to different domains of Fz7; however, a trimeric complex cannot be immunoprecipitated (Fig. 6, D and E). Instead, Fz7 forms discrete complexes with PAPC (AMCP) or with C-cadherin (AMCC). BiFC studies confirm that C-cadherin binds to the C terminus of Fz7 in vivo and that this interaction prevents cis-dimerization of C-cadherin. Thus, Wnt-11/Fz7 is not only required to form bipolar cells (Habas et al., 2001, 2003) and to reorientate the microtubules (Rigo-Watermeier et al., 2011), but, simultaneously, Wnt-11 decreases C-cadherin adhesion.
Wnt-11/Fz7 does not reduce cell adhesion by C-cadherin internalization
Cadherin endocytosis is a common mechanism during cell migration to lower cell adhesion (Troyanovsky, 2009). In Drosophila melanogaster, inhibition of Drosophila E-cadherin recycling leads to failures in the rearrangement of pentagonal into hexagonal cells of the wing (Classen et al., 2005). In zebrafish, the migration of the prechordal plate progenitors depends on cellular cohesion, which is dynamically regulated by endocytotic turnover of E-cadherin (Ulrich et al., 2005). In both processes, noncanonical Wnt signaling or Wnt-11 induces Rab5-dependent cadherin internalization. Conflicting reports exist about internalization of C-cadherin during Xenopus gastrulation. Ogata et al. (2007) reported that activin/nodal induces the expression of FLRT3 and Rnd1. Both form a complex with Unc5B, which leads to Dynamin1-dependent internalization of C-cadherin and a decrease in cell adhesion (Karaulanov et al., 2009). However, in the gastrulating Xenopus embryo, regional changes in cytoplasmic versus membrane pools of C-cadherin could not be confirmed (Chen and Gumbiner, 2006; this study). Here, we show that instead of C-cadherin, its regulator, PAPC, undergoes a dramatic change in subcellular localization, which is triggered by Wnt-11. Depletion of Wnt-11 resulted in a clathrin- and Dynamin1-dependent internalization of PAPC. Vice versa, overexpression of Wnt-11 stabilized PAPC at the membrane, which decreases C-cadherin–mediated adhesion. Interestingly, in zebrafish, Wnt-11 locally functions at the plasma membrane by accumulating Fz7 and the atypical cadherin Flamingo. However, these accumulations promote local cell contact persistence (Witzel et al., 2006).
AMCs interfere with cadherin-lateral dimerization and clustering
A key finding of this study is that the two AMCs, AMCC and AMCP, reduce adhesion by a loss of C-cadherin dimers and of lateral C-cadherin–β-catenin adhesion complexes. We assume that Wnt-11 prevents lateral clustering of C-cadherin by two independent mechanisms: (1) Wnt-11/Fz7 stabilizes PAPC at the membrane, most likely by triggering its oligomerization (Chen et al., 2007) and displacing C-cadherin–β-catenin adhesion complexes; and (2) Wnt-11 supports C-cadherin binding to Fz7 and thereby directly competes with the formation of C-cadherin dimers.
Both AMCs might act tissue specifically because PAPC is only expressed in the dorsal mesendoderm, whereas Fz7 and C-cadherin were found in all germ layers on the dorsal side (Medina et al., 2004). Fz7 and PAPC functions are required for the formation of Brachet’s cleft, the border between the ectoderm and the involuting mesendoderm (Wacker et al., 2000). In dorsal mesoderm, C-cadherin–mediated adhesion is reduced because the separation behavior against the ectoderm could be inhibited by overexpression of C-cadherin (Wacker et al., 2000). The reduction of cadherin function in the mesendoderm could therefore be achieved by the AMCP, which thereby could contribute to the tissue separation process.
PAPC sorting activity requires the extracellular and the transmembrane domain (Chen et al., 2007); therefore, the membrane-anchored extracellular domain of PAPC is able to disrupt PAPC–Fz7–TM1 interaction (BiFC; unpublished data). PAPC also forms homo- and eventually heterooligomers via disulfide bond formation. The latter seems to be required for lowering C-cadherin adhesion, as DTT treatment of blastomeres abolished the PAPC effect (Chen et al., 2007).
Wnt-11 also supports the formation of an Fz7/C–cadherin complex. This directly interferes with cis-dimerization of cadherins. The latter is discussed to be essential for strengthening adhesion forces. In a cell free system, Brieher et al. (1996) measured a strong increase in particle size when beads were covered with C-cadherin EC1-5 dimers instead of monomers. Using an elegant oligomerization assay, Yap et al. (1997) reported that lateral clustering of the extracellular domain of C-cadherin in living cells increased cell adhesion. They fused C-cadherin to FKBP12 and induced clustering by adding FK1012, a cell-permeant divalent that binds to the FKBP12 tag. Takeda et al. (1999) confirmed the formation of cadherin cis-dimers in cross-linking experiments. More recently, Harrison et al. (2011) identified a cis-interface formed by the concave side of EC1 and the convex site of the EC2 region conserved in all classical type I cadherins. Point mutations within the cis-interface resulted in extreme mobile and unstable E-cadherin junctions. They suggest that trans-interaction is the initial event in cadherin-mediated cell adhesion followed by cis-interactions required for stabilization (Wu et al., 2010). Here, we report for the first time that preventing cis-dimerization of cadherins is a physiological mechanism for local tuning of cell adhesion in mass cell movement; Wnt-11/Fz7 triggers this mechanism by capturing C-cadherin and PAPC into distinct AMCs during convergent extension. Future high-resolution microscopy studies are required to compare the dynamics in localization and stability of both AMCs.
Materials and methods
Plasmid, constructs, and in vitro mRNA transcription
Generating fluorescent xPAPC constructs, the stop codon of Fl-PAPC from pCS2+ Fl-xPAPC (Kim et al., 1998) was replaced by an NheI site via PCR site-directed mutagenesis, resulting in pCS2+ Fl-xPAPC/NheI. xPAPC-mCherry was generated by amplifying the coding sequence of mCherry using EB3-mCherry (a gift from V. Small, Institute of Molecular Biotechnology, Vienna, Austria) as a template. The PCR fragment was then subcloned into the NheI–XhoI site of Fl-xPAPC/NheI, resulting in pCS2+ xPAPC-mCherry. xPAPC-EGFP was generated by subcloning the EGFP coding sequence of pEGFP-N1 (Takara Bio Inc.) into the NheI–XhoI site of pCS2+ Fl-xPAPC/NheI, resulting in pCS2+ xPAPC-EGFP.
pCS2+ xWnt-11, pCS2+ xPAPC-GFP, and pCS2+ GAP43-GFP were gifts from E. DeRobertis (University of California Los Angeles, Los Angeles, CA). The coding sequence of mCherry was amplified by PCR and inserted into the SacI–XhoI site of pCS2+ GAP43-GFP to replace the GFP coding sequence (resulting in pCS2+ GAP43-mCherry). pCS2+ H2B-mCherry was a gift from R. Mayor (University College London, London, England, UK), pCS2+ xWnt-5a was a gift from R. Moon (University of Washington, Seattle, WA), pCS2+ dnDyn1 was a gift from A. Yap (University of Queensland, St. Lucia, Queensland, Australia), and pcDNA3.1(+) xC-cadherin–EGFP was a gift from K.W. Cho (University of California, Irvine, CA; Ogata et al., 2007).
pCS2+ xFz7 encodes the full-length xFz7 (Medina and Steinbeisser, 2000). pCS2+ ΔCxFz7 lacks the 26 C-terminal amino acids, pCS2+ NxFz7-fun comprises the N-terminal domain of xFz7 fused with a fun domain (Medina et al., 2000), and pCS2+ ΔNxFz7 encodes the seven transmembrane domains and the C tail of xFz7 (Winklbauer et al., 2001). The cytoplasmic domain of xFz7 was amplified via PCR from pCS2+ xFz7 and subcloned into the StuI–XbaI site of pCS2+ flag, resulting in CxFz7-flag.
For immunoprecipitation, PAPC was amplified from pCS2+ FlPAPC by PCR and ligated into the ClaI restriction site of pCS2+ 3xHA (a gift from R. Swain, Birmingham University Medical School, Birmingham, England, UK), resulting in pCS2+ xPAPC 3xHA. pCS2+ xFz7-myc consists of the full-length xFz7 fused to the myc tag (Medina and Steinbeisser, 2000).
To generate the constructs for BiFC, the N- or C-terminal part of YFP together with a linker region was amplified from pBIFC-bFosYC155 and pBIFC-bJunYN155 (a gift from T.K. Kerppola, University of Michigan Medical School, Ann Arbor, MI). The PCR fragments were ligated into the EcoRI–XhoI restriction site of pCS2+ vector, generating pCS2+ YN and pCS2+ YC. pCS2+ xPAPC-myc and pCS2+ xFz7-TM1-myc were generated by amplifying the coding sequence of pCS2+ PAPC or amino acids 1–262 of pCS2+ xFz7 and cloning them into the ClaI restriction site of pCS2+ myc tag (provided by R. Rupp [Ludwig-Maximilians-University Munich, Munich, Germany] and D. Turner [University of Michigan Medical School, Ann Arbor, MI]). Then, the coding sequences of pCS2+ xC-cadherin–myc (a gift from B. Gumbiner, University of Virginia, Charlottesville, VA), pCS2+ xPAPC-myc, pCS2+ xFz7-myc (Medina and Steinbeisser, 2000), and pCS2+ xFz7-TM1-myc were amplified by PCR, introducing AsuII restriction sites on both ends of the fragments. The PCR fragments were subcloned into the AsuII restriction site of pCS2+ YN and YC, resulting in pCS2+ xPAPC-myc-YN, pCS2+ xPAPC-myc-YC, xFz7-myc-YN, pCS2+ xFz7-TM1-myc-YC, pCS2+ xC-cadherin–myc-YN, and pCS2+ xC-cadherin–myc-YC. All constructs were tested in cell culture for functionality.
Capped mRNAs were synthesized from linearized plasmids using the mMESSAGE mMACHINE kit (Ambion). All used antisense morpholino oligonucleotides (MOs) were ordered by Gene Tools, LLC and were previously described: xWnt-11 MO (Pandur et al., 2002), xWnt-5a MO (Schambony and Wedlich, 2007), xFz7 MO (Winklbauer et al., 2001), and xPAPC MO1 and xPAPC MO2 (Unterseher et al., 2004).
Xenopus embryo manipulations
Xenopus eggs were obtained from females injected with 300 IU of human chorionic gonadotropin (Sigma-Aldrich) and fertilized in vitro. Then, they were dejellied with 2% cysteine hydrochloride, pH 8.2, and embryos were microinjected in 1× MBSH (88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 0.82 mM MgSO4, 0.41 mM CaCl2, 0.33 mM Ca(NO3)2, and 10 mM Hepes, pH 7.4). For Keller open-face (DMZ) explants, the two medial dorsal cells of 8- or 16-cell–stage embryos were equatorially injected, whereas for animal cap explants, the embryos were injected at the animal pole at the 8-cell stage. The injection amount varied depending on the constructs, on the used antisense MOs, and on the experiments and is mentioned in the figure legends. The embryos were cultured in 0.1× MBSH at 14–18°C and staged according to Nieuwkoop and Faber (1967).
Animal cap explants for BiFC analysis and blastomere adhesion assays were excised from stage-8.5 embryos. DMZ explants (area of ∼0.1 mm × 0.1 mm above the dorsal blastopore lip) were cut out at stage 10.25. They were cultured in 1× MBSH containing 10 µg/ml streptomycin sulfate and 10 µg/ml penicillin on Petri dishes or chambered coverglasses (VWR International) secured below a separate coverslip using Baysilone paste (Carl Roth GmbH). Petri dishes, chambered cover glasses, and cover slips were precoated overnight at 4°C with BSA (1% in 1× MBSH).
Fluorescence immunostainings of DMZ explants
For immunostainings, DMZ explants were cultured on BSA-coated Petri dishes until stage 11. Afterward, explants were fixed in 4% PFA buffered in 1× APBS (2.7 mM KCl, 0.15 mM KH2PO4, 103 mM NaCl, and 0.7 mM NaHPO4, pH 7.5) for 30 min at room temperature and washed with 100 mM NaCl and 100 mM Tris/HCl, pH 7.4, for 30 min at room temperature.
Excised DMZ explants were directly subjected to fluorescence immunohistochemistry, or they were embedded in cold water fish gelatin/sucrose and sliced longituditionally. For embedding, DMZ explants were first incubated in 15% cold water fish gelatin/15% sucrose overnight at room temperature and then transferred to 25% cold water fish gelatin/15% sucrose for equilibrating again overnight at room temperature. Subsequently, the explants were embedded in 20% cold water fish gelatin/15% sucrose and frozen in dry ice for at least 1 h. Frozen sections of 10-µm thickness were prepared using a cryostat (CM1900; Leica). The frozen sections were dried at room temperature for 1 h, incubated for 10 min in aceton, dried again at room temperature for 1 h, rehydrated in 1× APBS, and incubated with blocking buffer (20% horse serum in 1× APBS) for at least 30 min at room temperature. Incubation with the primary antibody α-clathrinHC (3.3 µg/ml; Abcam) in blocking buffer was performed overnight at 4°C. After washing three times for 10 min each in 1× APBS, the secondary antibody α–rabbit IgM-Cy3 (5 µg/ml; Dianova) in blocking buffer was applied for 1 h at room temperature. The sections were counterstained with DAPI to visualize the nuclei, washed three times for 10 min each in 1× APBS, and finally embedded with Mowiol-DABCO (Carl Roth GmbH).
Whole DMZ explants were permeabilized by incubation in 20% DMSO/80% methanol overnight at −20°C. Then, they were rehydrated with descending methanol concentrations, washed extensively with 1× APBS-T (0.5% Triton X-100), and incubated with blocking buffer (20% horse serum in 1× APBS-T) for 2 h at room temperature. Incubation with the primary antibody mouse α–xC-cadherin (6B6, undiluted supernatant; Developmental Studies Hybridoma Bank) and rat α–xβ-catenin (PGDS 7D12, undiluted supernatant; provided by R. Rupp) was performed overnight at 4°C. After washing extensively with 1× APBS and reincubation with blocking buffer (20% horse serum in 1× APBS) for 1 h at room temperature, the secondary antibody α–mouse IgM-Cy3 or α–rat IgM-Cy3 (5 µg/ml; Dianova) in blocking buffer was applied overnight at room temperature. The explants were counterstained with DAPI to visualize the nuclei, washed extensively in 1× APBS, and finally embedded with Mowiol-DABCO.
Cell dispersal assay
Embryos were injected into one blastomere at the 32-cell stage with mRNA coding for GAP43-GFP alone or in combination with xPAPC, xFz7, or xWnt-11, as indicated in the figure legends. At stage 12.5, the GAP43-GFP–expressing blastomeres were observed and recorded under a fluorescent microscope.
Blastomere adhesion assay
1 µg/ml CEC1-5_Fc (provided by C. Niessen, University of Cologne, Cologne, Germany) diluted in 1× MFM (88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, 7.5 mM Tris/HCl, and 1 mM CaCl2, pH 7.6) was spotted in a 6-cm Petri dish for 2 h in a humidified chamber at room temperature, and the coated Petri dish was blocked overnight with 0.5% BSA in 1× MFM at 4°C. Right before assay, the medium was exchanged to 1× MFM.
10 animal caps encoding H2B-mCherry and GAP43-GFP as a lineage tracer were excised in 1× CMFM (88 mM NaCl, 1 mM KCl, 2.4 mM NaHCO3, and 7.5 mM Tris/HCl, pH 7.6) at stage 8.5. Afterward, the epidermis was removed from the explants, and they were dissociated in 1× CMFM under rotation. A 15-µl cell suspension was pipetted to the CEC1-5-Fc spots and incubated for 30 min. Afterward, the Petri dish was flipped over in a chamber filled with 1× MFM for 10 min. Fluorescent pictures were taken before and after assay. The cells before and the remaining cells after assay were counted.
BiFC assay
For BiFC, injected animal caps and DMZ explants were dissected and cultured at 14°C (culture condition allowing reconstitution of YFP fluorescence) in 1× MBSH containing 10 µg/ml streptomycin sulfate and 10 µg/ml penicillin on BSA-coated chambered coverglasses until the animal cap explants reached stage 8.5 and the DMZ explants reached stage 11 and were recorded by confocal time-lapse microscopy.
Immunoprecipitation assay
60 DMZ explants were excised and lysed in NOP buffer (150 M NaCl, 10 mM Tris/HCl, pH 7.8, 1 mM MgCl2, 0.75 mM CaCl2, and 2% NP-40) supplemented with protease inhibitor (cOmplete; Roche). xFz7 was pulled down using mouse α-myc antibody (3 µg of purified 9E10; Developmental Studies Hybridoma Bank) and subjected to Western blotting.
Cell surface biotinylation assay and biotin pulse-chase experiments
For cell surface biotinylation assay and biotin pulse-chase experiments, 20 DMZ explants per construct and time point were excised and incubated in 1× MMR (88 mM NaCl, 20 mM Hepes, 10 mM KCl, 2.4 mM NaHCO3, 0.8 mM MgSO4, 0.4 mM CaCl2, and 0.33 mM CaNO3, pH 7.4) for 20 min. To specifically inhibit the different endocytosis pathways, DMZ explants were incubated with appropriate inhibitors (60 µM chlorpromazine, 300 µM monodansylcadaverine, 5 µg/ml filipin, and 200 µM genistein) in 1× MMR for 45 min at room temperature. For biotin labeling of the cell surface proteins, explants were incubated for 30 min with 1 mg/ml Sulfo-NHS-SS-Biotin (Thermo Fisher Scientific) in 1× MMR at 4°C. Unbound biotin was removed by washing with ice-cold 10 mM glycin/1× MMR. For cell surface biotinylation assay, the explants were lysed immediately in NOP buffer supplemented with protease inhibitor (cOmplete). For biotin pulse-chase experiments, the explants were further washed three times with ice-cold 1× APBS complemented with 0.5 mM CaCl2 and 0.33 mM MgCl2. Then, medium was exchanged to 1× MMR at room temperature to induce endocytosis. The endocytosis was stopped at distinct time points (0, 3, 7, 15, 30, 45, and 60 min) by lysis in NOP buffer supplemented with protease inhibitor (cOmplete). As negative control, 20 embryos (stage 11) were lysed in 1 mg/ml Sulfo-NHS-SS-Biotin/NOP buffer, and biotin labeling was immediately stopped by adding 10 mM glycin/NOP buffer. The surface protein fraction was obtained by precipitation of biotin-labeled protein with NeutrAvidin agarose beads (Thermo Fisher Scientific). Total, surface, and the nonbiotinylated cytoplasmic protein fractions were then subjected to Western blotting.
Western blotting and PonceauS staining
For Western blotting, proteins were transferred to polyvinylidene difluoride or nitrocellulose membrane and incubated with the corresponding primary antibodies: goat α-myc (Abcam), rabbit α-GFP (Invitrogen), rabbit α–xC-cadherin (provided by C. Niessen), mouse α-HA (Abcam), chicken α-GFP (Abcam), mouse α-tubulin (DM1A; Abcam), or mouse α-myc (9E10; Developmental Studies Hybridoma Bank) followed by incubation with corresponding alkaline phosphatase–labeled goat α–rabbit, goat α–mouse, or donkey α–goat IgG (all purchased from Dianova). Finally, membranes were developed using NBT/BCIP (Roche).
For PonceauS staining, membranes were incubated for 5 min in PonceauS staining solution (0.5% PonceauS in 1% acetic acid) and washed three times in distilled water under shaking before incubation with the corresponding primary antibodies.
Microscope image acquisition
For fluorescence time-lapse microscopy, DMZ explants were cultured in 1× MBSH containing 10 µg/ml streptomycin sulfate and 10 µg/ml penicillin at room temperature. Images were captured every 30 s from stage 10.5 onward for 30 min with an inverted computer-controlled microscope (DMIRE2; Leica) using the automator function of Openlab software (version 4.0; PerkinElmer) and a digital camera (model C4742-95; Hamamatsu Photonics). The DMIRE2 microscope was equipped with an objective lens (Plan-Apochromat 63×/1.32 Ph3 oil) and the filter cubes L5 (Alexa Fluor 488, FITC) and Y3 (Alexa Fluor 546, Cy3; both from Leica). Images were processed with Openlab software (version 4.0) and Photoshop (CS3 extended version 10.0.1; Adobe) using γ adjustments.
Analyses of fluorescence immunostainings were performed on an inverted laser confocal microscope (LSM 5; Carl Zeiss) equipped with an objective lens (LCI Plan-Neofluar 63×/1.3 immersion corrosion differential interference contrast; Carl Zeiss) and appropriate filter combinations. Optical sections (distance of 0.5 µm) were acquired at room temperature with a digital camera (AxioCam HR) and the LSM Image Browser (both from Carl Zeiss) followed by maximum intensity projection of the z stack. Images were processed with the LSM Image Browser (release 4.2) and Photoshop (CS3 extended version 10.0.1).
To analyze blastomere adhesion assay, optical sections (distance of 0.6 µm) were taken before and after assay at room temperature with a spinning-disk inverse microscope (Z1 Cell Observer; Carl Zeiss) equipped with the objective lens Plan-Apochromat 10×/0.45 differential interference contrast II and the appropriate emission filters using AxioVision software (release 4.8.2; Carl Zeiss). For analysis of BiFC experiments, optical sections (distance of 0.5 µm) were captured every 20 s for 10 min at room temperature with a spinning-disk inverse microscope (Z1 Cell Observer) equipped with the objective lens C Apochromat 63×/1.2 W differential interference contrast III and the appropriate emission filters using AxioVision software (release 4.8.2). The images were processed by extended focus projection and smoothing using AxioVision software (release 4.8.2) and Photoshop (CS3 extended version 10.0.1).
Statistics
To evaluate Western blotting, cell surface biotinylation assay, and biotin pulse-chase assay, the developed membranes were scanned with a resolution of 300 dpi in gray scale. With Photoshop (CS3 extended version 10.01), the mean gray values and pixel values of each protein band were determined calculating the signal intensity. Signal intensity was normalized against GAP43-GFP signal intensity or according to the total protein level. For relative quantification, the alteration of respective band intensity was determined by dividing the appropriate band intensity through the normalized intensity.
For biotin pulse-chase assay, comparison of band intensity was performed by ImageJ software (National Institutes of Health) using the gel analysis method (densitometry measurements). Band intensities of surface fraction were normalized against the corresponding total protein bands. Relative surface fraction was calculated by diving each normalized value through the normalized value of time point 0.
For cell dispersal assay, the intensity (mean gray value) and the size of GFP-expressing clusters (pixel) were determined by ImageJ software. To determine the cell sorting capacity, the relative ratio intensity was divided through the measured area. To determine the cell number before and after the blastomere adhesion assay, the cells were counted with the aid of ImageJ software and normalized to the controls.
Online supplemental material
Fig. S1 shows that xWnt-11 specifically influences the subcellular localization of xPAPC. Fig. S2 shows the influence of xPAPC on subcellular localization of xC-cadherin–EGFP. Fig. S3 shows the used criteria for evaluation of the cell dispersal assay. Fig. S4 shows the cell sorting capacity of different xFz7 deletion mutants. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.201110076/DC1.
Supplementary Material
Acknowledgments
We would like to thank K. Linsmeier for technical assistance and C. Niessen for the generous support by implementation of the blastomere adhesion assay as well as for providing xC-cadherin CEC1-5_Fc protein and the antiserum against endogenous xC-cadherin.
This work has been supported by German Research Foundation research grants given to H. Steinbeisser (STE 14/2) and D. Wedlich (1208/10-1).
Footnotes
Abbreviations used in this paper:
- AMC
- adhesion-modulating complex
- BiFC
- bimolecular fluorescence complementation
- DMZ
- dorsal marginal zone
- MO
- morpholino oligonucleotide
- PAPC
- paraxial protocadherin
- PCP
- planar cell polarity
References
- Abu-Elmagd M., Garcia-Morales C., Wheeler G.N. 2006. Frizzled7 mediates canonical Wnt signaling in neural crest induction. Dev. Biol. 298:285–298 10.1016/j.ydbio.2006.06.037 [DOI] [PubMed] [Google Scholar]
- Brieher W.M., Yap A.S., Gumbiner B.M. 1996. Lateral dimerization is required for the homophilic binding activity of C-cadherin. J. Cell Biol. 135:487–496 10.1083/jcb.135.2.487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Gumbiner B.M. 2006. Paraxial protocadherin mediates cell sorting and tissue morphogenesis by regulating C-cadherin adhesion activity. J. Cell Biol. 174:301–313 10.1083/jcb.200602062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Molino C., Liu L., Gumbiner B.M. 2007. Structural elements necessary for oligomerization, trafficking, and cell sorting function of paraxial protocadherin. J. Biol. Chem. 282:32128–32137 10.1074/jbc.M705337200 [DOI] [PubMed] [Google Scholar]
- Chen X., Koh E., Yoder M., Gumbiner B.M. 2009. A protocadherin-cadherin-FLRT3 complex controls cell adhesion and morphogenesis. PLoS ONE. 4:e8411 10.1371/journal.pone.0008411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H.A., Yamamoto T.S., Ueno N. 2007. ANR5, an FGF target gene product, regulates gastrulation in Xenopus. Curr. Biol. 17:932–939 10.1016/j.cub.2007.04.034 [DOI] [PubMed] [Google Scholar]
- Classen A.K., Anderson K.I., Marois E., Eaton S. 2005. Hexagonal packing of Drosophila wing epithelial cells by the planar cell polarity pathway. Dev. Cell. 9:805–817 10.1016/j.devcel.2005.10.016 [DOI] [PubMed] [Google Scholar]
- Damke H., Baba T., Warnock D.E., Schmid S.L. 1994. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol. 127:915–934 10.1083/jcb.127.4.915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich L.V., Strutt D. 2011. Principles of planar polarity in animal development. Development. 138:1877–1892 10.1242/dev.054080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habas R., Kato Y., He X. 2001. Wnt/Frizzled activation of Rho regulates vertebrate gastrulation and requires a novel Formin homology protein Daam1. Cell. 107:843–854 10.1016/S0092-8674(01)00614-6 [DOI] [PubMed] [Google Scholar]
- Habas R., Dawid I.B., He X. 2003. Coactivation of Rac and Rho by Wnt/Frizzled signaling is required for vertebrate gastrulation. Genes Dev. 17:295–309 10.1101/gad.1022203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerschmidt M., Wedlich D. 2008. Regulated adhesion as a driving force of gastrulation movements. Development. 135:3625–3641 10.1242/dev.015701 [DOI] [PubMed] [Google Scholar]
- Harrison O.J., Jin X., Hong S., Bahna F., Ahlsen G., Brasch J., Wu Y., Vendome J., Felsovalyi K., Hampton C.M., et al. 2011. The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure. 19:244–256 10.1016/j.str.2010.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskovits J.S., Burgess C.C., Obar R.A., Vallee R.B. 1993. Effects of mutant rat dynamin on endocytosis. J. Cell Biol. 122:565–578 10.1083/jcb.122.3.565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaulanov E., Böttcher R.T., Stannek P., Wu W., Rau M., Ogata S., Cho K.W., Niehrs C. 2009. Unc5B interacts with FLRT3 and Rnd1 to modulate cell adhesion in Xenopus embryos. PLoS ONE. 4:e5742 10.1371/journal.pone.0005742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola T.K. 2008. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys. 37:465–487 10.1146/annurev.biophys.37.032807.125842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.H., Yamamoto A., Bouwmeester T., Agius E., Robertis E.M. 1998. The role of paraxial protocadherin in selective adhesion and cell movements of the mesoderm during Xenopus gastrulation. Development. 125:4681–4690 [DOI] [PubMed] [Google Scholar]
- Lee C.H., Gumbiner B.M. 1995. Disruption of gastrulation movements in Xenopus by a dominant-negative mutant for C-cadherin. Dev. Biol. 171:363–373 10.1006/dbio.1995.1288 [DOI] [PubMed] [Google Scholar]
- Medina A., Steinbeisser H. 2000. Interaction of Frizzled 7 and Dishevelled in Xenopus. Dev. Dyn. 218:671–680 [DOI] [PubMed] [Google Scholar]
- Medina A., Reintsch W., Steinbeisser H. 2000. Xenopus frizzled 7 can act in canonical and non-canonical Wnt signaling pathways: implications on early patterning and morphogenesis. Mech. Dev. 92:227–237 10.1016/S0925-4773(00)00240-9 [DOI] [PubMed] [Google Scholar]
- Medina A., Swain R.K., Kuerner K.M., Steinbeisser H. 2004. Xenopus paraxial protocadherin has signaling functions and is involved in tissue separation. EMBO J. 23:3249–3258 10.1038/sj.emboj.7600329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mettlen M., Pucadyil T., Ramachandran R., Schmid S.L. 2009. Dissecting dynamin’s role in clathrin-mediated endocytosis. Biochem. Soc. Trans. 37:1022–1026 10.1042/BST0371022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller H.A., Kühl M., Finnemann S., Schneider S., van der Poel S.Z., Hausen P., Wedlich D. 1994. Xenopus cadherins: The maternal pool comprises distinguishable members of the family. Mech. Dev. 47:213–223 10.1016/0925-4773(94)90040-X [DOI] [PubMed] [Google Scholar]
- Niessen C.M., Gumbiner B.M. 2002. Cadherin-mediated cell sorting not determined by binding or adhesion specificity. J. Cell Biol. 156:389–399 10.1083/jcb.200108040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niessen C.M., Leckband D., Yap A.S. 2011. Tissue organization by cadherin adhesion molecules: Dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol. Rev. 91:691–731 10.1152/physrev.00004.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieuwkoop P.D., Faber J. 1967. Normal table of Xenopus laevis (Daudin). A systematical and chronological survey of the development from the fertilized egg till the end of metamorphosis. North Holland Publishing Co., Amsterdam. 260 pp. [Google Scholar]
- Ogata S., Morokuma J., Hayata T., Kolle G., Niehrs C., Ueno N., Cho K.W. 2007. TGF-beta signaling-mediated morphogenesis: Modulation of cell adhesion via cadherin endocytosis. Genes Dev. 21:1817–1831 10.1101/gad.1541807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandur P., Läsche M., Eisenberg L.M., Kühl M. 2002. Wnt-11 activation of a non-canonical Wnt signalling pathway is required for cardiogenesis. Nature. 418:636–641 10.1038/nature00921 [DOI] [PubMed] [Google Scholar]
- Rigo-Watermeier T., Kraft B., Ritthaler M., Wallkamm V., Holstein T., Wedlich D. 2011. Functional conservation of Nematostella Wnts in canonical and noncanonical Wnt-signaling. Biology Open. 1:43–51 10.1242/bio.2011021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schambony A., Wedlich D. 2007. Wnt-5A/Ror2 regulate expression of XPAPC through an alternative noncanonical signaling pathway. Dev. Cell. 12:779–792 10.1016/j.devcel.2007.02.016 [DOI] [PubMed] [Google Scholar]
- Takeda H., Shimoyama Y., Nagafuchi A., Hirohashi S. 1999. E-cadherin functions as a cis-dimer at the cell-cell adhesive interface in vivo. Nat. Struct. Biol. 6:310–312 10.1038/7542 [DOI] [PubMed] [Google Scholar]
- Troyanovsky S.M. 2009. Regulation of cadherin-based epithelial cell adhesion by endocytosis. Front Biosci (Schol Ed). 1:61–67 [DOI] [PubMed] [Google Scholar]
- Ulrich F., Krieg M., Schötz E.M., Link V., Castanon I., Schnabel V., Taubenberger A., Mueller D., Puech P.H., Heisenberg C.P. 2005. Wnt11 functions in gastrulation by controlling cell cohesion through Rab5c and E-cadherin. Dev. Cell. 9:555–564 10.1016/j.devcel.2005.08.011 [DOI] [PubMed] [Google Scholar]
- Unterseher F., Hefele J.A., Giehl K., De Robertis E.M., Wedlich D., Schambony A. 2004. Paraxial protocadherin coordinates cell polarity during convergent extension via Rho A and JNK. EMBO J. 23:3259–3269 10.1038/sj.emboj.7600332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker S., Grimm K., Joos T., Winklbauer R. 2000. Development and control of tissue separation at gastrulation in Xenopus. Dev. Biol. 224:428–439 10.1006/dbio.2000.9794 [DOI] [PubMed] [Google Scholar]
- Wang Y., Nathans J. 2007. Tissue/planar cell polarity in vertebrates: New insights and new questions. Development. 134:647–658 10.1242/dev.02772 [DOI] [PubMed] [Google Scholar]
- Wang Y., Janicki P., Köster I., Berger C.D., Wenzl C., Grosshans J., Steinbeisser H. 2008. Xenopus Paraxial Protocadherin regulates morphogenesis by antagonizing Sprouty. Genes Dev. 22:878–883 10.1101/gad.452908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winklbauer R., Medina A., Swain R.K., Steinbeisser H. 2001. Frizzled-7 signalling controls tissue separation during Xenopus gastrulation. Nature. 413:856–860 10.1038/35101621 [DOI] [PubMed] [Google Scholar]
- Witzel S., Zimyanin V., Carreira-Barbosa F., Tada M., Heisenberg C.P. 2006. Wnt11 controls cell contact persistence by local accumulation of Frizzled 7 at the plasma membrane. J. Cell Biol. 175:791–802 10.1083/jcb.200606017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Jin X., Harrison O., Shapiro L., Honig B.H., Ben-Shaul A. 2010. Cooperativity between trans and cis interactions in cadherin-mediated junction formation. Proc. Natl. Acad. Sci. USA. 107:17592–17597 10.1073/pnas.1011247107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K., Andreasson K.I., Sugiura H., Maru E., Dominique M., Irie Y., Miki N., Hayashi Y., Yoshioka M., Kaneko K., et al. 1999. Arcadlin is a neural activity-regulated cadherin involved in long term potentiation. J. Biol. Chem. 274:19473–1979 10.1074/jbc.274.27.19473 [DOI] [PubMed] [Google Scholar]
- Yamamoto A., Amacher S.L., Kim S.H., Geissert D., Kimmel C.B., De Robertis E.M. 1998. Zebrafish paraxial protocadherin is a downstream target of spadetail involved in morphogenesis of gastrula mesoderm. Development. 125:3389–3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap A.S., Brieher W.M., Pruschy M., Gumbiner B.M. 1997. Lateral clustering of the adhesive ectodomain: A fundamental determinant of cadherin function. Curr. Biol. 7:308–315 10.1016/S0960-9822(06)00154-0 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Sivasankar S., Nelson W.J., Chu S. 2009. Resolving cadherin interactions and binding cooperativity at the single-molecule level. Proc. Natl. Acad. Sci. USA. 106:109–114 10.1073/pnas.0811350106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.