

Highwire, a conserved axonal E3 ubiquitin ligase, regulates the initiation of axonal degeneration after injury in Drosophila by regulating the levels of the NAD+ biosynthetic enzyme, Nmnat, and the Wnd kinase.
Abstract
Axonal degeneration is a hallmark of many neuropathies, neurodegenerative diseases, and injuries. Here, using a Drosophila injury model, we have identified a highly conserved E3 ubiquitin ligase, Highwire (Hiw), as an important regulator of axonal and synaptic degeneration. Mutations in hiw strongly inhibit Wallerian degeneration in multiple neuron types and developmental stages. This new phenotype is mediated by a new downstream target of Hiw: the NAD+ biosynthetic enzyme nicotinamide mononucleotide adenyltransferase (Nmnat), which acts in parallel to a previously known target of Hiw, the Wallenda dileucine zipper kinase (Wnd/DLK) MAPKKK. Hiw promotes a rapid disappearance of Nmnat protein in the distal stump after injury. An increased level of Nmnat protein in hiw mutants is both required and sufficient to inhibit degeneration. Ectopically expressed mouse Nmnat2 is also subject to regulation by Hiw in distal axons and synapses. These findings implicate an important role for endogenous Nmnat and its regulation, via a conserved mechanism, in the initiation of axonal degeneration. Through independent regulation of Wnd/DLK, whose function is required for proximal axons to regenerate, Hiw plays a central role in coordinating both regenerative and degenerative responses to axonal injury.
Author Summary
Axons degenerate after injury and during neurodegenerative diseases, but we are still searching for the cellular mechanism responsible for this degeneration. Here, using a nerve crush injury assay in the fruit fly Drosophila, we have identified a role for a conserved molecule named Highwire (Hiw) in the initiation of axonal degeneration. Hiw is an E3 ubiquitin ligase thought to regulate the levels of specific downstream proteins by targeting their destruction. We show that Hiw promotes axonal degeneration by regulating two independent downstream targets: the Wallenda (Wnd) kinase, and the NAD+ biosynthetic enzyme nicotinamide mononucleotide adenyltransferase (Nmnat). Interestingly, Nmnat has previously been implicated in a protective role in neurons. Our findings indicate that Nmnat protein is down-regulated in axons by Hiw and that this regulation plays a critical role in the degeneration of axons and synapses. The other target, the Wnd kinase, was previously known for its role in promoting new axonal growth after injury. We propose that Hiw coordinates multiple responses to regenerate damaged neuronal circuits after injury: degeneration of the distal axon via Nmnat, and new growth of the proximal axon via Wnd.
Introduction
Axon degeneration can be induced by a variety of insults, including injury. When an axon is transected from the cell body, the distal axon “stump” degenerates through a regulated self-destruction process called Wallerian degeneration [1]. This process appears to be actively regulated in axons; however, the endogenous cellular machinery that regulates and executes this degeneration process is poorly understood.
Previous studies have implicated a role for the ubiquitin proteasome system (UPS) in Wallerian degeneration, since inhibition of UPS leads to a delay in the early stages of degeneration [2],[3]. One explanation for this result is that the UPS mediates bulk protein degradation via the combined action of many ubiquitin ligases. However an alternative model is that one or several specific E3 ligases target the destruction of key inhibitors of the degeneration process. Here, using an in vivo assay for Wallerian degeneration in Drosophila, we identify an essential role for a specific E3 ubiquitin ligase in promoting Wallerian degeneration.
The ligase, known as Highwire (Hiw) in Drosophila, Phr1 in mice, is well known from studies in multiple model organisms for its conserved functions in regulating axonal and synaptic morphology during development [4]–[12]. We found that mutations in hiw strongly inhibit the initiation of Wallerian degeneration in multiple neuronal types and developmental stages. Until recently [13],[14], such a strong loss-of-function phenotype has not been reported for this process.
Mutations in hiw also inhibit synaptic retraction caused by cytoskeletal mutations [15]. However the finding that Hiw promotes axonal degeneration was originally perplexing, since a known target of Hiw, the Wallenda (Wnd) MAP kinase kinase kinase (also known as dileucine zipper kinase [DLK]) [16],[17], was found to promote Wallerian degeneration in mouse DRG and Drosophila olfactory neurons [18]. In hiw mutants Wnd levels are increased [9],[16],[17], however degeneration is inhibited. A partial explanation for these opposing results is that Wnd plays a protective role in some neuronal types [19],[20]. However this alone could not account for the essential role of Hiw in Wallerian degeneration of all neuron types. These findings pointed to the existence of additional targets for Hiw.
Recent studies in vertebrate cultured neurons have suggested the NAD+ synthase enzyme nicotinamide mononucleotide adenyltransferase 2 (Nmnat2) as an attractive target of post-translational regulation in axons [21]. Nmnat2 is transported in axons, where it has a short protein half-life, and neurons depleted for Nmnat2 undergo axonal degeneration [21]. Moreover, many gain-of-function studies suggest that increasing the activity of an Nmnat enzyme in axons can effectively delay Wallerian degeneration [22],[23]. The most classic example of this comes from studies of the Wallerian degeneration Slow (WldS) gain-of-function mutation in the Nmnat1 locus, which causes a greater than 10-fold delay in the degeneration of injured axons [24]. However, despite the plethora of studies examining the effect of overexpressing Nmnat enzymes [23], very little is known about the role of the endogenous Nmnat enzymes in axons and how their activity may be regulated.
In contrast to the three isoforms in vertebrates, the Drosophila genome contains a single nmnat gene, for which two splice forms are annotated. nmnat is an essential gene, whose depletion in neurons causes neurodegeneration [25]–[27]. Here we find that Hiw and ubiquitination negatively regulate the levels of axonal Nmnat in vivo. Moreover endogenous Nmnat is required, in parallel to Wnd, for mutations in hiw to inhibit degeneration. By down-regulating the levels of Nmnat protein, Hiw promotes the initiation of Wallerian degeneration in axons and synapses. Moreover, through co-regulation of the Wnd/DLK kinase, whose function is required for proximal axons to initiate new axonal growth [28]–[32], Hiw coordinates both regenerative and degenerative responses to axonal injury.
Results
Highwire Plays an Essential Role in Wallerian Degeneration
We used a previously described nerve crush assay [20],[30] to study the degeneration of motoneuron and sensory neuron axons within segmental nerves in third instar Drosophila larvae. To quantify the degeneration of motoneuron axons, we used the m12-Gal4 driver to label only a subset of motoneurons with UAS-mCD8-GFP (Figure 1A, 1B, and Materials and Methods). In wild-type (WT) animals, these axons are completely fragmented within 24 h after injury (Figure 1A) [20].
Figure 1. Mutations in hiw strongly delay Wallerian degeneration of motoneuron axons and synapses after injury.

(A) In Drosophila third instar larvae, single axons are labeled by expression of UAS-mCD8::GFP with the m12-Gal4 driver. In a wild-type (WT) background, axons distal to the injury site have completely degenerated within 24 h after nerve crush injury, however axons remain intact in the hiw ΔN mutant background. (B) Quantification of axon degeneration index in different genotypes. (See Materials and Methods and [20] for quantification methods). The degeneration index (percent degenerated) is shown with white bars, while black bars show the complementary percentage (percent intact). The genotypes are: (UAS-mCD8::GFP/+; m12-Gal4/+), (hiw ΔN ;UAS-mCD8::GFP/+; m12-Gal4/+), (UAS-mCD8::GFP/+; m12-Gal4/UAS-hiw-ΔRING), (UAS-mCD8::GFP/UAS-UBP2; m12-Gal4/+), (UAS-mCD8::GFP/+; m12-Gal4/UAS-WldS). (C) Representative images of muscle 4 NMJs in wild-type (WT) or hiw ΔN mutants 24 h after injury. In WT animals, the presynaptic marker Futsch (green) completely disappears, while the neuronal membrane, labeled with antibodies to HRP (red), remain in discontinuous fragments. In contrast, NMJs in the hiw mutant (which are overgrown in an uninjured animal [8]) remain continuous and intact after injury. Of note, Futsch staining does not completely cover some synaptic branches in hiw mutant, but quantification of the extent of Futsch coverage (as in [20]) shows no significant difference between injured and uninjured hiw mutants (unpublished data). (D) Quantification of NMJ degeneration. White bars represent percentage of NMJs that have completely degenerated, defined by a complete loss of Futsch staining from the NMJ. Gray bars represent the percentage of NMJs that are partially degenerated, defined by a partial fragmentation Futsch staining and neuronal membrane (see Materials and Methods). Black bars represent the percentage of NMJs that are intact. The genotypes are: (Canton S), (hiw ΔN ), (hiwND8, BG380-Gal4; UAS-hiw/+), (BG380-Gal4;; UAS-hiw-ΔRING/+), (BG380-Gal4; UAS-UBP2/+), (BG380-Gal4;; UAS-WldS/+). (E) Representative traces of evoked and spontaneous neurotransmitter release recorded from wild-type (Canton S) and hiw mutant (hiw ΔN ) larvae before or 24 h after injury. Calibration: 200 ms, 2 mV for spontaneous release; 30 ms, 5 mV for evoked release. (F–H) Histograms showing (F) average spontaneous miniature EJP frequency, (G) spontaneous miniature EJP amplitude, and (H) evoked EJP amplitude, either from uninjured (black bars) or injured (24 h after injury, gray bars), in Canton S (WT) or hiw ΔN larvae. n = 10 recordings for each genotype. In WT injured larvae, only one single miniature event (amplitude 2 mV) was observed in all ten recordings. Of note, in uninjured larvae the amplitudes of evoked and miniature EJPs were smaller in hiw mutant, as previously reported [8]. Scale bars = 12.5 µm, error bars represent standard error; ***p<0.001; ns, not significant, p>0.05 in t-test.
Hiw is a large, highly conserved protein thought to function as an E3 ubiquitin ligase [17],[33]. Previous studies have suggested that Hiw regulates the ability of axons to regenerate after injury [28],[30]. Here we investigated whether Hiw plays a role in degeneration after injury.
In both hiw null (hiw ΔN ) and hypomorph (hiwND8) mutant animals, axonal degeneration was strongly inhibited. Even 48 h after injury (which is the latest time that can be visualized before pupation) the distal stump of injured axons remained intact in hiw mutants (Figure 1A and 1B). The protection from degeneration was also recapitulated in neurons that expressed the dominant negative mutation, hiw-ΔRING (Figure 1B), but not in adjacent neurons that did not express Gal4. These results strongly suggest that Hiw performs a cell-autonomous function in promoting axonal degeneration after injury. Similarly, we found that overexpression of the de-ubiquitinating enzyme UBP2 [34] delayed degeneration of Drosophila motoneuron axons and neuromuscular junctions (NMJs) (Figure 1B and 1D).
The hiw mutation also inhibited degeneration of the NMJ (Figure 1C). In wild-type animals, pre-synaptic proteins, such as the MAP1B homologue Futsch, disappeared completely from all NMJ boutons within 24 h after injury while the axonal membrane, detected with anti-HRP antibodies, fragmented into individual spheres (Figure 1C). In hiw mutants, all markers of NMJ structure remained intact (Figures 1C, 1D, and S1). Expression of hiw cDNA in motoneurons restored their ability to degenerate after injury (Figure 1D).
To test whether the distal stump of hiw mutants remained functional, NMJ synapses at muscle 6 were subjected to a standard electrophysiology recording paradigm either before or after injury (Figure 1E–1H). At 24 h after injury, wild-type NMJs were completely silent: no evoked excitatory junction potentials (EJPs) were observed (Figure 1H), and only one single spontaneous miniature event (mEJP) was observed in all ten recordings (Figure 1F). In contrast, at 24 h after injury, recordings in hiw mutant NMJs showed robust spontaneous mEJPs and evoked EJPs, resembling uninjured hiw NMJs [8]. Hence axons and synapses are functionally intact and resilient to degeneration in hiw mutants.
We then tested whether Hiw promotes axonal degeneration in other neuron types (Figure 2). The sensory neuron axons in larval segmental nerves were also injured in the nerve crush assay, and their distal axons also degenerated in a Hiw-dependent manner (Figure 2A). We then tested the role of Hiw in degeneration of adult neurons, which can be studied over a longer window of time. In wild-type animals, the distal stumps of olfactory neuron axons in the antennal lobe degenerated within 1 d after their cell bodies were removed by antennal lobe transection [2],[35]. In contrast, in hiw null mutants, olfactory neuron axons remained in the antennal lobe even 20 d after cell body removal (Figure 2B and 2C), which is comparable with the extent of protection by the WldS gain-of-function mutation [2],[35]. These dramatic phenotypes in multiple neuron types suggest that Hiw plays a fundamental role in the initiation of axonal degeneration after injury.
Figure 2. Wallerian degeneration in neurons of different neural types and developmental stages is strongly arrested in hiw mutants.

(A) The nerve terminals of class IV sensory neurons in the ventral nerve cord, visualized by driving UAS-mCD8::RFP with ppk-Gal4, are completely degenerated and cleared within 24 h after injury in wild-type (WT) animals, however these injured axons remain intact in the hiw ΔN mutant. (Because the site of injury was in segment A2, all axons whose terminals are to the right of the yellow arrows have been injured). (B) Olfactory neuron axons in adult flies are labeled by driving expression of UAS-mCD8::GFP with OR47b-Gal4. These axons degenerate within 1 d after antenna removal in wild-type flies, however in hiwΔN mutants these axons remain intact even 20 d after axotomy. (C) Quantification of the percentage of animals which retain GFP-labeled commissural axons (scored as described in [2],[35]), in a time course after axotomy. Scale bars = 12.5 µm.
The Wallenda MAPKKK Is Only Partially Required for the highwire Degeneration Phenotype
To understand the mechanism for Hiw in Wallerian degeneration we first considered a previously characterized target of Hiw regulation, the Wnd/DLK kinase. A previous study in mouse DRG and Drosophila olfactory neurons found that degeneration is delayed in wnd(dlk) mutants [18]. However, in larval motorneurons, we found the opposite result, since mutations in hiw lead to increased levels of Wnd kinase in axons [16], and overexpression of wnd in motoneuron axons can delay Wallerian degeneration [20]. Consistent with Wnd playing a protective role against degeneration downstream of Hiw, the protection from degeneration in hiw mutants was suppressed in hiw; wnd double mutants, although the suppression was only partial (Figure 3). In contrast, the synaptic overgrowth and overbranching phenotype in hiw mutants was completely suppressed in the hiw;wnd double mutants [16]. We also noticed that while hiw mutations inhibited degeneration in multiple neuron types, overexpression of wnd did not protect olfactory neuron and sensory neuron axons from degeneration [20]. Hence the degeneration phenotype for hiw mutants could not be accounted for by Wnd alone. This suggested the existence of additional downstream effectors of Hiw during axonal degeneration.
Figure 3. Role of the Wnd/DLK MAPKKK in Hiw-regulated degeneration.

(A) OK6-Gal4, UAS-mCD8-GFP labeled motoneuron axons (green) are severely fragmented in wild-type (WT) axons 24 h after injury, while they remain intact in hiw ΔN mutants (hiw ΔN ; OK6-Gal4/UAS-mCD8::GFP). Axons in hiw; wnd double mutants (hiw ΔN ; OK6-Gal4/UAS-mCD8-GFP; wnd1/wnd2) are only mildly fragmented 24 h after injury, implying that mutation of wnd only partially suppressed the hiw mutant degeneration phenotype. (B) Representative muscle 4 NMJs labeled by immunostaining for Futsch (green) and HRP (neuronal membrane, red) in hiw ΔN mutants or hiw; wnd double mutants (hiw ΔN ;;wnd1/wnd2). At 24 h after injury, NMJs have completely degenerated in wild-type (Figure 1) but are intact in hiw mutants. In hiw;wnd double mutants, some NMJs have completely degenerated (upper panel), while others remain intact (lower panel). (C) Quantification of the percentage of NMJs that are completely degenerated, partially degenerated, or intact (see Materials and Methods) for the following genotypes: (Canton S), (hiw ΔN ), (hiw ΔN ;;wnd1/wnd2), (BG380-Gal4; UAS-wnd/+). Scale bars = 12.5 µm.
Nmnat Is a Downstream Target of Highwire during Wallerian Degeneration
A well-known and intensively studied negative regulator of Wallerian degeneration is Nmnat [23]. An increased activity of this enzyme, first discovered in the WldS mutation, can strongly inhibit degeneration after injury [36]. This gain-of-function phenotype for nmnat bears a striking resemblance to the hiw loss-of-function phenotype in its ability to delay the onset of Wallerian degeneration.
There is only one nmnat gene in Drosophila and it has been shown to be required for neural integrity [25]–[27]. To disrupt expression of this essential gene post-embryonically, we used the Gal4/UAS system to express double-stranded RNA [37] targeting nmnat, (UAS-nmnat-RNAi), in neurons. Immunostaining with an anti-Nmnat antibody [25] indicated that the knockdown of Nmnat was effective (Figure S2A); however, it was unlikely to be complete, since neuronal clones that are homozygous mutant for Nmnat undergo spontaneous degeneration in uninjured animals [25],[26]. In contrast, RNAi-mediated knockdown of nmnat in larva motoneurons did not affect the development or stability of axons and synapses (Figure S2B), and only modestly affected the time course of degeneration after injury (Figure 4B). However knockdown of nmnat strongly suppressed the hiw protective phenotype, both in axons (Figure 4A and 4B) and NMJ synapses (Figure 4C and 4D). Similarly, reduction of Nmnat also suppressed the protection from degeneration caused by overexpression of UBP2 (Figure S3). These results suggest that Nmnat function is an important component of Hiw's role in the degeneration process. Interestingly the NMJ synaptic overgrowth phenotype of the hiw mutants was not suppressed by RNAi knockdown of nmnat (Figure 4C and 4E). This implies that Hiw regulates synaptic morphology independently of Nmnat function, or at least through a mechanism that is less sensitive to Nmnat function than degeneration. In contrast, Wnd is required for synaptic overgrowth in hiw mutants, and data presented below suggest that Nmnat and Wnd function independently.
Figure 4. Regulation of Wallerian degeneration by Hiw depends upon endogenous Nmnat.

(A) Nmnat is required for the protective phenotype of hiw. m12-Gal4, UAS-mCD8::GFP labeled axons (green) 24 h after injury in animals either mutant for hiw (hiwND8,UAS-Dcr2; UAS-mCD8::GFP/+; m12-Gal4/+) or mutant for in hiw mutant and depleted for nmnat by RNA interference (hiwND8, UAS-Dcr2; UAS-mCD8::GFP/UAS-nmnat-RNAi; m12-Gal4/+). (B) Degeneration index for the m12-Gal4, UAS-mCD8::GFP labeled single axons at different time points after injury in the following genotypes: (UAS-Dcr2; UAS-mCD8::GFP/+; m12-Gal4/+), (UAS-Dcr2; UAS-mCD8-GFP/UAS-nmnat-RNAi; m12-Gal4/+), (hiwND8, UAS-Dcr2; UAS-mCD8::GFP/+; m12-Gal4/+), (hiwND8, UAS-Dcr2; UAS-mCD8::GFP/UAS-nmnat-RNAi; m12-Gal4/+), (hiwND8, BG380-Gal4; UAS-nmnat/UAS-nmnat-RNAi; m12-Gal4,UAS-mCD8::GFP/+). RNAi depletion of nmnat alone only modestly affects the rate of degeneration (compare blue to black); however, it strongly inhibits the protection observed in the hiw mutant (compare orange to red). (C) Representative images of NMJs at muscle 4 24 h after injury in hiw mutants (hiwND8, BG380-Gal4; UAS-Dcr2/+) or hiw mutants depleted for nmnat in neurons (hiwND8, BG380-Gal4; UAS-Dcr2/UAS-nmnat-RNAi). Futsch staining in green labels cytoskeleton structure and HRP staining in red labels neuronal membrane. (D) Quantification of the percentage of NMJs that are completely degenerated, partially degenerated or intact in the above genotypes. (E) Quantification of average bouton numbers per NMJ at muscle 4 in the following genotypes: (BG380-Gal4, UAS-Dcr2), (BG380-Gal4, UAS-Dcr2; UAS-nmnat-RNAi/+), (hiwND8,BG380-Gal4; UAS-Dcr2/+), (hiwND8, BG380-Gal4; UAS-Dcr2/UAS-nmnat-RNAi). Scale bars = 12.5 µm, error bars represent standard error; *p<0.05; **p<0.01; ***p<0.001; ns, not significant, p>0.05 in t-test.
Wallenda and Nmnat Function in Parallel Downstream of Highwire
To further probe the relationship between Wnd and Nmnat, we conducted genetic epistasis analysis. Overexpression (O/E) of either wnd or nmnat cDNA can delay Wallerian degeneration in Drosophila motoneurons (Figure 5A–5D), so we tested whether the phenotype of O/E nmnat required wnd, and vice versa, whether the phenotype of O/E wnd required nmnat.
Figure 5. Wnd/DLK and Nmnat protect axons through parallel mechanisms downstream of Hiw.

(A) Representative muscle 4 NMJs at 24 h after injury immunostained for Futsch (green) and HRP (neuronal membrane, red) for the following genotypes: WT (Canton S), neuronally overexpressed nmnat (BG380-Gal4; UAS-HA-nmnat/+), or overexpressed nmnat in a wnd mutant background (BG380-Gal4; UAS-HA-nmnat/+; wnd1/wnd2). Overexpression of nmnat protected NMJs from degeneration and this protection was not compromised by mutations in wnd. (B) Quantification of NMJ degeneration in the above genotypes. (C) UAS-mCD8::GFP/+; m12-Gal4/+ labeled singles axons (green) 24 h after injury in WT (UAS-Dcr2; UAS-mCD8::GFP/+; m12-Gal4/+), when overexpressing wnd (UAS-Dcr2; UAS-mCD8-GFP/+; m12-Gal4/UAS-wnd), or when overexpressing wnd in conjunction with nmnat RNAi (UAS-Dcr2; UAS-mCD8-GFP/UAS-nmnat-RNAi; m12-Gal4/UAS-wnd). Reducing Nmnat levels by this method had no effect upon the protection caused by overexpression of wnd. (D) Degeneration index of the m12-Gal4, UAS-mCD8::GFP labeled single axons at different time points after injury in the above genotypes. (E) Representative images of NMJs at muscle 4 18 h after injury stained for Futsch (green) and HRP (red) in the following genotypes: (hiwND8,BG380-Gal4; UAS-Dcr2/+), (hiwND8, BG380-Gal4;UAS-Dcr2/+;UAS-wnd-RNAi/+), (hiwND8, BG380-Gal4; UAS-Dcr2/UAS-nmnat-RNAi), (hiwND8, BG380-Gal4; UAS-Dcr2/UAS-nmnat-RNAi; UAS-wnd-RNAi/+). (F) Quantification of the percentage of NMJs that are completely degenerated, partially degenerated, or intact at 12 h or 18 h after injury, for the genotypes described above. (G) Model: Wnd and Nmnat inhibit axonal degeneration through independent pathways downstream of Hiw. Scale bars = 12. 5 µm, error bars represent standard error; *p<0.05; ns, not significant, p>0.05 in t-test.
We found that disruption of wnd function had no effect upon the protection from degeneration by O/E nmnat (Figure 5A and 5B). For the converse experiment, we tested whether knockdown of nmnat by expression of UAS-nmnant-RNAi affected the protection by O/E wnd (Figure 5C and 5D). While this method for disrupting Nmnat suppressed the hiw degeneration phenotype (Figure 4), it had no effect upon the O/E wnd phenotype (Figure 5C and 5D). These observations suggest that Nmnat and Wnd protect axons from degeneration through independent mechanisms.
We then tested whether knockdown of nmnat and wnd by RNA interference had additive effects in suppressing the hiw degeneration phenotype (Figure 5E and 5F). Since nmnat-RNAi rescues the hiw phenotype very strongly on its own at 24 h after injury, we assayed earlier time points, 12 and 18 h after injury, for additive effects with wnd-RNAi. Expression of wnd-RNAi alone in the hiw mutant background caused 42% of the NMJs to degenerate (including complete degeneration and partial degeneration) within 18 h of injury, while expression of nmnat-RNAi alone caused 59% of the hiw mutant NMJs to degenerate at this time point. Combined knockdown of both nmnat and wnd led to a nearly complete suppression of the hiw degeneration phenotype, with 92% of the NMJs degenerating (Figure 5E and 5F). Together, these results suggest that Wnd and Nmnat function independently downstream of Hiw in the Wallerian degeneration process (Figure 5G).
Highwire Regulates the Levels of Nmnat Protein
Hiw and its homologues are known to function within an E3 ubiquitin ligase complex [17],[33],[38]–[41]. An attractive hypothesis is that Hiw promotes ubiquitination and protein turnover of endogenous Nmnat protein. Consistent with this hypothesis, we found that knockdown of nmnat suppressed the protection from degeneration caused by overexpression of the de-ubiquitinating enzyme UBP2 (Figure S3). We therefore asked whether mutation in hiw leads to an increase in the levels of Nmnat protein. Most strikingly, we noticed an appearance of Nmnat protein in the synapse and neurite-rich neuropil of hiw mutants, which was not detectable in a wild-type background (Figure 6A and 6B). We also observed complex changes in the distribution of Nmnat in neuronal nuclei and glia (Figure S2).
Figure 6. Hiw negatively regulates the levels of Nmnat protein in axons and synapses.

(A) Hiw regulates endogenous Nmnat protein in neuropil. In hiw ΔN mutants, Nmnat protein can be detected within the neuropil of the ventral nerve cord, denoted with asterisks. This area of the nerve cord is devoid of cell bodies and enriched in neurites and synapses. (B) Quantification (relative levels) of Nmnat staining in neuropil, for WT (w118), hiw mutant (hiw ΔN), hiw,Ctrl (hiw ΔN, BG380-Gal4), hiw, nmnat-RNAi (hiw ΔN, BG380-Gal4, UAS-nmnat-RNAi), and nmnat-RNAi (BG380-Gal4, UAS-nmnat-RNAi). See Materials and Methods. (C) Western blot with adult heads or larval brains to compare total protein levels of HA-Nmnat in wild-type (ctrl) and hiw mutant backgrounds. The UAS-HA::nmnat transgene is expressed in neurons with the BG380-Gal4 driver, and males are used for all experiments. (D–F) The UAS-HA-Nmnat transgene was expressed in motoneurons with the OK6-Gal4 driver, in wild-type (OK6-Gal4/UAS-HA::nmnat), hiw mutant (hiw ΔN;OK6-Gal4/UAS-HA::nmnat) and hiw; wnd double mutant (hiw ΔN ;OK6-Gal4/UAS-HA::nmnat;wnd1/wnd 2 ) backgrounds. HA-Nmnat protein is detected by immunostaining for HA. (D) Representative images of HA-Nmnat in motoneuron cell bodies, (E) segmental (peripheral) nerves, and (F) NMJ synapses, stained for anti-HA (green) and HRP (neuronal membrane, red). (G–I) Quantification of the average HA-Nmnat intensity for the above genotypes in (G) cell bodies, (H) segmental nerves, and (I) NMJ synapses at muscle 4. See Materials and Methods for details about quantification methods. In hiw mutants, Nmnat intensity is increased, particularly at NMJ synapses. Loss of wnd, in hiw;wnd double mutants, has no effect upon this increase. Scale bars = 12.5 µm, error bars represent standard error; *p<0.05; ***p<0.001; ns, not significant, p>0.05 in t-test.
To test whether Hiw regulates Nmnat in neurons via a post-transcriptional mechanism, we drove expression of transgenic HA-tagged nmnat cDNA in neurons via an ectopic Gal4/UAS promoter. In hiw mutants, the total level of HA-Nmnat protein, as detected on Western blots, increased in both larval brains (3.1±0.6-fold) and adult heads (5.2±1.1-fold) (Figure 6C). By immunocytochemistry, the HA-Nmnat protein (which represents a splice form that lacks the nuclear localization sequence) could readily be detected in motoneuron cell bodies (Figure 6D and 6G) and axons within segmental nerves (Figure 6E and 6H), but is barely detectable at NMJ synapses (Figure 6F and 6I). In hiw mutants, the levels of HA-Nmnat increased in all compartments, however the 5-fold increase quantified at NMJ synapses was most striking (Figure 6G–6I). The increase in Nmnat protein levels remained in the hiw;wnd double mutant background (Figure 6E–6I), adding further support to the model that Hiw regulates Nmnat protein independently of Wnd.
Highwire Regulates Nmnat Via Ubiquitination
The hiw mutation led to an increase in the levels of transgenic Nmnat, which was expressed via the ectopic Gal4/UAS promoter. We confirmed that the hiw mutation did not increase expression from the different Gal4 drivers used (ppk-Gal4, OK6-Gal4, and BG380Gal4, unpublished data). Hence the regulation of Nmnat by Hiw takes place post-transcriptionally. To test whether Nmnat is regulated by ubiquitination, we overexpressed the yeast ubiquitin protease UBP2 in neurons, which can counteract the function of ubiquitin ligases [34],[42]. We found that co-expression of UBP2 in neurons with the HA-nmnat transgene caused an increase in the levels of HA-Nmnat protein (Figure 7A and 7C), resembling the hiw mutant (Figure 6). This suggests that the levels of Drosophila Nmnat are controlled by ubiquitination.
Figure 7. Hiw and ubiquitination down-regulate Nmnat protein.

(A) The UAS-HA-nmnat transgene is expressed in motoneurons with OK6-Gal4 driver in a wild-type (WT)_ genetic background (OK6-Gal4,UAS-HA::nmnat/+) or when the yeast deubiquitinase UBP2 is co-expressed (OK6-Gal4,UAS-HA::nmnat/UAS-UBP2). Segmental nerves stained with anti-HA antibody (green). (B) Transgenic HA-nmnat is co-expressed with a control UAS- construct (BG380-Gal4; UAS-HA::nmnat/+; UAS-nls::GFP/+) or with the full-length hiw cDNA (BG380-Gal4; UAS-HA::nmnat/UAS-hiw). Segmental nerves stained for HA (green) and HRP (neuronal membrane, violet). (C) Quantification of the average HA-Nmnat intensity in segmental nerves in (A) and (B). The average intensity is normalized to the control for each experimental group. (D) Distal axons and axon terminals of ppk-Gal4,UAS-mCD8::RFP labeled sensory neurons stained for HA (green) and RFP (red). The UAS-HA-Nmnat transgene is co-expressed with a control UAS- construct (UAS-HA::nmnat/+; ppk-Gal4,UAS-mCD8::RFP/UAS-nls::GFP), or the full-length hiw cDNA (UAS-HA::nmnat/UAS-hiw; ppk-Gal4,UAS-mCD8::RFP/+), or a dominant negative hiw transgene mutated for conserved cysteines in the RING domain [67] (UAS-HA::nmnat/+; ppk-Gal4,UAS-mCD8::RFP/UAS-hiw-ΔRING). (E) Quantification of the average HA-Nmnat intensity in the sensory neuron axon terminals for the above genotypes. See Materials and Methods for the quantification method. (F) Hiw can down-regulate Nmnat protein in S2R+ cells. S2R+ cells were co-transfected with pUAST-HA::Nmnat, and either pUAST-GFP (control vector), pUAST-Hiw, or Hiw-ΔRING. All cells were co-transfected with pMT-Gal4 and induced with 0.5 mM copper sulfate for 24 h. Hiw is not expressed endogenously in S2R+ cells, and numerous breakdown products are observed for the ectopically expressed Hiw protein. The reduction in ectopic HA-Nmnat levels in lane 3 indicates that Hiw is capable of post-transcriptionally regulating Nmnat, and that the RING domain is required for this activity (lane 4). (G) Nmnat and Hiw-ΔRING form a physical interaction. Co-immunoprecipitation assays were performed from S2R+ cells lysate either co-transfected with pUAST-HA::Nmnat and pUAST-Hiw-ΔRING (lane 5) or mixed lysates from individual pUAST-HA::Nmnat and pUAST-Hiw-ΔRING transfections (lane 6). HA-Nmnat was immunoprecipitated by mouse anti-HA antibody against the HA tag on Nmnat. Despite the fact that Hiw-ΔRING was significantly degraded in S2R+ cell lysate (detected by Western blotting for Hiw antibody), a significant portion of Hiw-ΔRING protein co-immunoprecipitated with HA-Nmnat. The Input lanes (1–3) represent 1/25 of the total extract used for each immunoprecipitation. Scale bars = 12.5 µm; error bars represent standard error; ***p<0.001 in t-test.
We next tested whether the action of the Hiw E3 ubiquitin ligase is sufficient to modify Nmnat protein level in axons and synapses. Co-overexpression of hiw cDNA (O/E hiw) with HA-nmnat caused a strong decrease in HA-Nmnat protein in motoneuron axons (Figure 7B and 7C). Because Nmnat protein was difficult to detect at the NMJ (Figure 6F), we also examined the nerve terminals of class IV sensory neurons, whose concentrated location in the ventral nerve cord was easier to visualize. O/E hiw caused a reduction in HA-Nmnat protein in sensory axon terminals (Figure 7D and 7E). In contrast, co-expression of the dominant negative hiw-ΔRING mutation caused an increased level of HA-Nmnat in the sensory axon terminals (Figure 7D and 7E). Further evidence that Hiw function is sufficient to down-regulate Nmnat comes from studies in S2R+ cells, which do not express Hiw endogenously. Co-expression of Hiw, but not of Hiw-ΔRING, led to down-regulation of HA-Nmnat protein (Figures 7F and S4A). These findings suggest that Hiw plays a direct role in regulating the levels of Nmnat protein.
Curiously, we were unable to obtain evidence that Hiw down-regulates Nmnat via the UPS. Inhibition of the proteasome by addition of MG132, using several different concentrations and periods of time that affect known targets to the UPS (Materials and Methods) [43],[44], did not affect the down-regulation of Nmnat by Hiw in S2R+ cells (Figure S4A). To inhibit the proteasome in vivo we co-expressed dominant-negative proteasome subunit mutations, DTS5 and DTS7, which in previous studies had been shown to lead to allow targets of the UPS to accumulate [45]–[47]. This led to only minor (7%) changes in the levels of HA-Nmnat in sensory neuron terminals (Figure S4B). Surprisingly, inhibition of the proteasome had a much greater effect upon HA-Nmnat levels in a hiw null mutant than in a wild-type background (Figure S4C). This observation does not favor a simple model that Hiw regulates Nmnat via the UPS. Instead, the data suggest that additional ubiquitin ligases may regulate Nmnat, and that the regulation of Nmnat may be more sensitive to the UPS when hiw is absent.
While the above data indicate that ubiquitination is important for the regulation of Nmnat, the detailed mechanism by which Hiw regulates Nmnat remains to be determined. The mechanism may involve a direct interaction, since co-immunoprecipitation experiments indicate that Nmnat can robustly interact with the enzyme dead Hiw-ΔRING protein in S2R+ cells (Figure 7G).
Highwire Promotes Destruction of Nmnat in the Distal Stump of Injured Axons
A recent study using vertebrate cultured neurons suggested that the disappearance of Nmnat2, which has a short half-life, from the distal stump of axons may serve as a trigger for the Wallerian degeneration process [21]. This leads to an attractive hypothesis that Hiw promotes the disappearance of Nmnat protein from the distal stump. Supporting this model, we observed that HA-Nmnat levels become significantly reduced in axons (Figure S5A) and synapses (Figure 8) distal to the injury site. In contrast, HA-Nmnat levels increase in the proximal stump after injury (Figure S5A), consistent with the model that a cytoplasmic form of this enzyme is transported in axons from the cell body [21]. Within 4 h after injury, the majority of HA-Nmnat in sensory axon terminals had disappeared (Figure 8). By comparison, a significant amount of green fluorescent protein (GFP)-Hiw remained at this time point (Figure S5B).
Figure 8. Hiw promotes Nmnat protein turnover in the injured distal axons and synapses.

(A) Distal axons and synapses of ppk-Gal4,UAS-mCD8::RFP labeled sensory neurons, located in the ventral nerve cord, either before or 24 h after injury. Transgenic HA::nmnat is expressed in a wild-type (WT) genetic background (UAS-HA::nmnat/+; ppk-Gal4,UAS-mCD8::RFP/+), hiw ΔN mutant (hiw ΔN ; UAS-HA::nmnat/+; ppk-Gal4,UAS-mCD8::RFP/+) or co-expressed with UBP2 (UAS-HA::nmnat/UAS-UBP2; ppk-Gal4,UAS-mCD8::RFP/+). Consistent with observations in motoneurons (Figure 6), mutation of hiw or co-expression of UBP2 causes a dramatic elevation in HA-Nmnat protein, and this does not disappear after injury, in dramatic contrast to the disappearance of HA-Nmnat in wild-type (WT) animals. Of note, the nerve terminals of the ppk-Gal4 labeled axons appear to be overgrown in hiw and UBP2 expressing mutants, similar to previous descriptions in other neuron types [8],[41]. Hence the quantification shown in (B) is normalized to the size of the nerve terminals (labeled by mCD8-RFP). Due to the expression of HA-nmnat, no axons are degenerating in any of the above genotypes. (B) Quantification of the average HA-Nmnat intensity in ppk-Gal4 expressing sensory neuron axon terminals, within the most posterior four segments of the ventral nerve cord. Error bars represent standard error. ns, not significant, p>0.05. (C) Model: Hiw promotes multiple independent responses to injury, through independent pathways. Hiw promotes degeneration in the distal stump by down-regulating Nmnat, and concurrently regulates regeneration (and protection from degeneration [15],[30]) in the proximal stump by regulating Wnd. Since Hiw may localize and function in distal axons, injury may relieve the inhibition of Wnd by Hiw in the proximal stump. Scale bars = 12.5 µm, error bars represent standard error; ***p<0.001; ns, not significant, p>0.05 in t-test.
When hiw was mutant, the levels of HA-Nmnat in the distal stump did not decrease significantly below its starting level, even 24 h after injury (Figure 8A and 8B). Expression of UBP2 had a similar effect upon HA-Nmnat in the distal stump after injury (Figure 8A and 8B). These findings indicate that Hiw and the ubiquitination are required for the disappearance of Nmnat protein in the distal stump.
Highwire Can Specifically Down-regulate Mouse Nmnat2 Protein in Drosophila Neurons
Vertebrates utilize three distinct Nmnat enzymes, which localize to distinct subcellular locations. We tested whether Hiw was capable of influencing the levels of ectopically expressed mouse Nmnat1, which localizes to nuclei, mouse Nmnat2, which co-localizes with golgi and late endosome markers, or mouse Nmnat3, which localizes to mitochondria [48]–[50], by crossing UAS-mNmnat1::myc, UAS-mNmnat2::myc, and UAS-mNmnat3::myc transgenes [51],[52] into the hiw mutant background. Intriguingly, mutations in hiw resulted in increased levels of mNmant2-myc protein within axons and synaptic terminals of class IV sensory neurons (Figure 9). This finding implies that mNmant2-myc protein can be transported to distal axons and synapses, and that mouse Nmnat2 shares a conserved protein feature with Drosophila Nmnat that allows it to be regulated by Hiw. In contrast, loss of hiw had no effect upon the levels of mNmnat1 or mNmnat3. We interpret that the distinct subcellular localization of mNmnat2 may make this protein more susceptible to regulation by Hiw, and that that a conserved mechanism, involving Hiw homologues, may regulate Nmnat2 in vertebrate neurons.
Figure 9. Hiw is capable of regulating mouse Nmnat2, but not mouse Nmnat1 or Nmnat3, protein.
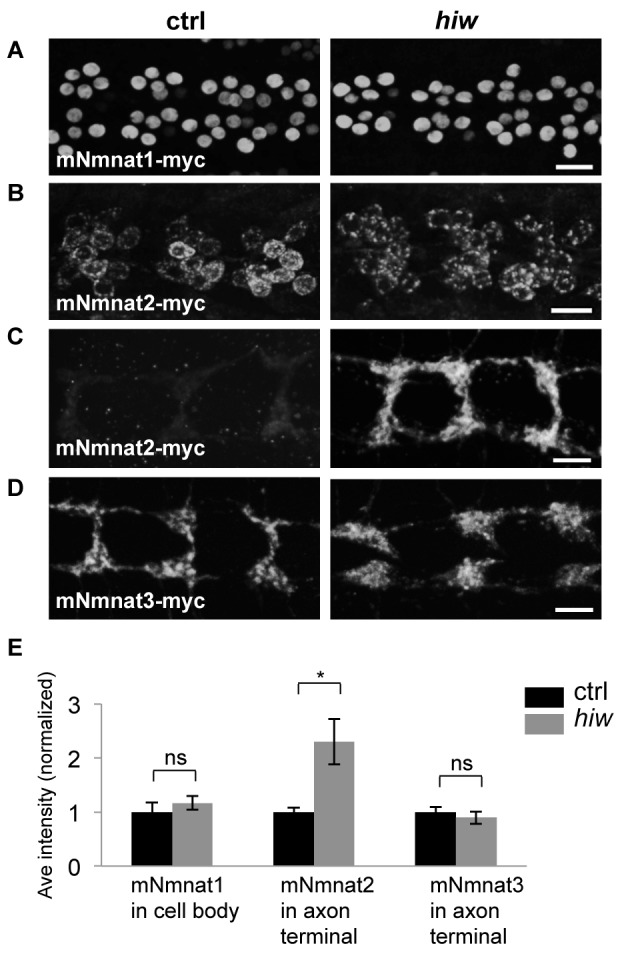
(A, B) UAS-mNmnat1::myc and UAS-mNmnat2::myc transgenes, expressed in motoneurons with the BG380-Gal4 driver, show no difference in cell body levels between control and hiwND8 mutants. (C–D) UAS-mNmnat2::myc, and UAS-mNmnat3::myc were expressed with ppk-Gal4 in order to visualize localization at sensory neuron axon terminal in the nerve cord. The levels of mNmnat2-myc protein at axon terminals were increased in hiwND8 mutants. In contrast, mNmant3-myc protein levels were similar in between control and hiwND8 mutant genotypes. (E) Quantification of average intensity of mNmnat1 in cell body, and mNmnat2 and mNmnat3 in axon terminals from (A–D). Scale bars = 12.5 µm, error bars represent standard error; *p<0.05; ns, not significant, p>0.05 in t-test.
Discussion
Highwire Promotes Degeneration by Down-regulating Nmnat Protein
Since the discovery of the dramatic inhibition of degeneration by the WldS mutation, many studies have focused upon the action of the NAD+ biosynthetic enzyme isoforms, Nmnat1, Nmnat2, and Nmnat3, which in some circumstances can confer protection against axonal degeneration (reviewed in [22],[23]). Most of these studies involve gain-of-function overexpression experiments; it has been difficult to address the role of endogenous Nmnat enzymes in this process. Recent observations indicate that endogenous Nmnat activity plays an essential role in neuronal survival, and its depletion leads to neurodegeneration [21],[25]–[27]. In addition, recent studies in vertebrate neurons suggest that the cytoplasmic isoform, Nmnat2, has a short half-life in neurons [21]. An attractive model proposes that Nmnat2 is rapidly turned over in axons, and that its loss in the distal stump of an axon, which has become disconnected from its cell body, leads to the initiation of Wallerian degeneration [21].
Some aspects of this model are supported by our current in vivo characterization in Drosophila. We have identified Hiw, a highly conserved protein with features of an E3 ubiquitin ligase, as an important regulator of Wallerian degeneration. Hiw's role in this process involves the Nmnat protein, whose levels in axons and synapses are regulated post-transcriptionally by Hiw function. In hiw mutants, Wallerian degeneration is strongly inhibited, and the increased level of Nmnat protein in hiw mutants is both required and sufficient to inhibit degeneration.
While the localization of endogenous Hiw in Drosophila is not known, homologues in mice and Caenorhabditis elegans have been detected in axons and at synapses [9],[53], so it is in the appropriate location to target the destruction of Nmnat in distal axons (Figure 8C). However, it remains to be determined whether the down-regulation of Nmnat in the distal stump per se is the trigger for Wallerian degeneration. When HA-Nmnat was overexpressed, axons were protected from degeneration long after the rapid disappearance of detectable protein in the distal stump. It is possible that even very low levels of Nmnat protein are sufficient to protect from degeneration. It is also formally possible that the basal levels of Nmnat before injury, rather than the disappearance of Nmnat after injury, is an important determinant of degeneration. We also acknowledge that axonal degeneration likely involves additional steps downstream or in parallel to the regulation of Nmnat by Hiw. While overexpression of Hiw can induce a reduction in HA-Nmnat levels (Figure 7), we were unable to observe an enhanced rate of degeneration when Hiw was overexpressed.
The Relationship of Highwire and the UPS
Studies almost a decade ago suggested a role for the UPS in the initiation of Wallerian degeneration [3]. It is tempting to propose that this role is manifested by the regulation of Nmnat by Hiw. However our observations caution against a simple interpretation that Hiw regulates Nmnat via the UPS, since Hiw can promote disappearance of Nmnat protein in cells in a manner unaffected by proteasome inhibitors (Figure S4A). Moreover, in vivo, inhibition of the proteasome had only a minor effect upon Nmnat levels in a wild-type background (Figure S4B and S4C). However in hiw mutants, Nmnat levels were very sensitive to the function of the proteasome (Figure S4C). We interpret that additional ubiquitin ligases and the UPS may regulate Nmnat independently of Hiw.
Regardless of the role of the proteasome, our observations suggest that ubiquitin plays an important role in Nmnat regulation. Overexpression of the yeast de-ubiquitinating protease UBP2 leads to increased levels of Nmnat protein and inhibition of Wallerian degeneration, in a manner that requires endogenous Nmnat (Figure S3). Future studies of the mechanism by which Hiw regulates Nmnat will therefore consider potential proteasome-independent roles of ubiquitination. Of note, in yeast UBP2 has been shown to preferentially disassemble polyubiquitin chains linked at Lys63 [54], which have been found to perform non-proteolytic functions in DNA repair pathways [55], kinase activation [56], and receptor endocytosis [57],[58]. We should also consider the possibility that Hiw regulates Nmnat indirectly: since we have thus far been unable to detect any ubiquitinated Nmnat species, it is possible that an intermediate, yet unknown, regulator of Nmnat may be the actual substrate of ubiquitination. Nevertheless, co-immunoprecipitation studies from S2R+ cells indicate that Hiw and Nmnat have the capacity to interact (Figure 7G).
Highwire Can Regulate Mouse Nmnat2
The mechanism and cellular location of Nmnat's protective action is a highly debated subject. Observations in the literature point to both NAD+-dependent and NAD+-independent models for the strong protection by the WldS mutation [23]. The location of its protective action may be the mitochondria, since mitochondrially localized Nmnat can protect axons from degeneration [51],[52],[59]. However golgi/endosomal localized Nmnat2 can also be protective [21],[27],[60],[61]. Our findings suggest that mutation of hiw leads to an increase in the pool of endogenous Nmnat that functionally impacts degeneration.
While the site of endogenous Nmnat function during axonal degeneration remains to be identified, we found that the levels of ectopically expressed mouse Nmnat2 were specifically increased in the hiw mutant background. In contrast, the levels of nuclearly localized mNmnat1 or mitochondrially localized mNmnat3 were unaffected by Hiw. Since Nmnat2 has a short half-life in vertebrate neurons [21], it is intriguing to propose that it is regulated by Hiw orthologs via an analogous mechanism.
Since Nmnat2 does not appear to localize to mitochondria, does this favor a non-mitochondrial activity, such as function as a chaperone [62],[63], for the protective action? It remains challenging to determine the exact location of protection, since the most apparent changes in Nmnat protein may not necessarily be the functionally relevant changes.
Multiple Roles of Highwire in Responses to Injury
A previously characterized target of Hiw regulation is the Wnd MAP kinase kinase kinase [16],[17]. This axonal kinase is also capable of inhibiting Wallerian degeneration in motoneurons [20]. The protective action of Wnd requires a downstream signaling cascade and changes in gene expression mediated by the Fos transcription factor [20]. Loss of nmnat does not affect this signaling cascade (unpublished data) nor does it affect the protective action of Wnd (Figure 5C and 5D). Conversely, loss of wnd does not affect the protection caused by overexpressing nmnat (Figure 5A and 5B). Importantly, the regulation of Nmnat by Hiw does not appear to require Wnd function, and Wnd and Nmnat can protect axons independently of each other. These findings favor the model that Wnd and Nmnat are both regulated by Hiw and influence axonal degeneration through independent mechanisms.
The Wnd kinase plays additional roles in neurons, which can be genetically separated from Nmnat function. These include regulation of synaptic growth: a dramatic synaptic overgrowth phenotype in hiw mutants is fully suppressed by mutation of wnd, but is not at all affected by knockdown of nmnat (Figure 4E). Wnd/DLK also promotes axonal sprouting in response to axonal injury [30], which is also unaffected by nmnat knockdown (unpublished data). It is therefore clear that by regulating both Wnd and Nmnat, Hiw regulates multiple independent pathways in neurons.
It is intriguing that the actions of both Wnd and Nmnat promote cellular responses to axonal injury. Axonal regeneration requires an initiation of a growth program within the axon, which depends upon the function of Wnd and its homologues [28]–[32]. Equally important is a clearance of the distal stump to make room for the regenerating axon [64]–[66]. Since both Wnd and Nmnat are transported in axons [21],[30], Figure 8C proposes a model in which Hiw function in the distal axon terminal could simultaneously promote destruction of Nmnat in the distal stump, and accumulation of Wnd in the proximal stump. The latter is observed after injury [30], and is required to promote new axonal growth. The actual location in which Hiw regulates Nmnat remains to be determined. As an upstream regulator of both sprouting in the proximal stump and degeneration of the distal stump, Hiw may play a central role in regulating the ability of a neuron to regenerate its connection after injury.
Importantly, the protective action of Nmnat may not be limited to Wallerian degeneration. The WldS mutation can protect neurons from degeneration in a wide variety of paradigms, from models of neurodegenerative disease, diabetic neuropathy, excitotoxity, and loss of myelination [22],[23]. These findings suggest that action and regulation of Nmnat function is broadly important for neuronal function and maintenance. As a critical regulator of Nmnat, the Hiw ubiquitin ligase and its vertebrate homologues deserve further scrutiny for potential roles in human health and disease.
Materials and Methods
Fly Stocks
The following strains were used in this study: Canton-S (wild-type), hiwND8 [8], hiw ΔN, UAS-hiw and UAS-hiw-ΔRING from [67], OK6-Gal4 [68], BG380-Gal4 [69] m12-Gal4 (P(GAL4)5053A) [70], ppk-Gal4 [71], Or47b-Gal4 [72], UAS-UBP2 [41], UAS-DTS5, and UAS-DTS7 from [45], wnd1, wnd3, and UAS-wnd from [16]. UAS-HA::nmnat [25], UAS-WldS [2], UAS-mNmnat1::myc, UAS-mNmnat2::myc, and UAS-mNmnat3::myc [51],[52], and UAS-Dcr2 were gifts from Grace Zhai, Liqun Luo, Marc Freeman, and Stephan Thor. UAS-wnd-RNAi (Construct ID 13786) and UAS-nmnat-RNAi (construct ID 32255) were acquired from the Vienna RNAi center [37].
Nerve Crush Assay
The segmental nerves of third instar larvae were visualized through the cuticle under a standard dissection stereomicroscope. While larvae were anesthetized with CO2 gas, the segmental nerves were pinched tightly through the cuticle for 5 s with Dumostar number 5 forceps. After successful injury, the posterior half of the larva was paralyzed. Larvae were then transferred to a grape plate and kept alive for varying periods of time at 25°C. Also see [30].
Immunocytochemistry
Larvae were dissected in PBS and fixed in 4% paraformaldehyde or formaldehyde in PBS for 25 min for the following antibodies used: ms anti-Futsch (1∶100), guinea pig (gp) anti-NMNAT [25], (gift from Hugo Bellen and Grace Zhai, 1∶1,000), rat anti-HA (Roche, 1∶100), rat anti-elav (1∶50), or fixed in Bouin's fixative for 15 min for the following antibodies: ms anti-Brp (1∶200), Rb anti-GluRIII (1∶1,000 [73]), Rb anti-DVLGUT (1∶10,000, [74]). Rat anti-elav (7E8A10) and ms anti-Brp (NC82) were obtained from Developmental Studies Hybridoma Bank, University of Iowa. The conjugated secondary antibodies are used and diluted as follows: Cy3-Gt anti-HRP and Cy5-Gt anti-HRP (from Jackson labs) at 1∶200, A488-Rb anti-GFP (from Molecular Probes) at 1∶1,000. For secondary antibodies Cy3 and Alexa-488 conjugated Goat anti-rb/mouse/rat/gp (from Invitrogen) were used at 1∶1,000. All antibodies were diluted in PBS-0.3%Triton with 5% normal goat serum.
Imaging
Confocal images were collected at room temperature on an Improvision spinning disk confocal system, consisting of a Yokagawa Nipkow CSU10 scanner, and a Hamamatsu C9100-50 EMCCD camera, mounted on a Zeiss Axio Observer with 25× (0.8 NA) multi and 40× (1.3NA), 63× (1.5NA), and 100× (1.46 NA) oil objectives. Similar settings were used to collect all compared genotypes and conditions. Volocity software (Perkin Elmer) was used for all measurements of average and total intensities.
For measurement of Nmnat intensity in the neuropil, the neuropil area was selected based on co-staining for the synaptic marker Brp. Objects meeting intensity criteria of >0.8 standard deviations above the mean were selected within a 140-µm long region of the ventral nerve cord and then summed for total intensity. The average intensity of the HA-Nmnat staining in muscle 4 NMJs was measured within the synaptic area defined by HRP staining after subtraction of background intensity for each image. The average intensity of the HA-Nmnat staining in motoneuron axons and sensory nerve terminus was measured with a similar protocol. Likewise for neuronal nuclei, the average intensity for Nmnat staining was measured in neuronal nuclei defined by staining for the neuronal marker Elav. Numbers are shown normalized to the average intensity of the control for each figure.
Quantification of Degeneration
To quantify axonal degeneration, we scored (while blind to genotype) the fragmentation of m12-Gal4, UAS-mCD8-GFP labeled axons within segmental nerves according to one of five categories between 0 and 100% (with 100% meaning completely degenerated) as described in [20]. All measurements indicate the average from >100 axons.
To quantify the degeneration of the NMJ, NMJs were stained for the MAP1B homologue Futsch and axonal membrane marker HRP, and were scored into one of three categories: (1) complete degeneration, defined by a complete loss of Futsch staining from the NMJ and fragmentation of the axonal membrane, (2) partial degeneration, defined by a partial loss of Futsch staining from the NMJ and partial membrane fragmentation, and (3) no degeneration, in which there was no fragmentation of the membrane or Futsch, similar to uninjured control animals. All quantifications shown represent the average scores from multiple NMJs from >six animals quantified in duplicate by two independent observers who were blind to the genotype.
Degeneration of ORN axons was quantified following the previously defined method [2],[35] by calculating the percentage of brains for each genotype in which contralateral axon projections could still be detected.
For all the statistical analysis, Student's t test was applied.
Electrophysiology
Intracellular recordings were made from muscle 6 in segments A3 and A4 of third-instar male larvae. The larvae were visualized with oblique illumination on an Olympus BX51W1 fixed stage upright microscope with a 10× water immersion objective. Sharp electrodes (15–25 MΩ), made of borosilicate glass (outer diameter 1.2 mm) were filled with 3 M KCl. The signal was amplified with a Geneclamp 500B (Molecular Devices), digitized with a Digidata 1322A interface (Molecular Devices), and stored on a PC with pClamp 10.2 (Molecular Devices). Recordings were performed in HL3 Stewart saline [75] that contained (in mM) 70 NaCl, 5 KCl, 20 MgCl2, 10 HCO3, 5 trehalose, 115 sucrose, 5 HEPES, 1 CaCl2,, the pH was adjusted to 7.2. For all genotypes the resting membrane potentials and input resistances were similar, with average resting potentials of −73±4 and input resistances of 6±0.2 MΩ. To elicit evoked EJPs, the nerve was drawn into a tight-fitting suction electrode and stimulated with brief (1 ms) depolarizing pulses controlled with Digidata interface. The stimulus amplitude was set to 125% of the amplitude necessary to activate the higher threshold of the two excitatory axons that innervate the muscle. For injured wild-type larvae (in which nerve stimulation did not produce evoked synaptic responses) the stimulus amplitude was set to double the amplitude used for un-injured larvae. However evoked responses were not observed, even at the largest stimulus amplitude that the equipment could produce. For analysis of evoked responses, 100 events per cell recorded at 0.2 Hz were measured using the cursor feature in Clampfit 10.2 (Molecular Devices) and then averaged. For analysis of spontaneous miniature EJPs, at least 50 consecutive events were measured per cell using MiniAnal (Synaptosoft). mEJP frequency was calculated from the first 30 s of recording time.
Cell Culture
S2R+ cells were cultured in Schneider's medium (Gibco) which contains 10% (v/v) FBS (Gibco) and 1% penicillin-streptomycin (Invitrogen). For plasmid transfection, cells were transfected using FuGENE 6 (Promega) following the manufacturer's instructions. Copper sulfate solution (0.5 mM) was added 6 h after transfection to induce plasmid expression. Cell lysates were collected after 24 h. Plasmids used for transfection were pMT-Gal4 [76], pUAST-eGFP [77], pUAST-GFP-Hiw [67], pUAST-HiwΔRING [67], and pUAST-HA-Nmnat [25].
To inhibit the UPS, cells were treated with MG132 (InSolution, Calbiochem) or DMSO as control using several different conditions: 25 µM for 6 h, 5 µM for 20 h, and 5 µm for 36 h. All of these conditions led to an increase in the levels of polyubiquitinated proteins, detected by Western blots probed with FK1 antibodies.
Biochemistry
The following antibodies were used for Western blotting: rb anti-Hiw (ref, 1∶2,000), rat anti-HA (Flourochem, 1∶2,500), ms anti-β-tubulin (1E7) and ms anti-β-catenin (armadillo, N27A1) from Developmental Studies Hybridoma bank (University of Iowa), ms anti-polyubiquitin, (FK1, Enzo Life Sciences, 1∶1,000), and ms anti-ubiquitin (P4D1, Cell Signaling, 1∶1,000). Westerns were probed with IRDye 800CW and 680RD conjugated secondary antibodies (LiCor biosciences, 1∶10,000) and imaged for quantitative analysis via a LiCor Odyssey imaging system.
S2R+ cells were transfected with either pUAST-HiwΔRING or pUAST-HiwΔRING and pUAST-HA-Nmnat. Cells from 6-cm dishes were harvested in 500-µl ice-cold lysis buffer (20 mM HEPES [pH 7.5]), 200 mM KCl, 0.05% Triton X-100, 2.5 mM EDTA, 5 mM DTT, 5% glycerol and Complete proteinase inhibitor [Promega]). 1.5 mg Protein G conjugated Dynabeads (Invitrogen) were used to capture 10 µl mouse monoclonal anti-HA antibody (HA-7 ascites fluid, Sigma) at room temperature for 30 min, and were then incubated with cell lysates for 30 min at room temperature. The immunoprecipitates were then washed three times with ice-cold lysis buffer and subjected to Western blotting analysis.
Supporting Information
Synaptic markers remain intact in hiw mutants after injury. Representative muscle 4 NMJs for WT (Canton S) or hiw (hiwΔN) mutants stained in (A) for Futsch (green), Dvglut (synaptic vesicles, red), and HRP (neuronal membrane, blue). In (B) NMJs are stained for GluRIII (post-synaptic GluR receptor subunit [49], green), Brp (pre-synaptic active zones, red), and HRP (axonal membrane, blue), before or 24 h after injury. While hiw mutants have reduced Dvglut staining ([12] and A) and smaller synaptic Brp and GluRIII puncta (B), there is no noticeable difference between the uninjured and injured NMJs. Scale bars = 12.5 µm.
(TIF)
Endogenous Nmnat in Drosophila motoneurons. Depletion of Nmnat in larval motoneurons by expression of nmnat-RNAi. Expression of UAS-nmnat-RNAi with a pan-neuronal Gal4 driver (BG380-Gal4, UAS-Dcr2; UAS-nmnat-RNAi/+) depletes Nmnat staining (green) in neuronal nuclei (marked by co-staining with Elav, red) but not in neighboring glial cells (for which Nmnat staining increased). Quantification of the reduced staining in neuronal nuclei suggested that the Nmnat levels were reduced to 49.3% of wild-type levels in motoneurons (p<0.01, n = 6.7). (A) Depletion of nmnat by expression of nmnat-RNAi in neurons does not affect NMJ morphology. Representative muscle 4 NMJs stained for Futsch (green), Dvglut (synaptic vesicles, red), and HRP (neuronal membrane, blue). We did not observe spontaneous axonal or synaptic degeneration when nmnat was depleted by RNAi, probably because the depletion was not complete. (B) In hiw (hiw ΔN ) mutants, endogenous Nmnat (green) is reduced in neuronal nuclei. Similarly to the nmnat RNAi knockdown in (A), Nmnat staining increases in neighboring glial cells. (C) Injury signaling via Wnd may down-regulate nuclear Nmnat. Nmnat protein disappears from neuronal nuclei and appears in neighboring glial cells 24 h after injury. A similar change occurs in hiw mutants (C), and when Wnd is overexpressed in neurons (BG380-Gal4; UAS-wnd/+). Conversely, wnd loss-of-function mutants (wnd-1/wnd-2) have increased levels of nuclear Nmnat. Because Wnd becomes activated by axonal injury, we expect that these changes in nuclear Nmnat are mediated by a common mechanism. The functional relevance of these changes is not yet clear. (D) Quantification of average nuclear Nmnat intensity, normalized for wild type, for experiments in (D). Scale bars = 12.5 µm.
(TIF)
Inhibition of degeneration by UBP2 requires Nmnat function. (A) m12-Gal4, UAS-mCD8::GFP labeled single axons (green) 24 h after injury in animals co-expressing UBP2 with a control UAS line (UAS-Dcr2; UAS-UBP2/+; UAS-nls::DsRed-(Ctrl)/m12-Gal4, UAS-mCD8::GFP) or co-expressing UBP2 when with UAS-nmnat RNAi to reduce endogenous Nmnat (UAS-Dcr2; UAS-UBP2/UAS-nmnatRNAi; m12-Gal4, UAS-mCD8::GFP/+). (B) Degeneration index for the m12-Gal4, UAS-mCD8::GFP labeled axons for genotypes in (A). Scale bars = 12.5 µm; error bars represent standard error; ***p<0.001 in t-test.
(TIF)
Hiw down-regulates Nmnat independently of the UPS. (A) The decrease in Nmnat levels promoted by Hiw induction in S2R+ cells was not diminished when the proteasome was inhibited. S2R+ cells co-transfected with pUAST-HA::Nmnat and pUAST-GFP or pUAST-GFP::Hiw, then were incubated with DMSO vehicle or 5 µM MG132 for 20 h. The levels of HA-Nmnat are compared by Western blotting for the HA epitope. Relative levels compared to the β-tubulin standard were measured on the LiCor Odyssey system. Similar results were observed for additional concentrations and time points (25 µM MG132 for 6 h, and 5 µM MG132 for 12 h, unpublished data). (B) Inhibition of the proteasome in sensory neurons only modestly changes HA-Nmnat levels. Average HA-Nmnat intensity in ppk sensory neuron axon terminals was compared between wild-type (UAS-HA::nmnat/+; ppk-Gal4,UAS-mCD8::RFP/+), and animals co-expressing DTS5 and DTS7 to inhibit the proteasome (UAS-HA::nmnat/UAS-DTS5; ppk-Gal4,UAS-mCD8::RFP/UAS-DTS7). Data are shown for two conditions: flies raised continuously at 25°C, and flies raised at 25°C, then shifted to 30°C for 2 d. Error bars represent standard error. *p<0.05. (C) Hiw and the UPS may influence Nmnat levels cooperatively. Total protein from third instar larval brains or young adult heads processed for Western blot from animals co-expressing DTS5 and DTS7 [45] to inhibit the proteasome (raised at 25°C), and compared to animals co-expressing two control UAS-PKC transgenes. Relative levels of HA-Nmnat protein, compared to the β-catenin standard, were measured on the LiCor Odyssey system. Combination of the hiw mutation with inhibition of the proteasome leads to much higher levels of HA-Nmnat, suggesting that Hiw and the UPS may potentially influence HA-Nmnat independently, rather than through the same pathway. Error bars represent standard error; *p<0.05 in t-test.
(TIF)
Changes in HA-Nmnat protein level in proximal and distal stumps, contrasted with GFP-Hiw after injury. (A) Quantification of average HA-Nmnat intensity in motoneurons axons for wild-type (OK6-Gal4/UAS-HA::nmnat) or hiw mutants (hiwΔN;OK6-Gal4/UAS-HA::nmnat) before (black) or 8 h after (gray) injury. HA-Nmnat levels in both proximal and distal axons were quantified and normalized to the average HA-Nmnat intensity in uninjured WT animals as described in Materials and Methods. Injury induces an increase of HA-Nmnat in the proximal stump in both wild-type and hiw mutant backgrounds. However, in the distal stump, the levels of HA-Nmnat reduced by 60.7% within 8 h in WT animals, but remained constant in hiw mutants. (B) Quantification of average HA-Nmnat and GFP-Hiw intensity in axon terminals of ppk-Gal4,UAS-mCD8::RFP labeled sensory neurons before or 4 h after injury. Error bars represent standard error; **p<0.01; ***p<0.001; ns, not significant, p>0.05 in t-test.
(TIF)
Acknowledgments
We thank Grace Zhai, Hugo Bellen, Liqun Luo, and Marc Freeman for generous gifts of reagents, and, the Bloomington Stock Center (Indiana University), Developmental Studies Hybridoma bank (University of Iowa) and the Vienna Drosophila RNAi center for Drosophila resources. We thank Jennifer Diep, Travis Washington, Leni Truong, Emily Han, and Nicolette Ognjanovski for technical assistance.
Abbreviations
- DLK
dileucine zipper kinase
- EJP
excitatory junction potential
- GFP
green fluorescent protein
- Hiw
Highwire
- NMJ
neuromuscular junction
- Nmnat
nicotinamide mononucleotide adenyltransferase
- O/E
overexpression
- UPS
ubiquitin proteasome system
- Wnd
Wallenda
Funding Statement
This work was supported by grants NS069844 (to CAC) and NS07092 (to CW) from the National Institutes of Health (http://www.ninds.nih.gov/). XX was the recipient of a Rackham Predoctoral Fellowship at the University of Michigan. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript
References
- 1. Waller A (1850) Experiments on the section of the glossopharyngeal and hypoglossal nerves of the frog, and observations of the alterations produced thereby in the structure of their primitive fibres. Philos Trans R Soc Lond 140: 423–429. [PMC free article] [PubMed] [Google Scholar]
- 2. Hoopfer ED, McLaughlin T, Watts RJ, Schuldiner O, O'Leary DD, et al. (2006) Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron 50: 883–895. [DOI] [PubMed] [Google Scholar]
- 3. Zhai Q, Wang J, Kim A, Liu Q, Watts R, et al. (2003) Involvement of the ubiquitin-proteasome system in the early stages of wallerian degeneration. Neuron 39: 217–225. [DOI] [PubMed] [Google Scholar]
- 4. D'Souza J, Hendricks M, Le Guyader S, Subburaju S, Grunewald B, et al. (2005) Formation of the retinotectal projection requires Esrom, an ortholog of PAM (protein associated with Myc). Development 132: 247–256. [DOI] [PubMed] [Google Scholar]
- 5. Schaefer AM, Hadwiger GD, Nonet ML (2000) rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron 26: 345–356. [DOI] [PubMed] [Google Scholar]
- 6. Burgess RW, Peterson KA, Johnson MJ, Roix JJ, Welsh IC, et al. (2004) Evidence for a conserved function in synapse formation reveals Phr1 as a candidate gene for respiratory failure in newborn mice. Mol Cell Biol 24: 1096–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhen M, Huang X, Bamber B, Jin Y (2000) Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron 26: 331–343. [DOI] [PubMed] [Google Scholar]
- 8. Wan HI, DiAntonio A, Fetter RD, Bergstrom K, Strauss R, et al. (2000) Highwire regulates synaptic growth in Drosophila. Neuron 26: 313–329. [DOI] [PubMed] [Google Scholar]
- 9. Lewcock JW, Genoud N, Lettieri K, Pfaff SL (2007) The ubiquitin ligase Phr1 regulates axon outgrowth through modulation of microtubule dynamics. Neuron 56: 604–620. [DOI] [PubMed] [Google Scholar]
- 10. Bloom AJ, Miller BR, Sanes JR, DiAntonio A (2007) The requirement for Phr1 in CNS axon tract formation reveals the corticostriatal boundary as a choice point for cortical axons. Genes Dev 21: 2593–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Po MD, Hwang C, Zhen M (2010) PHRs: bridging axon guidance, outgrowth and synapse development. Curr Opin Neurobiol 20: 100–107. [DOI] [PubMed] [Google Scholar]
- 12. Hendricks M, Jesuthasan S (2009) PHR regulates growth cone pausing at intermediate targets through microtubule disassembly. J Neurosci 29: 6593–6598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Osterloh JM, Yang J, Rooney TM, Fox AN, Adalbert R, et al. (2012) dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science 337: 481–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wakatsuki S, Saitoh F, Araki T (2011) ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat Cell Biol 13: 1415–1423. [DOI] [PubMed] [Google Scholar]
- 15. Massaro CM, Pielage J, Davis GW (2009) Molecular mechanisms that enhance synapse stability despite persistent disruption of the spectrin/ankyrin/microtubule cytoskeleton. J Cell Biol 187: 101–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Collins CA, Wairkar YP, Johnson SL, DiAntonio A (2006) Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron 51: 57–69. [DOI] [PubMed] [Google Scholar]
- 17. Nakata K, Abrams B, Grill B, Goncharov A, Huang X, et al. (2005) Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell 120: 407–420. [DOI] [PubMed] [Google Scholar]
- 18. Miller BR, Press C, Daniels RW, Sasaki Y, Milbrandt J, et al. (2009) A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nat Neurosci 12: 387–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fang Y, Bonini NM (2012) Axon degeneration and: insights from Drosophila drosophila models of nerve injury. Annu Rev Cell Dev Biol 28: 575–597. [DOI] [PubMed] [Google Scholar]
- 20. Xiong X, Collins CA (2012) A conditioning lesion protects axons from degeneration via the Wallenda/DLK MAPkinase signaling cascade. J Neurosci 32: 610–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gilley J, Coleman MP (2010) Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol 8: e1000300 doi:10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang JT, Medress ZA, Barres BA (2012) Axon degeneration: molecular mechanisms of a self-destruction pathway. J Cell Biol 196: 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Coleman MP, Freeman MR (2010) Wallerian degeneration, wld(s), and nmnat. Annu Rev Neurosci 33: 245–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, et al. (2001) Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci 4: 1199–1206. [DOI] [PubMed] [Google Scholar]
- 25. Zhai RG, Cao Y, Hiesinger PR, Zhou Y, Mehta SQ, et al. (2006) Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol 4: e416 doi:10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wen Y, Parrish JZ, He R, Zhai RG, Kim MD (2011) Nmnat exerts neuroprotective effects in dendrites and axons. Mol Cell Neurosci 48: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fang Y, Soares L, Teng X, Geary M, Bonini NM (2012) A novel Drosophila model of nerve injury reveals an essential role of nmnat in maintaining axonal integrity. Curr Biol 22: 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hammarlund M, Nix P, Hauth L, Jorgensen EM, Bastiani M (2009) Axon regeneration requires a conserved MAP kinase pathway. Science 323: 802–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yan D, Wu Z, Chisholm AD, Jin Y (2009) The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell 138: 1005–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xiong X, Wang X, Ewanek R, Bhat P, Diantonio A, et al. (2010) Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. J Cell Biol 191: 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Itoh A, Horiuchi M, Bannerman P, Pleasure D, Itoh T (2009) Impaired regenerative response of primary sensory neurons in ZPK/DLK gene-trap mice. Biochem Biophys Res Commun 383: 258–262. [DOI] [PubMed] [Google Scholar]
- 32. Shin JE, Cho Y, Beirowski B, Milbrandt J, Cavalli V, et al. (2012) Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron 74: 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han S, Witt RM, Santos TM, Polizzano C, Sabatini BL, et al. (2008) Pam (Protein associated with Myc) functions as an E3 ubiquitin ligase and regulates TSC/mTOR signaling. Cell Signal 20: 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Baker RT, Tobias JW, Varshavsky A (1992) Ubiquitin-specific proteases of Saccharomyces cerevisiae. Cloning of UBP2 and UBP3, and functional analysis of the UBP gene family. J Biol Chem 267: 23364–23375. [PubMed] [Google Scholar]
- 35. MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, et al. (2006) The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron 50: 869–881. [DOI] [PubMed] [Google Scholar]
- 36. Lunn ER, Perry VH, Brown MC, Rosen H, Gordon S (1989) Absence of Wallerian Degeneration does not Hinder Regeneration in Peripheral Nerve. Eur J Neurosci 1: 27–33. [DOI] [PubMed] [Google Scholar]
- 37. Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, et al. (2007) A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 448: 151–156. [DOI] [PubMed] [Google Scholar]
- 38. Wu C, Daniels RW, DiAntonio A (2007) DFsn collaborates with Highwire to down-regulate the Wallenda/DLK kinase and restrain synaptic terminal growth. Neural Dev 2: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liao EH, Hung W, Abrams B, Zhen M (2004) An SCF-like ubiquitin ligase complex that controls presynaptic differentiation. Nature 430: 345–350. [DOI] [PubMed] [Google Scholar]
- 40. Saiga T, Fukuda T, Matsumoto M, Tada H, Okano HJ, et al. (2009) Fbxo45 forms a novel ubiquitin ligase complex and is required for neuronal development. Mol Cell Biol 29: 3529–3543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. DiAntonio A, Haghighi AP, Portman SL, Lee JD, Amaranto AM, et al. (2001) Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature 412: 449–452. [DOI] [PubMed] [Google Scholar]
- 42. Kee Y, Lyon N, Huibregtse JM (2005) The Rsp5 ubiquitin ligase is coupled to and antagonized by the Ubp2 deubiquitinating enzyme. EMBO J 24: 2414–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Swarup S, Verheyen EM (2011) Drosophila homeodomain-interacting protein kinase inhibits the Skp1-Cul1-F-box E3 ligase complex to dually promote Wingless and Hedgehog signaling. Proc Natl Acad Sci U S A 108: 9887–9892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wojcik C, Yano M, DeMartino GN (2004) RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome-dependent proteolysis. J Cell Science 117: 281–292. [DOI] [PubMed] [Google Scholar]
- 45. Belote JM, Fortier E (2002) Targeted expression of dominant negative proteasome mutants in Drosophila melanogaster. Genesis 34: 80–82. [DOI] [PubMed] [Google Scholar]
- 46. Speese SD, Trotta N, Rodesch CK, Aravamudan B, Broadie K (2003) The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Curr Biol 13: 899–910. [DOI] [PubMed] [Google Scholar]
- 47. Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, et al. (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447: 859–863. [DOI] [PubMed] [Google Scholar]
- 48. Berger F, Lau C, Dahlmann M, Ziegler M (2005) Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem 280: 36334–36341. [DOI] [PubMed] [Google Scholar]
- 49. Zhang X, Kurnasov OV, Karthikeyan S, Grishin NV, Osterman AL, et al. (2003) Structural characterization of a human cytosolic NMN/NaMN adenylyltransferase and implication in human NAD biosynthesis. J Biol Chem 278: 13503–13511. [DOI] [PubMed] [Google Scholar]
- 50. Mayer PR, Huang N, Dewey CM, Dries DR, Zhang H, et al. (2010) Expression, localization, and biochemical characterization of nicotinamide mononucleotide adenylyltransferase 2. J Biol Chem 285: 40387–40396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Avery MA, Rooney TM, Pandya JD, Wishart TM, Gillingwater TH, et al. (2012) WldS prevents axon degeneration through increased mitochondrial flux and enhanced mitochondrial Ca2+ buffering. Curr Biol 22: 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Avery MA, Sheehan AE, Kerr KS, Wang J, Freeman MR (2009) Wld S requires Nmnat1 enzymatic activity and N16-VCP interactions to suppress Wallerian degeneration. J Cell Biol 184: 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abrams B, Grill B, Huang X, Jin Y (2008) Cellular and molecular determinants targeting the Caenorhabditis elegans PHR protein RPM-1 to perisynaptic regions. Dev Dyn 237: 630–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kee Y, Munoz W, Lyon N, Huibregtse JM (2006) The deubiquitinating enzyme Ubp2 modulates Rsp5-dependent Lys63-linked polyubiquitin conjugates in Saccharomyces cerevisiae. J Biol Chem 281: 36724–36731. [DOI] [PubMed] [Google Scholar]
- 55. Bergink S, Jentsch S (2009) Principles of ubiquitin and SUMO modifications in DNA repair. Nature 458: 461–467. [DOI] [PubMed] [Google Scholar]
- 56. Deng L, Wang C, Spencer E, Yang L, Braun A, et al. (2000) Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103: 351–361. [DOI] [PubMed] [Google Scholar]
- 57. Galan JM, Haguenauer-Tsapis R (1997) Ubiquitin lys63 is involved in ubiquitination of a yeast plasma membrane protein. EMBO J 16: 5847–5854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Soetens O, De Craene JO, Andre B (2001) Ubiquitin is required for sorting to the vacuole of the yeast general amino acid permease, Gap1. J Biol Chem 276: 43949–43957. [DOI] [PubMed] [Google Scholar]
- 59. Yahata N, Yuasa S, Araki T (2009) Nicotinamide mononucleotide adenylyltransferase expression in mitochondrial matrix delays Wallerian degeneration. J Neurosci 29: 6276–6284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ljungberg MC, Ali YO, Zhu J, Wu CS, Oka K, et al. (2012) CREB-activity and nmnat2 transcription are down-regulated prior to neurodegeneration, while NMNAT2 over-expression is neuroprotective, in a mouse model of human tauopathy. Hum Mol Genet 21: 251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Feng Y, Yan T, Zheng J, Ge X, Mu Y, et al. (2010) Overexpression of Wld(S) or Nmnat2 in mauthner cells by single-cell electroporation delays axon degeneration in live zebrafish. J Neurosci Res 88: 3319–3327. [DOI] [PubMed] [Google Scholar]
- 62. Ali YO, Ruan K, Zhai RG (2012) NMNAT suppresses tau-induced neurodegeneration by promoting clearance of hyperphosphorylated tau oligomers in a Drosophila model of tauopathy. Hum Mol Genet 21: 237–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhai RG, Zhang F, Hiesinger PR, Cao Y, Haueter CM, et al. (2008) NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature 452: 887–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Martin SM, O'Brien GS, Portera-Cailliau C, Sagasti A (2010) Wallerian degeneration of zebrafish trigeminal axons in the skin is required for regeneration and developmental pruning. Development 137: 3985–3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bisby MA, Chen S (1990) Delayed wallerian degeneration in sciatic nerves of C57BL/Ola mice is associated with impaired regeneration of sensory axons. Brain Res 530: 117–120. [DOI] [PubMed] [Google Scholar]
- 66. Brown MC, Lunn ER, Perry VH (1992) Consequences of slow Wallerian degeneration for regenerating motor and sensory axons. J Neurobiol 23: 521–536. [DOI] [PubMed] [Google Scholar]
- 67. Wu C, Wairkar YP, Collins CA, DiAntonio A (2005) Highwire function at the Drosophila neuromuscular junction: spatial, structural, and temporal requirements. J Neurosci 25: 9557–9566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. McCabe BD, Hom S, Aberle H, Fetter RD, Marques G, et al. (2004) Highwire regulates presynaptic BMP signaling essential for synaptic growth. Neuron 41: 891–905. [DOI] [PubMed] [Google Scholar]
- 69. Budnik LT, Mukhopadhyay AK (1996) Modulatory action of epidermal growth factor on differentiated human granulosa lutein cells: cross-talk between ligand activated receptors for EGF and gonadotropin. Mol Cell Endocrinol 124: 141–150. [DOI] [PubMed] [Google Scholar]
- 70. Ritzenthaler S, Suzuki E, Chiba A (2000) Postsynaptic filopodia in muscle cells interact with innervating motoneuron axons. Nat Neurosci 3: 1012–1017. [DOI] [PubMed] [Google Scholar]
- 71. Kuo CT, Jan LY, Jan YN (2005) Dendrite-specific remodeling of Drosophila sensory neurons requires matrix metalloproteases, ubiquitin-proteasome, and ecdysone signaling. Proc Natl Acad Sci U S A 102: 15230–15235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vosshall LB, Wong AM, Axel R (2000) An olfactory sensory map in the fly brain. Cell 102: 147–159. [DOI] [PubMed] [Google Scholar]
- 73. Marrus SB, Portman SL, Allen MJ, Moffat KG, DiAntonio A (2004) Differential localization of glutamate receptor subunits at the Drosophila neuromuscular junction. J Neurosci 24: 1406–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Daniels RW, Collins CA, Gelfand MV, Dant J, Brooks ES, et al. (2004) Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci 24: 10466–10474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Stewart BA, Atwood HL, Renger JJ, Wang J, Wu CF (1994) Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol A 175: 179–191. [DOI] [PubMed] [Google Scholar]
- 76. Klueg KM, Alvarado D, Muskavitch MA, Duffy JB (2002) Creation of a GAL4/UAS-coupled inducible gene expression system for use in Drosophila cultured cell lines. Genesis 34: 119–122. [DOI] [PubMed] [Google Scholar]
- 77. Parker L, Gross S, Alphey L (2001) Vectors for the expression of tagged proteins in Drosophila. Bio Techniques 31: 1280–1282, 1284, 1286. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Synaptic markers remain intact in hiw mutants after injury. Representative muscle 4 NMJs for WT (Canton S) or hiw (hiwΔN) mutants stained in (A) for Futsch (green), Dvglut (synaptic vesicles, red), and HRP (neuronal membrane, blue). In (B) NMJs are stained for GluRIII (post-synaptic GluR receptor subunit [49], green), Brp (pre-synaptic active zones, red), and HRP (axonal membrane, blue), before or 24 h after injury. While hiw mutants have reduced Dvglut staining ([12] and A) and smaller synaptic Brp and GluRIII puncta (B), there is no noticeable difference between the uninjured and injured NMJs. Scale bars = 12.5 µm.
(TIF)
Endogenous Nmnat in Drosophila motoneurons. Depletion of Nmnat in larval motoneurons by expression of nmnat-RNAi. Expression of UAS-nmnat-RNAi with a pan-neuronal Gal4 driver (BG380-Gal4, UAS-Dcr2; UAS-nmnat-RNAi/+) depletes Nmnat staining (green) in neuronal nuclei (marked by co-staining with Elav, red) but not in neighboring glial cells (for which Nmnat staining increased). Quantification of the reduced staining in neuronal nuclei suggested that the Nmnat levels were reduced to 49.3% of wild-type levels in motoneurons (p<0.01, n = 6.7). (A) Depletion of nmnat by expression of nmnat-RNAi in neurons does not affect NMJ morphology. Representative muscle 4 NMJs stained for Futsch (green), Dvglut (synaptic vesicles, red), and HRP (neuronal membrane, blue). We did not observe spontaneous axonal or synaptic degeneration when nmnat was depleted by RNAi, probably because the depletion was not complete. (B) In hiw (hiw ΔN ) mutants, endogenous Nmnat (green) is reduced in neuronal nuclei. Similarly to the nmnat RNAi knockdown in (A), Nmnat staining increases in neighboring glial cells. (C) Injury signaling via Wnd may down-regulate nuclear Nmnat. Nmnat protein disappears from neuronal nuclei and appears in neighboring glial cells 24 h after injury. A similar change occurs in hiw mutants (C), and when Wnd is overexpressed in neurons (BG380-Gal4; UAS-wnd/+). Conversely, wnd loss-of-function mutants (wnd-1/wnd-2) have increased levels of nuclear Nmnat. Because Wnd becomes activated by axonal injury, we expect that these changes in nuclear Nmnat are mediated by a common mechanism. The functional relevance of these changes is not yet clear. (D) Quantification of average nuclear Nmnat intensity, normalized for wild type, for experiments in (D). Scale bars = 12.5 µm.
(TIF)
Inhibition of degeneration by UBP2 requires Nmnat function. (A) m12-Gal4, UAS-mCD8::GFP labeled single axons (green) 24 h after injury in animals co-expressing UBP2 with a control UAS line (UAS-Dcr2; UAS-UBP2/+; UAS-nls::DsRed-(Ctrl)/m12-Gal4, UAS-mCD8::GFP) or co-expressing UBP2 when with UAS-nmnat RNAi to reduce endogenous Nmnat (UAS-Dcr2; UAS-UBP2/UAS-nmnatRNAi; m12-Gal4, UAS-mCD8::GFP/+). (B) Degeneration index for the m12-Gal4, UAS-mCD8::GFP labeled axons for genotypes in (A). Scale bars = 12.5 µm; error bars represent standard error; ***p<0.001 in t-test.
(TIF)
Hiw down-regulates Nmnat independently of the UPS. (A) The decrease in Nmnat levels promoted by Hiw induction in S2R+ cells was not diminished when the proteasome was inhibited. S2R+ cells co-transfected with pUAST-HA::Nmnat and pUAST-GFP or pUAST-GFP::Hiw, then were incubated with DMSO vehicle or 5 µM MG132 for 20 h. The levels of HA-Nmnat are compared by Western blotting for the HA epitope. Relative levels compared to the β-tubulin standard were measured on the LiCor Odyssey system. Similar results were observed for additional concentrations and time points (25 µM MG132 for 6 h, and 5 µM MG132 for 12 h, unpublished data). (B) Inhibition of the proteasome in sensory neurons only modestly changes HA-Nmnat levels. Average HA-Nmnat intensity in ppk sensory neuron axon terminals was compared between wild-type (UAS-HA::nmnat/+; ppk-Gal4,UAS-mCD8::RFP/+), and animals co-expressing DTS5 and DTS7 to inhibit the proteasome (UAS-HA::nmnat/UAS-DTS5; ppk-Gal4,UAS-mCD8::RFP/UAS-DTS7). Data are shown for two conditions: flies raised continuously at 25°C, and flies raised at 25°C, then shifted to 30°C for 2 d. Error bars represent standard error. *p<0.05. (C) Hiw and the UPS may influence Nmnat levels cooperatively. Total protein from third instar larval brains or young adult heads processed for Western blot from animals co-expressing DTS5 and DTS7 [45] to inhibit the proteasome (raised at 25°C), and compared to animals co-expressing two control UAS-PKC transgenes. Relative levels of HA-Nmnat protein, compared to the β-catenin standard, were measured on the LiCor Odyssey system. Combination of the hiw mutation with inhibition of the proteasome leads to much higher levels of HA-Nmnat, suggesting that Hiw and the UPS may potentially influence HA-Nmnat independently, rather than through the same pathway. Error bars represent standard error; *p<0.05 in t-test.
(TIF)
Changes in HA-Nmnat protein level in proximal and distal stumps, contrasted with GFP-Hiw after injury. (A) Quantification of average HA-Nmnat intensity in motoneurons axons for wild-type (OK6-Gal4/UAS-HA::nmnat) or hiw mutants (hiwΔN;OK6-Gal4/UAS-HA::nmnat) before (black) or 8 h after (gray) injury. HA-Nmnat levels in both proximal and distal axons were quantified and normalized to the average HA-Nmnat intensity in uninjured WT animals as described in Materials and Methods. Injury induces an increase of HA-Nmnat in the proximal stump in both wild-type and hiw mutant backgrounds. However, in the distal stump, the levels of HA-Nmnat reduced by 60.7% within 8 h in WT animals, but remained constant in hiw mutants. (B) Quantification of average HA-Nmnat and GFP-Hiw intensity in axon terminals of ppk-Gal4,UAS-mCD8::RFP labeled sensory neurons before or 4 h after injury. Error bars represent standard error; **p<0.01; ***p<0.001; ns, not significant, p>0.05 in t-test.
(TIF)