

Abstract
Introduction
Dystonia is a neurological disorder associated with twisting motions and abnormal postures, which compromise normal movements and can be both painful and debilitating. It can affect a single body part (focal), several contiguous regions (segmental), or the entire body (generalized), and can arise as a result of numerous causes, both genetic and acquired. Despite the diversity of causes and manifestations, shared clinical features suggest that common mechanisms of pathogenesis may underlie many dystonias.
Areas Covered
This review identifies shared themes in etiologically-diverse dystonias on several biological levels. At the cellular level, abnormalities in the dopaminergic system, mitochondrial function, and calcium regulation are discussed. At the anatomical level, the roles of the basal ganglia and the cerebellum in dystonia are described. Global central nervous system dysfunction, with regard to aberrant neuronal plasticity, inhibition, and sensorimotor integration is also discussed. Using clinical data and data from animal models, this article seeks to highlight shared pathways that may be critical in understanding mechanisms and identifying novel therapeutic strategies in dystonia.
Expert Opinion
Identifying shared features of pathogenesis can provide insight into the biological processes that underlie etiologically-diverse dystonias, and can suggest novel targets for therapeutic intervention that may be effective in a broad group of affected individuals.
Keywords: dystonia, dopamine, mitochondria, calcium, basal ganglia, cerebellum, plasticity
1. Introduction
Dystonia is a neurological disorder characterized by involuntary muscle contractions that cause debilitating twisting movements and postures. The causes of dystonia are heterogeneous. Dystonia is classified as primary (idiopathic) when it occurs in the absence of other features, or secondary, when it occurs in response to an identifiable cause such as trauma or disease. In addition to etiological heterogeneity, dystonia can be categorized according to the scope of impairment: focal dystonias affect isolated regions of the body, segmental dystonias involve two or more contiguous regions and generalized dystonias involve broader regions, often including the trunk.
While these distinctions are useful clinically, the estimated prevalence of each individual type of dystonia is modest, leading to the perception that dystonia is a rare or “orphan” disorder. However the similarities that define the clinical diagnosis of dystonia suggest that common mechanisms may exist. It is therefore possible that the pathology underlying the spectrum of individual dystonias may ultimately be attributed to abnormalities in a few shared pathways. From a treatment perspective, the identification of shared features of pathogenesis may promote the development of broadly effective therapies. This approach has been used successfully in a number of etiologically diverse disorders. Chemotherapeutics, for example, are based on the understanding that all cancers are characterized at the most basic level by rapidly dividing cells. Administering pharmacologic agents that interrupt the cell cycle is therefore effective across a broad range of cancers, regardless of the diverse molecular etiologies. Similarly, while the causes and manifestations of epilepsy are diverse and in many cases unknown, current treatments targeted to presumed common pathways are effective in a broad range of patients.
Unlike other movement disorders, dystonia is not consistently associated with anatomical markers or obvious neurodegeneration. For this reason, the information that can be attained from studies in human patients is limited. Animal models of dystonia are therefore critical in determining the underlying pathogenesis. In the past decade, a number of animal models have become available which can be generally divided into two categories: etiologic or phenotypic. Animal models that mimic the inciting event or genetic predisposition (etiologic models) are useful for tracing the progression of downstream consequences that ultimately produce dystonia. In contrast, animal models that exhibit motor syndromes comparable to those seen in the human disorder (phenotypic models) can be used to work backwards through the pathophysiology. This review incorporates data from human subjects and data from animal models, to identify shared pathways that may be critical for understanding mechanisms and therapeutic strategies.
2. Biological Levels of Dysfunction
2.1 Cellular
2.1.1 Dopamine signaling
Abnormalities in the brain dopamine (DA) system are observed across etiologically diverse dystonias, suggesting DA dysfunction may be critical in the pathophysiology of dystonia [1]. Consistent with this idea, dystonia has been reported in Parkinson's disease (PD) [2,3], a disorder associated with frank degeneration of nigrostriatal DA neurons. Though relatively uncommon, focal dystonias have been reported in several cases as the presenting symptom prior to the onset of PD [3-5]. This is particularly true in the case of young onset PD, in which foot dystonia is typical [2,6]. More commonly, dystonia arises in advanced stages of PD as a complication of treatment with L-DOPA, the precursor to dopamine that is prescribed to increase brain dopamine concentrations [7,8]. In these patients, dystonia occurs when L-DOPA reaches a minimum threshold (off-period dystonia) [9,10]. That dystonia can arise as a result of daily fluctuations in DA agents suggests an acute mechanism of dysfunction. However, because dystonia occurs only after chronic L-DOPA treatment, long-term adaptations in the DA system may be involved as well. Consistent with this idea, drug-induced (tardive) dystonia can occur after either acute or chronic treatment with DA antagonists in patients suffering from psychiatric or gastrointestinal conditions [11,12]. In these individuals, dystonia can persist even after neuroleptic therapy is terminated [11,13]. Together, these data implicate both acute and long-term changes in DA function in dystonia.
Dystonia also occurs in a number of developmental disorders that are associated with DA dysfunction. Segawa disease, or DOPA-responsive dystonia (DRD) [14-20] is characterized by childhood-onset generalized dystonia, in which symptoms are mild in the morning and worsen throughout the course of the day (diurnal fluctuation) [21,22]. This disorder is defined by the dramatic improvement in symptoms upon administration of low doses of L-DOPA. Mutations in genes associated with DA synthesis underlie the majority of cases of DRD (Figure 1). The most common form of DRD is caused by mutations in GTP cyclohydrolase (GCH), the rate-limiting enzyme in tetrahydrobiopterin (BH4) synthesis [15,20]. Tetrahydrobiopterin is an obligatory co-factor for several enzymes, including tyrosine hydroxylase (TH), the rate-limiting enzyme in catecholamine synthesis. Mutations that result in DRD reduce GCH enzyme activity to 2-20% of normal [23], which in turn reduces TH activity, ultimately leading to nigrostriatal DA concentrations of <20% of normal values [15,23]. Mutations in other genes encoding enzymes critical in BH4 synthesis such as 6-pyruvoyltetrahydropterin synthase (PTPS) [16,20] and sepiapterin reductase (SPD) [14,19] have comparable results, reducing, but not abolishing TH activity. Mutations in TH itself also cause DRD. Here again TH activity is reduced to 2-20%, resulting in reduced but not abolished DA concentration. [17,18,24]. Overall, it appears that a significant reduction in dopamine during development, as opposed to a total loss, is sufficient to produce dystonia.
Figure 1. Mutations in the dopamine synthesis pathway associated with Dopa-Responsive Dystonia.
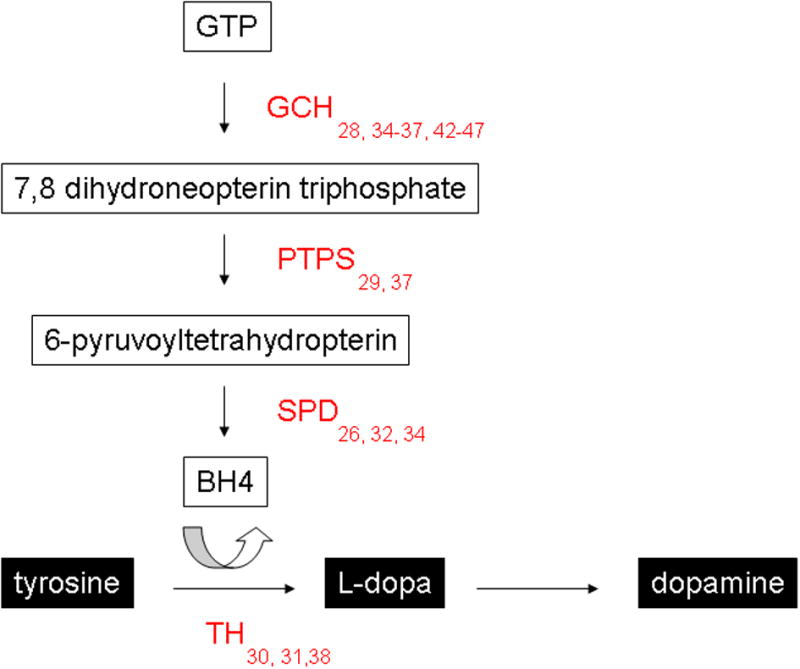
(GCH = GTP cyclohydrolase; PTPS = 6-pyruvoyl-tetrahydropterin synthase; SPD = sepiapterin reductase; TH = tyrosine hydroxylase)
Dystonia is also a prominent feature of Lesch-Nyhan disease (LND), a developmental syndrome that is also characterized by self-injurious behaviors [25,26]. LND is caused by mutations in hypoxanthine-guanine phosphoribosyl transferase (HPRT), which encodes the HPRT enzyme involved in the purine salvage pathway. In addition to HPRT hypofunction or complete deficiency, LND is associated with 70-90% loss of DA in the basal ganglia, as determined on autopsy [27,28]. As in DRD, DA is significantly reduced, but not abolished, and is not associated with cell death or degeneration. Studies in HPRT-deficient mice have demonstrated that this effect is restricted to the DA system, as concentrations of other neurotransmitters, including norepinephrine, serotonin and gamma aminobutyric acid (GABA) are comparable to controls [29]. The early-onset of LND, combined with the lack of overt cell death or degeneration, suggests that abnormalities in the functional development of the brain DA system may underlie this disorder. Data from a recent study suggests a mechanism whereby HPRT may directly affect the development of the DA system. HPRT-deficient cell lines express higher levels of engrailed 1 and 2, transcription factors that are critical in DA neuron differentiation and survival [30]. This was accompanied by a reduction in neurite outgrowth. This phenomenon was also shown in primary fibroblasts from LND patients, and correlated with disease severity. Together, these data suggest that HPRT deficiency may affect the initial wiring of the brain DA system during early development, which may ultimately result in the dystonic component of LND.
Functional changes in dopaminergic neurotransmission have also been demonstrated in other forms of dystonia. For example, in a patient suffering from paroxysmal exercise-induced dystonia cerebrospinal fluid levels of the DA metabolite homovanillic acid (HVA) were elevated twofold following an attack [31]. Data from the dystonic (sz) hamster, which exhibits paroxysmal dystonia, is consistent with this finding, whereby DA and its metabolites are specifically elevated in the striatum during stress-induced dystonic attacks [32]. In contrast, in DYT1 mutant mice [33,34], which express a mutation in human torsinA, a protein that is implicated in early-onset torsion dystonia (DYT1 dystonia), extracellular dopamine concentrations are markedly reduced in response to amphetamine suggesting impaired dopamine release or reuptake mechanisms. Likewise, extracellular dopamine is reduced during dystonic attacks in the tottering mouse model of generalized dystonia and after pharmacologically inducing generalized dystonia in normal mice by applying glutamate agonists to the cerebellum [35].
Reduced striatal D2 receptor binding is also frequently observed in dystonia and has been demonstrated in a number of human idiopathic dystonias, including blepharospasm [36], rotational torticollis [37], and cervical dystonia [38]. Imaging studies in individuals suffering from spinocerebellar ataxia type 2 (SCA2), in which head and neck dystonia were prominent reveal significant reductions in D2 dopamine receptor binding compared to control subjects [39]. A significant reduction in striatal D2 receptor availability has also been observed in DYT1 dystonia patients [38]. Experiments using animal models of dystonia have provided additional insight into the role of D2 receptors in the pathology of dystonia. Consistent with the results of imaging D2 dopamine receptors in DYT1 patients, functional changes in striatal D2 receptors have also been found in DYT1 mutant mice (hMT) [33,34], which express a mutation in human torsinA. Under physiologic conditions, D2 receptors act presynaptically to modulate DA release and reuptake, but are also present post-synaptically, where they modulate striatal GABA release. In hMT mice, postsynaptic D2R function is reduced, as demonstrated by impaired physiological response to D2 dopamine receptor agonists, a reduction in D2 dopamine receptor protein and a reduction in G-protein activation. Interestingly, a recent study demonstrated that biofeedback-based sensorimotor training significantly increased striatal D2 binding in individuals suffering from focal hand dystonia (FHD), as determined by [123I]iodobenzamide single-photon emission computed tomography [40]. D2 binding, which was restored to nearly normal levels (based on age/sex-matched healthy subjects) correlated with clinical improvement, suggesting it may be possible to correct the underlying molecular dysfunction with non-invasive interventions.
Despite the strong link to dystonia, pharmacological agents that enhance or inhibit DA signaling provide only modest relief for most dystonias, and are accompanied by significant risk of adverse side effects [41-43]. For example, while L-DOPA therapy leads to dramatic improvement in DRD patients, studies in other patient populations have yielded conflicting results, with some reporting improvement, and others reporting worsening of symptoms [42,44]. As discussed, DRD is associated with mutations in genes that are critical to DA synthesis. It is therefore not surprising that individuals with DRD consistently respond to L-DOPA treatment. The contradictory results in other studies are likely a consequence of the presence of mixed patient populations. Several reports have indicated that dopamine receptor antagonists may be effective in ameliorating symptoms in certain types of dystonia. For example, risperidone, an atypical antipsychotic with high affinity to the D2 DA receptor, has been shown to be efficacious in both idiopathic and secondary dystonias [45]. Other neuroleptics, including quetipine and clozapine have also been found to reduce symptom severity, however side effects, including sedation and orthostatic hypotension were noted [43,46]. It is paradoxical that both stimulation (via biofeedback-based sensorimotor training) and antagonism (via antipsychotic agents) of the D2 receptor are both effective in alleviating symptoms in different forms of dystonia. Though further investigation is needed, this suggests that divergence, in either direction, from an “optimal” level of DA signaling may contribute to the dystonic phenotype. Regardless, open label trials of neuroleptics for the treatment of dystonia have yielded mixed results [47], blinded trials have not been conducted, and the side effects of currently available dopaminergic compounds preclude their use in dystonia. Further research into the nature of DA dysfunction in this disorder is therefore warranted, and may lead to the development of treatments that more effectively address the complex relationship between DA and dystonia.
2.1.2 Mitochondrial function
Accidental exposure to the mitochondrial mycotoxin 3-nitropropionic acid (3-NPA) can cause dystonia in otherwise healthy individuals [48-50]. Following an outbreak of non-inflammatory encephalopathy caused by ingesting moldy sugarcane, the inciting agent was identified as 3-NPA, a potent neurotoxin produced by arthrinium fungus. 3-NPA poisoning is characterized by severe gastrointestinal symptoms, convulsions and coma. Upon recovering from acute symptoms, approximately 25% of adolescent patients develop segmental or generalized dystonia [48,49]. Imaging and autopsy studies have found that these individuals exhibit severe, selective degeneration of the basal ganglia, particularly within the striatum and globus pallidus [49,51]. The extent of the damage is correlated with the severity of dystonia. It is thought that free radicals produced by 3-NPA react with DA to produce damaging quinone compounds [52], which may account for the selective lesioning of the basal ganglia. Peripheral administration of 3-NPA to rodents and non-human primates results in a motor disorder that is comparable to that seen in humans [51,53,54]. In both cases, 3-NPA causes delayed-onset truncal and limb dystonia. In non-human primates, dystonic movements begin several weeks after treatment and progress until the symptoms become generalized [54,55]. As in humans, the severity of dystonia is directly related to the degree of striatal damage [53-55]. Histological studies in animals have revealed that the most significant damage occurs in striatal projection neurons [53,55]. This is accompanied by an increase in striatal dopamine, consistent with reduced striatonigral inhibition.
Dystonia is also found in a number of inherited mitochondrial disorders [56,57]. Leber's hereditary optic neuropathy (LHON) was the first neurodegenerative disorder to be linked to mitochondrial dysfunction. Genetic analysis of affected individuals revealed a mutation in the mtDNA complex I gene MTND6 [58]. LHON is characterized by bilateral degeneration of the optic nerve and the basal ganglia, which causes sudden-onset blindness and generalized dystonia. Another movement disorder in which mitochondrial dysfunction is implicated is Leigh disease. Leigh disease is associated with a number of motor symptoms, including dystonia, chorea and ataxia [57]. Mutations in several different mitochondrial genes are associated with this disorder, however between 10 and 20% of individuals exhibit point mutations in ATP synthase F0 subunit 6 (MT-ATP6), a gene associated with proton transport across the mitochondrial membrane [59,60]. Multiple focal regions of demyelination, capillary proliferation, and gliosis are observed in the basal ganglia, brainstem, and thalamus in this disorder. Unlike LHON, neuronal cell death is minimal in Leigh disease. Deafness-dystonia-optic neuropathy (DDON) syndrome, also known as Mohr-Tranebjærg syndrome is another mitochondrial disorder in which dystonia is a prominent feature. This disorder is caused by a mutation in the deafness-dystonia peptide (DDP1) gene, which encodes the mitochondrial translocase subunit Tim8 A [61]. Mutations in DDP lead to abnormal protein transport within the mitochondria, however it is not yet known how this affects mitochondrial function or how it contributes to the pathophysiology of the disorder. Individuals with DDON exhibit general brain atrophy and gliosis [62] and reduced activity in the striatum and parietal cortex [63,64]. Research in these types of disorders suggests that strategies designed to enhance existing mitochondrial energy production may be effective in ameliorating symptoms. For example, increasing riboflavin, a cofactor for mitochondrial complex I and II, was associated with improvement in several mitochondrial disorders in a number of small trials [65-67]. Recognizing mitochondrial dysfunction as a shared theme across dystonias may therefore be critical in identifying novel pathways for targeted therapeutic intervention.
In the absence of additional data, it is difficult to determine the exact relationship between mitochondrial dysfunction and dystonia in inherited mitochondrial disorders, which are often characterized by a number of neuropathologies. It is also difficult to determine whether the pathology associated with 3-NPA neurotoxicity can be extrapolated to other types of dystonias. It is therefore important to note that mitochondrial abnormalities have been observed in a number of idiopathic dystonias [68-71]. For example, decreased complex I activity has been reported in patients with focal [68,70] as well as segmental and generalized dystonia [68]. In one study the severity of the complex I deficit was found to be more pronounced in individuals with segmental or generalized forms than in those with focal dystonia [68]. Several instances of adult-onset dystonia have been linked to mutations in the gene encoding the ND1 subunit of the mitochondrial complex I enzyme [71]. ND1 is critical for complex I activity, and is highly expressed in cholinergic neurons in the striatum [72]. A mutation in the ND6 subunit, which like ND1 is also important for enzyme activity, has also been demonstrated in idiopathic dystonia [73]. In this case, the resulting reduction in complex I activity was accompanied by bilateral lesions of the striatum. Significant necrosis, particularly within the putamen, was noted in several other dystonic patients in which mitochondrial dysfunction was suspected [69,74], Importantly however, mitochondrial energy impairment and oxidative damage have been shown to occur in the absence of nigrostriatal degeneration in a genetic mouse model of PD [75], indicating that abnormal mitochondrial activity may also be involved in primary dystonias which are not accompanied by neurodegeneration. Although the affected circuitry may be the same in both cases, it may be necessary to consider whether degeneration is present to determine the most effective therapeutic intervention.
2.1.3 Calcium Regulation
Abnormal calcium homeostasis is also implicated in the pathophysiology of dystonia. Several dominantly inherited neurological disorders in humans are associated with mutations to the CACNA1A gene, which encodes the α12.1 pore-forming subunit of CaV2.1 (P/Q-type) voltage-gated calcium channels. For example, aberrant CaV2.1 channel activity has been implicated in both episodic ataxia type 2 (EA2) and spinocerebellar ataxia type 6 (SCA6) [76-79]. These conditions are characterized by cerebellar ataxia with prominent dystonia. EA2 mutations primarily result in protein instability and loss of function [80], while CAG repeats in the P-type channel underlie a toxic gain of function in SCA6 [81]. In both cases, the resulting change in calcium channel function is believed to underlie the motor disorders [82]. The presence of dystonic movements in these disorders firmly establishes dystonia as part of the clinical spectrum associated with CaV2.1 dysfunction [76-79].
Dystonia is also associated with mutations to the mouse Cacna1a gene. Two different targeted Cacna1a disruptions that completely abolish CaV2.1 activity result in a severe motor disorder that is reminiscent of generalized dystonia in humans [83,84]. Several other recessively inherited dystonic syndromes have been described in mice bearing spontaneous Cacna1a mutations, including in tottering, rocker, leaner and wobbly mice [85,86]. Leaner mice exhibit a severe and chronic generalized dystonia that resembles that of the null mice [87]. The leaner mutation disrupts normal splicing of the Cacna1a gene and causes an ~65% reduction in CaV2.1 currents and abnormal channel kinetics [87,88]. Both tottering and rocker mice exhibit episodes of dystonia superimposed on a mild baseline ataxia, though the episodes in tottering mice are always generalized and those in rocker mice exhibit a range of anatomical distributions [89]. The missense mutations expressed by tottering and rocker mice occur within highly-conserved pore residues of the α12.1 protein [90]. Electrophysiological studies demonstrate that CaV2.1 channels bearing the tottering mutation exhibit moderate ~40% reduction in calcium currents [91]. On the other hand, lethargic mouse mutants, which bear a spontaneous mutation within the auxiliary β4 Cacnab4 gene, exhibit relatively milder episodes of dystonia [89] that are associated with minor CaV2.1 calcium channel abnormalities [92]. Overall, the studies of calcium channel mutant mice suggest that a relationship exists between the extent of the underlying abnormal calcium signaling and the severity dystonia.
Several models also implicate abnormal CaV1.2 calcium channel (L-type) signaling in dystonia. In normal mice, systemic or central administration of L-type Ca2+ channel agonists (±Bay K 8644 and FPL 64176) induces a motor syndrome that closely resembles generalized dystonia in humans [93]. In addition, tottering mouse dystonia is extremely sensitive to CaV1.2 activity, as low-doses of L-type calcium channel agonists induces dystonia and antagonists block dystonia [94]. Moreover, CaV1.2 antagonists also ameliorate the dystonia in a hamster model (sz) that does not arise from genetic defects in calcium signaling dystonic [95].
Several other studies in humans have implicated calcium dysfunction in dystonia as well. For example, Wszolek and colleagues recently described an inherited syndrome characterized by extensive calcium deposition in the basal ganglia, thalamus, cerebral white matter, and cerebellum, which is accompanied by dystonic movements [96]. While no additional histological abnormalities were observed, PET imaging in two of the individuals demonstrated a reduction in D1 and D2 receptor binding and reduced 6-[18F]fluoro-L-dopa uptake. An association between a calcium-sensitive potassium channel (BK) and paroxysmal dyskinesias has also been recently demonstrated. In this report, a mutation in the alpha subunit of the BK channel was found to increase Ca2+ sensitivity three- to five-fold [97]. The authors propose that this increase in sensitivity may permit a faster rate of firing by inducing rapid repolarization, and that this may increase neuronal excitability, ultimately producing dyskinetic movements.
Together, this evidence suggests that abnormal calcium signaling is involved in dystonia. Calcium signaling plays a pivotal and complex role in neuronal excitability, however the mechanism whereby disruptions in normal calcium homeostasis cause dystonia remains unclear. Regardless, several compounds that regulate calcium signaling have been used to successfully treat neurological disorders, including dystonia, suggesting that targeting calcium channels is a novel therapeutic approach that should be explored.
2.2 Anatomical
A comprehensive examination of the brain substrates implicated in dystonia has recently been published and is outside of the scope of this review [98]. However several brain regions are consistently implicated across a number of etiologically-diverse dystonia, suggesting they may represent neuroanatomical correlates of a shared mechanism. These structures are discussed in greater detail below.
2.2.1 Basal Ganglia
Lesions of the basal ganglia (BG) and its connections are frequently accompanied by dystonia [99-103]. Structural defects are most commonly reported in the caudate, the putamen, and the thalamus, manifesting as mild focal dystonia [100,101] to severe generalized dystonia [103], depending on the extent of the lesion. Lesions of the globus pallidus have also been reported in patients with focal or segmental dystonia [102]. Functional imaging studies often reveal abnormalities in the BG of dystonic patients, even when lesions are not observed [99]. As in cases with overt structural abnormalities, changes in blood flow and metabolic activity are most commonly reported in the caudate, putamen, and thalamus. For example, hyperperfusion of both the putamen and the thalamus, as determined by fluorodeoxyglucose PET, has been observed in cases of cervical dystonia [104] and DYT1 dystonia [105,106], while hyperperfusion of the caudate has been noted in blepharospasm [107]. This is consistent with data from fMRI studies, which found increased activity in all three regions in individuals with FHD [108-110].
The comorbidity of dystonia and PD further implicates the BG in dystonia [5,6,111]. Derangement of basal ganglia pathways is the primary pathology in PD. It is therefore presumed that this phenomenon contributes to the expression of dystonic symptoms as well. Dystonia can also present in Huntington's disease (HD) [112,113], which is characterized by degeneration of the striatum. Interestingly, in young-onset HD, dystonia may be predominant over choreic movements, which are typical in adult-onset HD [114]. On autopsy, individuals with juvenile HD exhibit more severe degeneration, particularly within the caudate and putamen, than seen in adult-onset HD [115]. This may account for the difference in symptomology, providing further support for a role for the BG in dystonia.
The efficacy of neurosurgical interventions, including pallidotomy and deep brain stimulation (DBS), further implicate the basal ganglia in dystonia. For example, bilateral surgical ablation of the globus pallidus significantly improves generalized dystonia [116,117]. In recent years pallidotomy has been rejected in favor of non-ablative DBS, however a review of the current literature suggests that the efficacy and safety of both procedures are comparable [118]. Regardless, DBS, primarily in the globus pallidus or the subthalamic nuclei, has proven effective in some primary and secondary dystonias, ranging from focal [81,119] to generalized [81,120-122], including tardive forms [123-125]. The successful amelioration of symptoms in a wide-range of dystonias is particularly compelling evidence that BG dysfunction represents a shared component of the pathophysiology of dystonia.
2.2.2 Cerebellum
In addition to the BG, a growing body of evidence implicates cerebellar dysfunction in the pathophysiology of dystonia. Like the BG, cerebellar injury has been associated with the onset of dystonic symptoms in a number of cases [126-130]. Cerebellar stroke, for example, has been implicated in reports of acquired focal dystonias including cervical spasmodic torticollis [126,128,129], blepharospasm [129], and oro-mandibular dystonia [127]. Segmental hemidystonia was reported following vertebral artery occlusion in one case report [130]. Secondary dystonia has also been demonstrated in patients with cerebellar tumors [131-133]. In one patient suffering from cerebellar tuberculoma, focal limb dystonia resolved following tuberculostatic treatment [134], providing further support for a direct relationship between the cerebellum and the presentation of dystonic symptoms.
Structural imaging studies have revealed abnormalities in the cerebellum in primary dystonias as well. Using voxel-based morphometry, several reports have shown changes in gray matter density in the cerebellum in idiopathic cervical dystonia [135,136], benign essential blepharospasm [136], and in individuals with FHD [137]. Functional abnormalities, as indicated by changes in regional blood flow, have been demonstrated as well. Hyperperfusion of the cerebellum has been shown in exercise-induced paroxysmal dystonia [138,139], generalized dystonia [140], and FHD [141]. In one patient with generalized dystonia, differences in blood flow were noted in the right versus left deep cerebellar nuclei (DCN), and between the right cerebellar cortex and the right DCN [140], suggesting communication within the cerebellum itself may be compromised. Using [18F]fluorodeoxyglucose and positron emission tomography, increased metabolic activity has also been demonstrated in the cerebellum in essential blepharospasm [142] and DYT1 dystonia [106].
A recent study demonstrated abnormalities in cerebellar circuitry in DYT1 and DYT6 patients and non-manifesting carriers, [143]. Using diffusion tensor imaging (DTI), this study demonstrated that both manifesting and non-manifesting carriers exhibited reduced integrity of cerebellothalamocortical fiber tracts suggesting that abnormalities in cerebellar fiber tracts may represent an endophenotype in dystonias with a known genetic component. Interestingly, non-manifesting carriers displayed an additional area of fiber disruption along the thalamocortical portion of the pathway. Clinical penetrance could be predicted from the difference in connectivity measured in these two regions. This suggests that the second interruption may represent a compensatory mechanism whereby dystonic symptoms could be suppressed. In addition to providing insight into the incomplete penetrance of DYT1 and DYT6 dystonia, these data suggest that abnormalities in cerebellar signaling may lead to broader motor network dysfunction.
Animal models also implicate the cerebellum in the pathophysiology of dystonia. For example, robust induction of c-fos expression is observed in the cerebellum of tottering mice following dystonic attacks [144]. Dystonia is also associated with low-frequency oscillations in the cerebellar cortex in the tottering mouse, a phenomenon that is not observed in normal mice [145]. These oscillations are accompanied by increasingly coherent oscillations in electromyographic (EMG) activity in the face and hindlimb, providing strong support for an association between dystonic movements and abnormal activity in the cerebellum. Functional imaging studies have also revealed abnormalities in cerebellar activity in genetically dystonic (dt) rats [146] and hamsters [147]. Local application of kainic acid, a glutamate receptor agonist, to the cerebellar cortex causes robust, generalized dystonia in C57BL/6J mice [148], demonstrating that direct manipulation of cerebellar signaling can produce dystonic symptoms in otherwise normal mice. In addition to demonstrating a role for the cerebellum in dystonia, the tottering mouse model is associated with aberrant calcium signaling, suggesting a potential link between these two themes. Finally, perhaps the most compelling evidence in support of a role for the cerebellum in dystonia is that surgical removal of the cerebellum has been shown to eliminate dystonia in the dt rat [149], as well as the tottering [35] and lethargic [150] mouse mutants. Selective destruction of Purkinje cells in the tottering mouse also ameliorates dystonic symptoms [151], suggesting that Purkinje cell dysfunction is critical in mediating the dystonic phenotype.
While these findings provide strong support for a role for cerebellar dysfunction in dystonia, a number of recent studies indicate that combined dysfunction in the cerebellum and the BG may underlie the expression of dystonic movements. In a mouse model of rapid-onset dystonia-Parkinsonism, pharmacological manipulation of cerebellar signaling resulted in aberrant activity in the BG, which was accompanied by robust generalized dystonia [152]. Selective lesioning of the deep cerebellar nuclei, the main source of cerebellar output, or the centrolateral nucleus of the thalamus, which represents the di-synaptic link between the cerebellum and the basal ganglia, ameliorated the dystonia, providing further evidence that communication within the larger motor network is critical for the expression of dystonic movements. The idea that both the BG and the cerebellum are involved in dystonia is consistent with other reports, which have demonstrated that striatal levels of extracellular DA are significantly reduced during dystonic attacks in both tottering mice and normal mice which received microinjections of kainic acid into the cerebellar cortex [35]. In this study, the authors also showed that subclinical lesions of the striatum, using either 6-OHDA or quinolinic acid, exaggerated the “cerebellar” dystonia in tottering and kainic acid-treated mice. Additional studies are needed to determine the relationship between these structures in dystonia, distinguishing between primary and secondary changes in network dysfunction. It will also be important to determine whether secondary changes are compensatory, and whether they are adaptive or maladaptive.
2.3 Systems
2.3.1 Plasticity
Plasticity within motor circuits is implicated as a key component of motor learning and memory. However, emerging evidence suggests that plasticity may also be a critical component of the pathophysiology of dystonia [153-157]. In humans, direct measurement of long term potentiation (LTP) and long term depression (LTD), the physiological correlates of synaptic plasticity, is largely impractical. There has been only one reported case in which cortical output was directly recorded from electrodes implanted in the upper cervical spinal cord of a patient with cervical dystonia [158]. In this study, cortical inhibition was found to be significantly impaired in the dystonic patient, as compared to non-dystonic individuals with electrode implants for pain management. Human studies implicating maladaptive plasticity have primarily relied upon the ability of transcranial magnetic stimulation (TMS) of the motor cortex to evoke detectable responses in a target muscle. TMS has been used clinically for many years to test the integrity of the corticospinal tract. Because TMS stimulates the axons of neurons that synapse on pyramidal efferents, the size of the response produced by a stimulus reflects the intrinsic excitability of the cortex. Motor evoked potentials recorded via electromyography are therefore indirect indicators of plasticity. Using a paired associative stimulation (PAS) paradigm, which combines TMS with median nerve stimulation to produce LTP in the sensorimotor system, one study demonstrated that cranial and cervical dystonia is associated with enhanced corticospinal excitability as compared to healthy controls [156]. Importantly, these abnormalities were not observed in patients suffering from hemi-facial spasm, a non-dystonic condition, which suggests that it is not caused by the movement itself. A significant increase in excitability has also been observed in the S1 cortex of patients with focal hand dystonia [157].
The excitation evoked by TMS is limited to superficial layers of the brain, meaning it is most useful for providing information about cortical plasticity. Evidence from animal models suggests that aberrant synaptic remodeling is also present in subcortical structures, particularly within the striatum [159-161]. For example, LTP and LTD is altered in cortico-striatal synapses in slice preparations from a mouse model of DYT1 dystonia [160]. Specifically, it was shown that LTD was significantly attenuated in striatal medium spiny neurons, while the amplitude of LTP was significantly enhanced. As discussed previously, functional changes in striatal D2 DA receptors have been observed in the DYT1 mouse [33,34]. D2 DA receptors are critical in mediating LTD [162], and may therefore represent the anatomical correlate of aberrant plasticity in DYT1 dystonia. Interestingly, pharmacological blockade of adenosine A2A receptors (A2AR) can counteract the deficit in D2R-mediated neurotransmission in hMT mice [33]. Pharmacological blockade of A2ARs has been shown to mimic striatal postsynaptic D2R activation in MSNs [163], suggesting that the deficit in D2 function in hMT mice may be compensated for by eliminating the opposing action of A2ARs. A similar increase in LTP in the cortico-striatal pathway has also been observed in the dt(sz) hamster, a model of paroxysmal dystonia [159]. Interestingly, in slice preparations from the DYT1 mouse, these abnormalities could be corrected by reducing acetylcholine, a major modulator of MSN plasticity [161]. This may explain the efficacy of anti-cholinergic drugs in the treatment of some dystonias.
Two potential mechanisms may underlie maladaptive synaptic remodeling in dystonia. It may either arise from an inherent defect in one or more of the components that drive plasticity, or from “over-training” via repetition of a specific movement. The fact that exaggerated plasticity is most often seen in task-specific dystonias such as FHD and musician's dystonia is consistent with the latter theory [153,154,164]. Prospective studies in human patients provide indirect support for this mechanism, demonstrating that grey matter volume in the contralateral primary motor hand area is reduced following sustained immobilization of the hand and increased following repetitive training of a manual activity within individuals suffering from FHD [165]. In another study, practice-dependent plasticity, defined by the acceleration of movement in a thumb abduction task, was measured in individuals with focal hand dystonia and healthy controls [166]. Healthy controls, but not those with FHD, exhibited homeostatic suppression of practice-dependent plasticity. Further, the degree to which suppression was impaired correlated with clinical severity of individual FHD. This is consistent with a mechanism that is dependent upon abnormal motor learning. Direct evidence of remodeling in response to over-training has also been shown in non-human primates [167-169]. For example, electrophysiologic mapping of the primary somatosensory cortex following over-training in a repetitive motor task showed dramatic remodeling characterized by enlarged and overlapping receptive field topography [167]. These changes were accompanied by motor symptoms comparable to focal hand dystonia in humans. Overall, over-training in some individuals causes dystonia while others are unaffected suggesting that it is the combination of genes plus environment that predisposes some individuals to respond with abnormal plasticity.
Determining the pathological process that leads to enhanced plasticity in the motor circuit may also be key in understanding the relatively low penetrance characteristic of many dystonias with a known genetic component. DYT1 dystonia, for example, is inherited in an autosomal dominant pattern. However, between 60 and 70% of carriers are asymptomatic [170]. DYT1 dystonia patients exhibit prolonged corticospinal excitability following TMS, as compared to non-manifesting carriers of the DYT1 mutation [155], suggesting that a propensity for enhanced synaptic plasticity may be critical in the pathogenesis of dystonia. On the other hand, non-manifesting DYT1 carriers exhibit sub-clinical abnormalities in movement representation in the brain, as defined by changes in corticospinal excitability following TMS, when compared to healthy controls [155]. This implies that the ability to compensate for aberrant plasticity, as opposed to a predisposition towards enhanced plasticity, may be the differentiating factor between manifesting and non-manifesting individuals. In either case, maladaptive plasticity may represent an endophenotype that could prove to be useful for diagnostic purposes.
A number of current and emerging therapies may improve dystonic symptoms by affecting pathological plasticity. For example, botulinum toxin, the current standard of care, has recently been shown to reduce plasticity in the primary motor cortex in patients with primary dystonia [171]. In this study, PAS was administered to individuals with cervical dystonia before they received botulinum injections, as well as 1 and 3 months post-injection. Before the injection, PAS significantly facilitated MEPs in hand muscles. One month after the injection, MEPs were comparable to control levels, with partial recovery after 3 months. The authors propose that the attenuation of afferent input from the neck caused by the botulinum toxin results in reorganization of the motor cortex representation of hand muscles. Other traditional interventions, including limb immobilization [172] and constraint therapy [173], may also restore normal levels of synaptic connectivity. Data from several recent studies indicates that both deep brain stimulation and TMS-induced improvement in dystonia may be the result of “normalized” plasticity in motor circuits as well [174-176]. Repetitive TMS of the somatosensory cortex, for example, has been shown to improve FHD and enhance cortical activity [176]. Comparable changes in cortical excitability have been observed following stimulation of subcortical structures [174,175], suggesting cortical plasticity can be modulated indirectly. This may ultimately prove to be an essential part of the mechanism underlying the efficacy of therapeutic stimulation in dystonia.
2.3.2 Inhibition
The suppression of extraneous signaling is critical for the initiation and execution of movement. It is therefore not surprising that impaired inhibitory function has been implicated in many dystonias. Deficits in reciprocal inhibition in the spinal circuitry, for example, have been demonstrated in FHD, spasmodic torticollis, generalized dystonia and blepharospasm [177,178]. Impaired reciprocal inhibition is believed to be the proximal cause of the characteristic co-contractions which define the clinical diagnosis of dystonia. Consistent with this theory, current therapies such as botulinum toxin and pallidal DBS have been shown to restore reciprocal inhibition in dystonia patients [179,180].
A loss of inhibitory function has also been demonstrated in higher levels of the nervous system, particularly within the cortex. Like plasticity, cortical inhibition can be measured using TMS. For example, short intracortical inhibition (SICI), which reflects interneuron activity, can be determined by pairing a conditioning stimulus which is sufficient to activate cortical neurons, but does not activate descending pathways, with a subsequent supratheshold stimulus. The intracortical activity evoked by the conditioning stimulus directly affects the amplitude of the MEP produced by the second stimulus, and is therefore an indirect measure of SICI. Deficits in SICI have been demonstrated in the motor cortex in individuals with focal hand dystonia [181]. Interestingly, this impairment was not restricted to the contralateral cortex, but was observed in both hemispheres. Deficits in interhemispheric inhibition have also been demonstrated in mirror dystonia, a type of focal hand dystonia in which dystonic posturing occurs in one hand when the other hand is moved [182].
The ability to suppress excitability in an area surrounding an activated region (surround inhibition) is another type of inhibitory process that is believed to be compromised in dystonia. Although the concept of surround inhibition has not been widely investigated in the motor system, it is logical to hypothesize that as in the sensory system, where it contributes to precise perceptions, surround inhibition may contribute to the selective execution of desired movements. Indirect support for deficient surround inhibition in dystonia has been provided in several studies. For example, TMS of the M1 cortex paired with self-initiated flexion of the index finger normally results in significant suppression of MEP amplitudes in the little finger. In patients with focal hand dystonia, this paradigm has been shown to produce an increase in MEP amplitude in the little finger [183]. Similar deficits in inhibition in the premotor cortex have been documented in individuals with focal hand dystonia [184].
A direct link between cortical inhibition and dystonia has been demonstrated in a series of studies in non-human primates. In the first report, the local application of bicuculline, a GABA-A antagonist, onto the motor cortex severely impaired fine motor control [185]. Comparable deficits were also observed in a visual reaction-time task in which the subjects were trained to press or release a lever in response to a visual cue. These impairments were attributed to co-contractions of agonist and antagonist muscles in the arm during the behavior. In a follow-up study, the authors demonstrated that bicuculline increased the firing rate of active cells, provoked activity in “silent” cells, and converted unidirectional cells (cells that responded to either pressing or releasing the lever) to bidirectional cells (which responded to pressing and releasing the lever) during the execution of the reaction-time task [186]. These findings provide greater insight into a number of molecular mechanisms that may underlie the phenomenon of reduced inhibition in dystonia.
2.3.3 Sensorimotor Integration
Another theme in the pathophysiology of dystonia is the impairment of sensorimotor integration, although it is unclear whether this is driven by aberrant sensory input, or by a misrepresentation of sensory information at higher levels in the nervous system. While the precise location of signal derangement is not known, a number of clinical observations indicate that communication within the sensorimotor feedback circuit is compromised in dystonia.
The efficacy of the geste antagonistique, or sensory trick, in improving abnormal postures is compelling evidence for abnormal sensorimotor integration in dystonia. Sensory tricks involve touching the affected body part or an adjacent region in order to reduce or ameliorate the aberrant muscle contractions. For example, placing a hand on the chin or face can reduce muscle contractions in up to 70% of patients with cervical dystonia [187]. While sensory tricks were originally believed to work by distracting the individual's attention, physiological studies have since demonstrated that they produce measurable changes in muscle recruitment, as determined by EMG [188,189]. The mechanism underlying this phenomenon is still unknown, however PET imaging has revealed that sensory tricks reduce the activation of the supplementary motor area and the primary sensorimotor cortex in individuals with cervical dystonia [190], suggesting they may modulate central integration of sensory information. Sensory tricks are also effective in other forms of focal dystonia, including blepharospasm, oro-mandibular dystonia and FHD [191-193].
Inherent sensory and perceptual abnormalities have been observed in dystonic patients as well. For example, the perception of the vibration-induced illusion of movement is abnormal in patients with focal dystonia [194]. This illusion is associated with similar perceptual abnormalities in asymptomatic first degree relatives [195], suggesting that subtle deficits in sensory processing may represent an endophenotypic marker for dystonia. In another study, Kaji and colleagues found that vibration of the affected arm can induce dystonic posturing in individuals with focal hand dystonia [196]. In normal individuals, vibration of a muscle leads to increased MEP amplitude and reduced intracortical inhibition, however these changes are not observed in dystonic patients [197]. A number of studies have identified abnormalities in somatotopic representations in the sensorimotor cortex in dystonia [198-200], suggesting the defect in integration may be localized to this region. However it is not clear whether these abnormalities are causative, or reactionary. Future studies are needed to determine the nature of the relationship between impaired sensorimotor integration and dystonia.
3. Expert Opinion
In addition to providing insight into the biological processes that underlie etiologically-diverse dystonias, identifying shared features of pathogenesis can suggest novel targets for therapeutic intervention that have the potential to be effective in a broad group of affected individuals (Figure 2). This approach has been successfully applied to epilepsy, another heterogeneous neurologic disorder. In this case, the identification of abnormalities in GABA neurotransmission and enhanced cortical excitability in a number of epileptic conditions has lead to the development of drugs that are effective in broad groups of patients, regardless of specific etiologies. The idea of shared pathways as targets for drug discovery is also being investigated in a number of other CNS disorders characterized by diverse molecular etiologies, including ataxia [201], PD [202], and depression [203]. The use of botulinum toxin and other muscle relaxers represents such a strategy, as excessive muscle contractions are present, by definition, in all dystonias. Likewise, the basal ganglia and the cerebellum have both been shown to regulate cortical excitability, suggesting that aberrant cortical plasticity may represent another final common pathway in dystonia. The fact that stimulation-based interventions such as TMS and DBS, which are thought to directly affect plasticity, are broadly effective across many forms of dystonia, is consistent with this idea. The success of this approach thus far suggests that it may be advantageous to follow this strategy for future drug discovery efforts as well.
Figure 2. Schematic representation of convergent pathological mechanisms in dystonia.
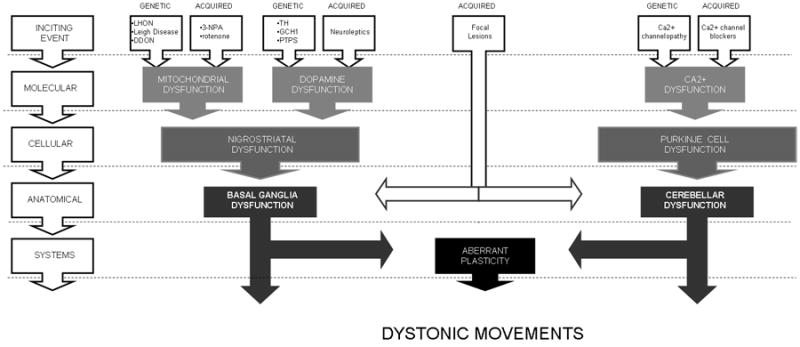
Pathological changes associated with an array of initial insults may converge to produce dysfunction in shared downstream pathways. Intervention in these downstream processes may effectively interrupt the expression of dystonic symptoms, regardless of the cause. (LHON = Leber's hereditary optic neuropathy; DDON = deafness-dystonia optic neuropathy; 3-NPA = 3-nitropropionic acid; TH = tyrosine hydroxylase GCH1 = GTP cyclohydrolase I; PTPS = 6-pyruvoyl-tetrahydropterin synthase)
The trend towards focusing on “pure” dystonias, such as DYT1 dystonia, to the exclusion of secondary and/or dystonia-plus syndromes represents a potential barrier in identifying relevant pathological mechanisms. The known causes for “pure” dystonias are limited in comparison to secondary and dystonia-plus syndromes. In addition, the notion of focusing on a ‘pure’ syndrome has never been a requirement in studies of other movement disorders such as PD or ataxias, which are likewise characterized by diverse etiologies and manifestations. While detailed studies of primary dystonias will undoubtedly provide insight into the mechanisms of pathogenesis in both primary and secondary dystonias, the information gained from “mixed” syndromes has already proven invaluable to the understanding and treatment of dystonia. For example, focal lesions of the basal ganglia and cerebellum first implicated these structures in dystonia. Similarly, the link between dystonia and DA dysfunction was, in part, derived from the presence of dystonic movements in PD and in patients on chronic neuroleptic therapies. Finally, in addition to the many phenotypic manifestations of primary dystonias, the combination of structural and physiological anomalies present in individual cases is rarely uniform. This argues against the very existence of a “pure” dystonic syndrome, suggesting that the current definition may not, in itself, be particularly useful.
On a similar note, it is also critical that a variety of animal models be studied to avoid potential idiosyncrasies in individual models. The pitfalls of using a single model have been clearly illustrated in pre-clinical studies of amyotropic lateral sclerosis (ALS) and multiple sclerosis (MS), where the pathological processes in the most commonly used models differ from that seen in most human cases [204,205]. Thus, despite identifying a number of promising drug candidates, studies using these models have failed to lead to an effective treatment for human patients. Fortunately, a number of models of dystonia, both etiologic and phenotypic, have become available in the last decade. The choice of model, as always, will depend on the overall goal of the research. For example, pharmacologically-induced dystonia is advantageous for high through-put screening of potential therapeutics, but may fail to replicate dysfunction in pathways that are activated when dystonia arises “naturally” (i.e. etiologic models). On the other hand, a number of attempts to develop etiologic models by introducing gene variants that are strongly correlated with dystonia in humans have failed to produce a behavioral phenotype.
While this review illustrates the value of investigating shared mechanisms of pathogenesis, the importance of identifying divergent mechanisms in distinct subtypes of dystonia cannot be overstated. Both approaches will likely provide unique insight into the underlying pathology, and will contribute in different ways to the development of effective therapeutics.
The ultimate objective of basic and clinical research in the field of dystonia is to identify the underlying pathological mechanisms, and to develop therapeutic strategies that prevent, alleviate, and/or resolve dystonic symptoms. The identification of shared features of pathogenesis addresses both goals by providing insight into the biological processes that underlie etiologically-diverse dystonias, and suggesting novel targets for broadly effective therapeutic intervention. Thus comparing similarities across different types of dystonia or different dystonia models may be more fruitful than extraordinarily detailed studies of a single type of dystonia or model.
Highlights.
Biological levels of dysfunction in dystonia:
-
Cellular:
Dopamine signaling
Mitochondrial function
Calcium regulation
-
Anatomical:
Basal Ganglia
Cerebellum
-
Systems:
Plasticity
Inhibition
Sensorimotor Integration
Acknowledgments
Declaration of Interest: This work was supported by the Bachmann-Strauss Dystonia and Parkinson Foundation, National Institutes of Health R01 NS33592, and National Institutes of Health U54 NS067501.
Abbreviations
- DA
dopamine
- PD
Parkinson's disease
- DRD
dopa responsive dystonia
- GCH
GTP cyclohydrolase
- BH4
tetrahydrobiopterin
- TH
tyrosine hydroxylase
- PTPS
6-pyruvoyltetrahydropterin
- SPD
sepiapterin reductase
- LND
Lesch Nyhan disease
- HPRT
hypoxanthine guanine phosphoribosyl transferase
- GABA
gamma-aminobutyric acid
- HVA
homovanillic acid
- SCA2
spinocerebellar ataxia type 2
- FHD
focal hand dystonia
- 3-NPA
3-nitropropionic acid
- LHON
Leber's hereditary optic neuropathy
- DDON
deafness-dystonia optic neuropathy
- DPP
deafness-dystonia peptide
- Ca2+
calcium
- EA2
episodic ataxia type 2
- SCA6
spinocerebellar ataxia type 6
- CACNA1A
calcium channel, voltage-dependent, P/Q type alpha 1A subunit
- BG
basal ganglia
- PET
positron emission tomography
- fMRI
functional magnetic resonance imaging
- 6-OHDA
6-hydroxydopamine
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- HD
Huntington's disease
- DBS
deep brain stimulation
- DCN
deep cerebellar nuclei
- DTI
diffusion tensor imaging
- LTP
long term potentiation
- LTD
long term depression
- TMS
transcranial magnetic stimulation
- MEP
motor evoked potential
- PAS
paired associative stimulation
- MSN
medium spiny neuron
- SICI
short intracortical inhibition
- ALS
amyotropic lateral sclerosis
- MS
multiple sclerosis
Contributor Information
Valerie B. Thompson, Emory University School of Medicine, Department of Pharmacology, Woodruff Memorial Research Building, Suite 6000, 101 Woodruff Circle, Atlanta, GA 30322.
H. A. Jinnah, Emory University School of Medicine, Departments of Neurology, Human Genetics & Pediatrics, Woodruff Memorial Research Building, Suite 6000, 101 Woodruff Circle, Atlanta, GA 30322.
Ellen J. Hess, Email: ejhess@emory.edu, Emory University School of Medicine, Department of Pharmacology, Woodruff Memorial Research Building, Suite 6000, 101 Woodruff Circle, Atlanta, GA 30322, Phone: (404) 727-4911, Fax: (404) 712-8576.
References
- 1*.Perlmutter JS, Mink JW. Dysfunction of dopaminergic pathways in dystonia. Adv Neurol. 2004;94:163–70. Excellent overview of the role of dopamine in dystonia. [PubMed] [Google Scholar]
- 2.Bruno MK, Ravina B, Garraux G, et al. Exercise-induced dystonia as a preceding symptom of familial Parkinson's disease. Mov Disord. 2004;19:228–30. doi: 10.1002/mds.10626. [DOI] [PubMed] [Google Scholar]
- 3.McKeon A, Matsumoto JY, Bower JH, et al. The spectrum of disorders presenting as adult-onset focal lower extremity dystonia. Parkinsonism Relat Disord. 2008;14:613–9. doi: 10.1016/j.parkreldis.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 4.Papapetropoulos S, Lundy DS, Casiano RR, et al. Laryngeal dystonia as a presenting symptom of young-onset Parkinson's disease. Mov Disord. 2007;22:1670–1. doi: 10.1002/mds.21184. [DOI] [PubMed] [Google Scholar]
- 5.Katchen M, Duvoisin RC. Parkinsonism following dystonia in three patients. Mov Disord. 1986;1:151–7. doi: 10.1002/mds.870010210. [DOI] [PubMed] [Google Scholar]
- 6.Lees AJ, Hardie RJ, Stern GM. Kinesigenic foot dystonia as a presenting feature of Parkinson's disease. J Neurol Neurosurg Psychiatry. 1984;47:885. doi: 10.1136/jnnp.47.8.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klawans HL, Paleologos N. Dystonia-Parkinson syndrome: differential effects of levodopa and dopamine agonists. Clin Neuropharmacol. 1986;9:298–302. [PubMed] [Google Scholar]
- 8.Lopez-Ariztegui N, Arevalo MA, de Ceballos ML, et al. Central levodopa influx and the clinical motor response to levodopa in patients with Parkinson disease complicated with motor fluctuations and dyskinesias. Clin Neuropharmacol. 2009;32:321–5. doi: 10.1097/WNF.0b013e3181b40378. [DOI] [PubMed] [Google Scholar]
- 9.Bravi D, Mouradian MM, Roberts JW, et al. End-of-dose dystonia in Parkinson's disease. Neurology. 1993;43:2130–1. doi: 10.1212/wnl.43.10.2130. [DOI] [PubMed] [Google Scholar]
- 10.Cubo E, Gracies JM, Benabou R, et al. Early morning off-medication dyskinesias, dystonia, and choreic subtypes. Arch Neurol. 2001;58:1379–82. doi: 10.1001/archneur.58.9.1379. [DOI] [PubMed] [Google Scholar]
- 11.Burke RE, Fahn S, Jankovic J, et al. Tardive dystonia: late-onset and persistent dystonia caused by antipsychotic drugs. Neurology. 1982;32:1335–46. doi: 10.1212/wnl.32.12.1335. [DOI] [PubMed] [Google Scholar]
- 12.Tonini M, Cipollina L, Poluzzi E, et al. Review article: clinical implications of enteric and central D2 receptor blockade by antidopaminergic gastrointestinal prokinetics. Aliment Pharmacol Ther. 2004;19:379–90. doi: 10.1111/j.1365-2036.2004.01867.x. [DOI] [PubMed] [Google Scholar]
- 13.Kiriakakis V, Bhatia KP, Quinn NP, et al. The natural history of tardive dystonia. A long-term follow-up study of 107 cases. Brain. 1998;121:2053–66. doi: 10.1093/brain/121.11.2053. [DOI] [PubMed] [Google Scholar]
- 14.Abeling NG, Duran M, Bakker HD, et al. Sepiapterin reductase deficiency an autosomal recessive DOPA-responsive dystonia. Mol Genet Metab. 2006;89:116–20. doi: 10.1016/j.ymgme.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 15.Furukawa Y, Shimadzu M, Rajput AH, et al. GTP-cyclohydrolase I gene mutations in hereditary progressive amd dopa-responsive dystonia. Ann Neurol. 1996;39:609–17. doi: 10.1002/ana.410390510. [DOI] [PubMed] [Google Scholar]
- 16.Hanihara T, Inoue K, Kawanishi C, et al. 6-Pyruvoyl-tetrahydropterin synthase deficiency with generalized dystonia and diurnal fluctuation of symptoms: a clinical and molecular study. Mov Disord. 1997;12:408–11. doi: 10.1002/mds.870120321. [DOI] [PubMed] [Google Scholar]
- 17.Knappskog PM, Flatmark T, Mallet J, et al. Recessively inherited L-DOPA-responsive dystonia caused by a point mutation (Q381K) in the tyrosine hydroxylase gene. Hum Mol Genet. 1995;4:1209–12. doi: 10.1093/hmg/4.7.1209. [DOI] [PubMed] [Google Scholar]
- 18.Ludecke B, Knappskog PM, Clayton PT, et al. Recessively inherited L-DOPA-responsive parkinsonism in infancy caused by a point mutation (L205P) in the tyrosine hydroxylase gene. Hum Mol Genet. 1996;57:1023–8. doi: 10.1093/hmg/5.7.1023. [DOI] [PubMed] [Google Scholar]
- 19.Neville BG, Parascandalo R, Farrugia R, et al. Sepiapterin reductase deficiency: a congenital dopa-responsive motor and cognitive disorder. Brain. 2005;128:2291–6. doi: 10.1093/brain/awh603. [DOI] [PubMed] [Google Scholar]
- 20.Thony B, Blau N. Mutations in the BH4-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteridine reductase. Hum Mutat. 2006;279:870–8. doi: 10.1002/humu.20366. [DOI] [PubMed] [Google Scholar]
- 21.Muller K, Homberg V, Lenard HG. Motor control in childhood onset dopa-responsive dystonia (Segawa syndrome) Neuropediatrics. 1989;20:185–91. doi: 10.1055/s-2008-1071289. [DOI] [PubMed] [Google Scholar]
- 22.Boyd K, Patterson V. Dopa responsive dystonia: a treatable condition misdiagnosed as cerebral palsy. BMJ. 1989;298:1019–20. doi: 10.1136/bmj.298.6679.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ichinose H, Ohye T, Takahashi E, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet. 1994;8:236–42. doi: 10.1038/ng1194-236. [DOI] [PubMed] [Google Scholar]
- 24.van den Heuvel LP, Luiten B, Smeitink JA, et al. A common point mutation in the tyrosine hydroxylase gene in autosomal recessive L-DOPA-responsive dystonia in the Dutch population. Hum Genet. 1998;102:644–6. doi: 10.1007/s004390050756. [DOI] [PubMed] [Google Scholar]
- 25.Ernst M, Zametkin AJ, Matochik JA, et al. Presynaptic dopaminergic deficits in Lesch-Nyhan disease. N Engl J Med. 1996;334:1568–72. doi: 10.1056/NEJM199606133342403. [DOI] [PubMed] [Google Scholar]
- 26.Nyhan WL. Dopamine function in Lesch-Nyhan disease. Environ Health Perspect. 2000;108:409–11. doi: 10.1289/ehp.00108s3409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lloyd KG, Hornykiewicz O, Davidson L, et al. Biochemical evidence of dysfunction of brain neurotransmitters in the Lesch-Nyhan syndrome. N Engl J Med. 1981;305:1106–11. doi: 10.1056/NEJM198111053051902. [DOI] [PubMed] [Google Scholar]
- 28.Saito Y, Ito M, Hanaoka S, et al. Dopamine receptor upregulation in Lesch-Nyhan syndrome: a postmortem study. Neuropediatrics. 1999;30:66–71. doi: 10.1055/s-2007-973462. [DOI] [PubMed] [Google Scholar]
- 29.Jinnah HA, Wojcik BE, Hunt M, et al. Dopamine deficiency in a genetic mouse model of Lesch-Nyhan disease. J Neurosci. 1994;14:1164–75. doi: 10.1523/JNEUROSCI.14-03-01164.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ceballos-Picot I, Mockel L, Potier MC, et al. Hypoxanthine-guanine phosphoribosyl transferase regulates early developmental programming of dopamine neurons: implications for Lesch-Nyhan disease pathogenesis. Hum Mol Gen. 2009;18:2317–27. doi: 10.1093/hmg/ddp164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barnett MH, Jarman PR, Heales SJ, et al. Further case of paroxysmal exercise-induced dystonia and some insights into pathogenesis. Mov Disord. 2002;17:1386–7. doi: 10.1002/mds.10291. [DOI] [PubMed] [Google Scholar]
- 32.Hamann M, Richter A. Striatal increase of extracellular dopamine levels during dystonic episodes in a genetic model of paroxysmal dyskinesia. Neurobiol Dis. 2004;16:78–84. doi: 10.1016/j.nbd.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 33.Napolitano F, Pasqualetti M, Usiello A, et al. Dopamine D2 receptor dysfunction is rescued by adenosine A2A receptor antagonism in a model of DYT1 dystonia. Neurobiol Dis. 2010;38:434–45. doi: 10.1016/j.nbd.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sciamanna G, Bonsi P, Tassone A, et al. Impaired striatal D2 receptor function leads to enhanced GABA transmission in a mouse model of DYT1 dystonia. Neurobiol Dis. 2009;34:133–45. doi: 10.1016/j.nbd.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neychev VK, Fan X, Mitev VI, et al. The basal ganglia and cerebellum interact in the expression of dystonic movement. Brain. 2008;131:2499–509. doi: 10.1093/brain/awn168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horie C, Suzuki Y, Kiyosawa M, et al. Decreased dopamine D-2 receptor binding in essential blepharospasm. Acta Neurol Scand. 2009;119:49–54. doi: 10.1111/j.1600-0404.2008.01053.x. [DOI] [PubMed] [Google Scholar]
- 37.Hierholzer J, Cordes M, Schelosky L, et al. Dopamine D2 receptor imaging with iodine-123-iodobenzamide SPECT in idiopathic rotational torticollis. J Nuc Med. 1994;35:1921–7. [PubMed] [Google Scholar]
- 38.Naumann M, Pirker W, Reiners K, et al. Imaging the pre- and postsynaptic side of striatal dopaminergic synapses in idiopathic cervical dystonia: a SPECT study using [123I] epidepride and [123I] beta-CIT. Mov Disord. 1998;13:319–23. doi: 10.1002/mds.870130219. [DOI] [PubMed] [Google Scholar]
- 39.Boesch SM, Donnemiller E, Muller J, et al. Abnormalities of dopaminergic neurotransmission in SCA2: a combined 123I-betaCIT and 123I-IBZM SPECT study. Mov Disord. 2004;19:1320–5. doi: 10.1002/mds.20159. [DOI] [PubMed] [Google Scholar]
- 40.Berger HJ, van der Werf SP, Horstink CA, et al. Writer's cramp: restoration of striatal D2-binding after successful biofeedback-based sensorimotor training. Parkinsonism Relat Disord. 2007;13:170–3. doi: 10.1016/j.parkreldis.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 41.Lang AE. Dopamine agonists and antagonists in the treatment of idiopathic dystonia. Adv Neurol. 1988;50:561–70. [PubMed] [Google Scholar]
- 42.Mandell S. The treatment of dystonia with L-dopa and haloperidol. Neurology. 1970;20:103–6. doi: 10.1212/wnl.20.11_part_2.103. [DOI] [PubMed] [Google Scholar]
- 43.Thiel A, Dressler D, Kistel C, et al. Clozapine treatment of spasmodic torticollis. Neurology. 1994;44:957–8. doi: 10.1212/wnl.44.5.957. [DOI] [PubMed] [Google Scholar]
- 44.Barrett RE, Yahr MD, Duvoisin RC. Torsion dystonia and spasmodic torticollis--results of treatment with L-dopa. Neurology. 1970;20:107–13. doi: 10.1212/wnl.20.11_part_2.107. [DOI] [PubMed] [Google Scholar]
- 45.Grassi E, Latorraca S, Piacentini S, et al. Risperidone in idiopathic and symptomatic dystonia: preliminary experience. Neurol Sci. 2000;21:121–3. doi: 10.1007/s100720070108. [DOI] [PubMed] [Google Scholar]
- 46.Gourzis P, Polychronopoulos P, Papapetropoulos S, et al. Quetiapine in the treatment of focal tardive dystonia induced by other atypical antipsychotics: a report of 2 cases. Clin Neuropharmacol. 2005;28:195–6. doi: 10.1097/01.wnf.0000174933.89758.c9. [DOI] [PubMed] [Google Scholar]
- 47.Greene P, Shale H, Fahn S. Analysis of open-label trials in torsion dystonia using high dosages of anticholinergics and other drugs. Mov Disord. 1988;3:46–60. doi: 10.1002/mds.870030107. [DOI] [PubMed] [Google Scholar]
- 48.Ming L. Moldy sugarcane poisoning--a case report with a brief review. J Toxicol Clin Toxicol. 1995;33:363–7. doi: 10.3109/15563659509028924. [DOI] [PubMed] [Google Scholar]
- 49.He F, Zhang S, Qian F, et al. Delayed dystonia with striatal CT lucencies induced by a mycotoxin (3-nitropropionic acid) Neurology. 1995;45:2178–83. doi: 10.1212/wnl.45.12.2178. [DOI] [PubMed] [Google Scholar]
- 50.Peraica M, Radic B, Lucic A, et al. Toxic effects of mycotoxins in humans. Bull World Health Organ. 1999;77:754–66. [PMC free article] [PubMed] [Google Scholar]
- 51.Beal MF, Brouillet E, Jenkins BG, et al. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–92. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Herrera-Mundo N, Sitges M. Mechanisms underlying striatal vulnerability to 3-nitropropionic acid. J Neurochem. 2010;114:597–605. doi: 10.1111/j.1471-4159.2010.06789.x. [DOI] [PubMed] [Google Scholar]
- 53.Fernagut PO, Diguet E, Stefanova N, et al. Subacute systemic 3-nitropropionic acid intoxication induces a distinct motor disorder in adult C57Bl/6 mice: behavioural and histopathological characterisation. Neuroscience. 2002;114:1005–17. doi: 10.1016/s0306-4522(02)00205-1. [DOI] [PubMed] [Google Scholar]
- 54.Palfi S, Leventhal L, Goetz CG, et al. Delayed onset of progressive dystonia following subacute 3-nitropropionic acid treatment in Cebus apella monkeys. Mov Disord. 2000;15:524–30. [PubMed] [Google Scholar]
- 55.Brouillet E, Hantraye P, Ferrante RJ, et al. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci U S A. 1995;92:7105–9. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Vries DD, Went LN, Bruyn GW, et al. Genetic and biochemical impairment of mitochondrial complex I activity in a family with Leber hereditary optic neuropathy and hereditary spastic dystonia. Am J Hum Genet. 1996;58:703–11. [PMC free article] [PubMed] [Google Scholar]
- 57.Sarzi E, Brown MD, Lebon S, et al. A novel recurrent mitochondrial DNA mutation in ND3 gene is associated with isolated complex I deficiency causing Leigh syndrome and dystonia. Am J Med Genet A. 2007;143:33–41. doi: 10.1002/ajmg.a.31565. [DOI] [PubMed] [Google Scholar]
- 58.Huoponen K, Vilkki J, Aula P, et al. A new mtDNA mutation associated with Leber hereditary optic neuroretinopathy. Am J Hum Genet. 1991;48:1147–53. [PMC free article] [PubMed] [Google Scholar]
- 59.Makino M, Horai S, Goto Y, et al. Confirmation that a T-to-C mutation at 9176 in mitochondrial DNA is an additional candidate mutation for Leigh's syndrome. Neuromuscular Disord. 1998;8:149–51. doi: 10.1016/s0960-8966(98)00017-0. [DOI] [PubMed] [Google Scholar]
- 60.Santorelli FM, Shanske S, Macaya A, et al. The mutation at nt 8993 of mitochondrial DNA is a common cause of Leigh's syndrome. Ann Neurol. 1993;34:827–34. doi: 10.1002/ana.410340612. [DOI] [PubMed] [Google Scholar]
- 61.Roesch K, Curran SP, Tranebjaerg L, et al. Human deafness dystonia syndrome is caused by a defect in assembly of the DDP1/TIMM8a-TIMM13 complex. Hum Mol Genet. 2002;11:477–86. doi: 10.1093/hmg/11.5.477. [DOI] [PubMed] [Google Scholar]
- 62.Tranebjaerg L, Jensen PK, Van Ghelue M, et al. Neuronal cell death in the visual cortex is a prominent feature of the X-linked recessive mitochondrial deafness-dystonia syndrome caused by mutations in the TIMM8a gene. Ophthalmic Genet. 2001;22:207–23. doi: 10.1076/opge.22.4.207.2220. [DOI] [PubMed] [Google Scholar]
- 63.Binder J, Hofmann S, Kreisel S, et al. Clinical and molecular findings in a patient with a novel mutation in the deafness-dystonia peptide (DDP1) gene. Brain. 2003;126:1814–20. doi: 10.1093/brain/awg174. [DOI] [PubMed] [Google Scholar]
- 64.Ezquerra M, Campdelacreu J, Munoz E, et al. A novel intronic mutation in the DDP1 gene in a family with X-linked dystonia-deafness syndrome. Arch Neurol. 2005;62:306–8. doi: 10.1001/archneur.62.2.306. [DOI] [PubMed] [Google Scholar]
- 65.Arts WFM, Scholte HR, Bogaard JM, et al. NADH-CoQ reductase deficient myopathy – Successful treatment with riboflavin. Lancet. 1983;2:581–2. doi: 10.1016/s0140-6736(83)90618-9. [DOI] [PubMed] [Google Scholar]
- 66.Bernsen P, Gabreels FJM, Ruitenbeek W, et al. Treatment of complex-I deficiency with riboflavin. J Neurol Sci. 1993;118:181–7. doi: 10.1016/0022-510x(93)90108-b. [DOI] [PubMed] [Google Scholar]
- 67.Penn AMW, Lee JWK, Thuillier P, et al. Melas Syndrome with mitochondrial transfer RNALue(UUR) mutation – correlation of clinical state, nerve-conduction, and muscle P-31 magnetic-resonance spectroscopy during treatment with nicotinamide and riboflavin. Neurology. 1992;42:2147–52. doi: 10.1212/wnl.42.11.2147. [DOI] [PubMed] [Google Scholar]
- 68.Benecke R, Strumper P, Weiss H. Electron transfer complex I defect in idiopathic dystonia. Ann Neurol. 1992;32:683–6. doi: 10.1002/ana.410320512. [DOI] [PubMed] [Google Scholar]
- 69.Nigro MA, Martens ME, Awerbuch GI, et al. Partial cytochrome b deficiency and generalized dystonia. Pediatr Neurol. 1990;6:407–10. doi: 10.1016/0887-8994(90)90010-x. [DOI] [PubMed] [Google Scholar]
- 70.Schapira AH, Warner T, Gash MT, et al. Complex I function in familial and sporadic dystonia. Ann Neurol. 1997;41:556–9. doi: 10.1002/ana.410410421. [DOI] [PubMed] [Google Scholar]
- 71.Simon DK, Friedman J, Breakefield XO, et al. A heteroplasmic mitochondrial complex I gene mutation in adult-onset dystonia. Neurogenetics. 2003;4:199–205. doi: 10.1007/s10048-003-0150-3. [DOI] [PubMed] [Google Scholar]
- 72.Pettus EH, Betarbet R, Cottrell B, et al. Immunocytochemical characterization of the mitochondrially encoded ND1 subunit of complex I (NADH: ubiquinone oxidoreductase) in rat brain. J Neurochem. 2000;75:383–92. doi: 10.1046/j.1471-4159.2000.0750383.x. [DOI] [PubMed] [Google Scholar]
- 73.Solano A, Roig M, Vives-Bauza C, et al. Bilateral striatal necrosis associated with a novel mutation in the mitochondrial ND6 gene. Ann Neurol. 2003;54:527–30. doi: 10.1002/ana.10682. [DOI] [PubMed] [Google Scholar]
- 74.Bruyn GW, Vielvoye GJ, Went LN. Hereditary spastic dystonia: a new mitochondrial encephalopathy? Putaminal necrosis as a diagnostic sign. J Neurol Sci. 1991;103:195–202. doi: 10.1016/0022-510x(91)90164-3. [DOI] [PubMed] [Google Scholar]
- 75.Palacino JJ, Sagi D, Goldberg MS, et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–22. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 76.Cuenca-Leon E, Banchs I, Serra SA, et al. Late-onset episodic ataxia type 2 associated with a novel loss-of-function mutation in the CACNA1A gene. J Neurol Sci. 2009;280:10–4. doi: 10.1016/j.jns.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 77.Giffin NJ, Benton S, Goadsby PJ. Benign paroxysmal torticollis of infancy: four new cases and linkage to CACNA1A mutation. Dev Med Child Neurol. 2002;44:490–3. doi: 10.1017/s0012162201002407. [DOI] [PubMed] [Google Scholar]
- 78.Roubertie A, Echenne B, Leydet J, et al. Benign paroxysmal tonic upgaze, benign paroxysmal torticollis, episodic ataxia and CACNA1A mutation in a family. J Neurol. 2008;255:1600–2. doi: 10.1007/s00415-008-0982-8. [DOI] [PubMed] [Google Scholar]
- 79.Spacey SD, Materek LA, Szczygielski BI, et al. Two novel CACNA1A gene mutations associated with episodic ataxia type 2 and interictal dystonia. Arch Neurol. 2005;62:314–6. doi: 10.1001/archneur.62.2.314. [DOI] [PubMed] [Google Scholar]
- 80.Vesper J, Klostermann F, Funk T, et al. Deep brain stimulation of the globus pallidus internus (GPI) for torsion dystonia--a report of two cases. Acta Neurochir Suppl. 2002;79:83–8. doi: 10.1007/978-3-7091-6105-0_19. [DOI] [PubMed] [Google Scholar]
- 81.Bereznai B, Steude U, Seelos K, et al. Chronic high-frequency globus pallidus internus stimulation in different types of dystonia: a clinical, video, and MRI report of six patients presenting with segmental, cervical, and generalized dystonia. Mov Disord. 2002;17:138–44. doi: 10.1002/mds.1250. [DOI] [PubMed] [Google Scholar]
- 82.McIntyre CC, Grill WM, Sherman DL, et al. Cellular effects of deep brain stimulation: model-based analysis of activation and inhibition. J Neurophysiol. 2004;91:1457–69. doi: 10.1152/jn.00989.2003. [DOI] [PubMed] [Google Scholar]
- 83.Jun K, Piedras-Renteria ES, Smith SM, et al. Ablation of P/Q-type Ca(2+) channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the alpha(1A)-subunit. Proc Natl Acad Sci U S A. 1999;96:15245–50. doi: 10.1073/pnas.96.26.15245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fletcher CF, Tottene A, Lennon VA, et al. Dystonia and cerebellar atrophy in Cacna1a null mice lacking P/Q calcium channel activity. FASEB Journal. 2001;15:1288–90. doi: 10.1096/fj.00-0562fje. [DOI] [PubMed] [Google Scholar]
- 85.Raike RS, Jinnah HA, Hess EJ. Animal models of generalized dystonia. NeuroRx. 2005;2:504–12. doi: 10.1602/neurorx.2.3.504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xie G, Clapcote SJ, Nieman BJ, et al. Forward genetic screen of mouse reveals dominant missense mutation in the P/Q-type voltage-dependent calcium channel, CACNA1A. Genes Brain Behav. 2007;6:717–27. doi: 10.1111/j.1601-183X.2007.00302.x. [DOI] [PubMed] [Google Scholar]
- 87.Doyle J, Ren X, Lennon G, et al. Mutations in the Cacnl1a4 calcium channel gene are associated with seizures, cerebellar degeneration, and ataxia in tottering and leaner mutant mice. Mammalian Genome. 1997;8:113–20. doi: 10.1007/s003359900369. [DOI] [PubMed] [Google Scholar]
- 88.Lorenzon NM, Lutz CM, Frankel WN, et al. Altered calcium channel currents in Purkinje cells of the neurological mutant mouse leaner. J Neurosci. 1998;18:4482–9. doi: 10.1523/JNEUROSCI.18-12-04482.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shirley TL, Rao LM, Hess EJ, et al. Paroxysmal dyskinesias in mice. Mov Disord. 2008;23:259–64. doi: 10.1002/mds.21829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zwingman TA, Neumann PE, Noebels JL, et al. Rocker is a new variant of the voltage-dependent calcium channel gene Cacna1a. J Neurosci. 2001;21:1169–78. doi: 10.1523/JNEUROSCI.21-04-01169.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wakamori M, Yamazaki K, Matsunodaira H. Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. J Biol Chem. 1998;273:34857–67. doi: 10.1074/jbc.273.52.34857. [DOI] [PubMed] [Google Scholar]
- 92.Pietrobon D. CaV2.1 channelopathies. Pflugers Arch. 2010;460:375–93. doi: 10.1007/s00424-010-0802-8. [DOI] [PubMed] [Google Scholar]
- 93.Jinnah HA, Sepkuty JP, Ho T, et al. Calcium channel agonists and dystonia in the mouse. Mov Disord. 2000;15:542–51. doi: 10.1002/1531-8257(200005)15:3<542::AID-MDS1019>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 94.Fureman BE, Jinnah HA, Hess EJ. Triggers of paroxysmal dyskinesia in the calcium channel mouse mutant tottering. Pharmacol Biochem Behav. 2002;73:631–7. doi: 10.1016/s0091-3057(02)00854-7. [DOI] [PubMed] [Google Scholar]
- 95.Richter A, Loscher W. Antidystonic effects of L-type Ca2+ channel antagonists in a hamster model of idiopathic dystonia. Eur J Pharmacol. 1996;300:197–202. doi: 10.1016/0014-2999(95)00878-0. [DOI] [PubMed] [Google Scholar]
- 96.Wszolek ZK, Baba Y, Mackenzie IR, et al. Autosomal dominant dystonia-plus with cerebral calcifications. Neurology. 2006;67:620–5. doi: 10.1212/01.wnl.0000230141.40784.09. [DOI] [PubMed] [Google Scholar]
- 97.Du W, Bautista JF, Yang H, et al. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat Genet. 2005;37:733–8. doi: 10.1038/ng1585. [DOI] [PubMed] [Google Scholar]
- 98*.Neychev VK, Gross RE, Lehericy S, et al. The functional neuroanatomy of dystonia. Neurobiol Dis. 2011;42:185–201. doi: 10.1016/j.nbd.2011.01.026. Thorough overview of the circuitry implicated in dystonia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bhatia KP, Marsden CD. The behavioural and motor consequences of focal lesions of the basal ganglia in man. Brain. 1994;117:859–76. doi: 10.1093/brain/117.4.859. [DOI] [PubMed] [Google Scholar]
- 100.Kurita H, Sasaki T, Suzuki I, et al. Basal ganglia arteriovenous malformation presenting as “writer's cramp”. Childs Nerv Syst. 1998;14:285–7. doi: 10.1007/s003810050227. [DOI] [PubMed] [Google Scholar]
- 101.Mahant N, Cordato DJ, Fung VS. Focal myoclonus-dystonia of the leg secondary to a lesion of the posterolateral putamen: clinical and neurophysiological features. Mov Disord. 2003;18:452–5. doi: 10.1002/mds.10382. [DOI] [PubMed] [Google Scholar]
- 102.Munchau A, Mathen D, Cox T, et al. Unilateral lesions of the globus pallidus: report of four patients presenting with focal or segmental dystonia. J Neurol Neurosurg Psychiatry. 2000;69:494–8. doi: 10.1136/jnnp.69.4.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Walker RH, Purohit DP, Good PF, et al. Severe generalized dystonia due to primary putaminal degeneration: case report and review of the literature. Mov Disord. 2002;17:576–84. doi: 10.1002/mds.10098. [DOI] [PubMed] [Google Scholar]
- 104.Galardi G, Perani D, Grassi F, et al. Basal ganglia and thalamo-cortical hypermetabolism in patients with spasmodic torticollis. Acta Neurol Scand. 1996;94:172–6. doi: 10.1111/j.1600-0404.1996.tb07049.x. [DOI] [PubMed] [Google Scholar]
- 105.Carbon M, Su S, Dhawan V, et al. Regional metabolism in primary torsion dystonia: effects of penetrance and genotype. Neurology. 2004;62:1384–90. doi: 10.1212/01.wnl.0000120541.97467.fe. [DOI] [PubMed] [Google Scholar]
- 106.Eidelberg D, Moeller JR, Antonini A, et al. Functional brain networks in DYT1 dystonia. Ann Neurol. 1998;44:303–12. doi: 10.1002/ana.410440304. [DOI] [PubMed] [Google Scholar]
- 107.Kerrison JB, Lancaster JL, Zamarripa FE, et al. Positron emission tomography scanning in essential blepharospasm. Am J Ophthalmol. 2003;136:846–52. doi: 10.1016/s0002-9394(03)00895-x. [DOI] [PubMed] [Google Scholar]
- 108.Peller M, Zeuner KE, Munchau A, et al. The basal ganglia are hyperactive during the discrimination of tactile stimuli in writer's cramp. Brain. 2006;129:2697–708. doi: 10.1093/brain/awl181. [DOI] [PubMed] [Google Scholar]
- 109.Hu XY, Wang L, Liu H, et al. Functional magnetic resonance imaging study of writer's cramp. Chin Med J (Engl) 2006;119:1263–71. [PubMed] [Google Scholar]
- 110.Blood AJ, Flaherty AW, Choi JK, et al. Basal ganglia activity remains elevated after movement in focal hand dystonia. Ann Neurol. 2004;55:744–8. doi: 10.1002/ana.20108. [DOI] [PubMed] [Google Scholar]
- 111.Tolosa E, Compta Y. Dystonia in Parkinson's disease. J Neurol. 2006;253(7):VII7–13. doi: 10.1007/s00415-006-7003-6. [DOI] [PubMed] [Google Scholar]
- 112.Louis ED, Anderson KE, Moskowitz C, et al. Dystonia-predominant adult-onset Huntington disease: association between motor phenotype and age of onset in adults. Arch Neurol. 2000;57:1326–30. doi: 10.1001/archneur.57.9.1326. [DOI] [PubMed] [Google Scholar]
- 113.Louis ED, Lee P, Quinn L, et al. Dystonia in Huntington's disease: prevalence and clinical characteristics. Mov Disord. 1999;14:95–101. doi: 10.1002/1531-8257(199901)14:1<95::aid-mds1016>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 114.Oliver J, Dewhurst K. Childhood and adolescent forms of Huntington's disease. J Neurol Neurosurg Psychiatry. 1969;32:455–9. doi: 10.1136/jnnp.32.5.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Byers RK, Gilles FH, Fung C. Huntington's disease in children. Neuropathologic study of four cases. Neurology. 1973;23:561–9. doi: 10.1212/wnl.23.6.561. [DOI] [PubMed] [Google Scholar]
- 116.Lozano AM, Kumar R, Gross RE, et al. Globus pallidus internus pallidotomy for generalized dystonia. Mov Disord. 1997;12:865–70. doi: 10.1002/mds.870120606. [DOI] [PubMed] [Google Scholar]
- 117.Ford B. Pallidotomy for generalized dystonia. Adv Neurol. 2004;94:287–99. [PubMed] [Google Scholar]
- 118.Gross RE. What happened to posteroventral pallidotomy for Parkinson's disease and dystonia? Neurotherapeutics. 2008;5:281–93. doi: 10.1016/j.nurt.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ostrem JL, Racine CA, Glass GA, et al. Subthalamic nucleus deep brain stimulation in primary cervical dystonia. Neurology. 2011;76:870–8. doi: 10.1212/WNL.0b013e31820f2e4f. [DOI] [PubMed] [Google Scholar]
- 120.Isaias IU, Alterman RL, Tagliati M. Deep brain stimulation for primary generalized dystonia: long-term outcomes. Arch Neurol. 2009;66:465–70. doi: 10.1001/archneurol.2009.20. [DOI] [PubMed] [Google Scholar]
- 121.Bittar RG, Yianni J, Wang S, et al. Deep brain stimulation for generalised dystonia and spasmodic torticollis. J Clin Neurosci. 2005;12:12–6. doi: 10.1016/j.jocn.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 122.Krauss JK, Loher TJ, Weigel R, et al. Chronic stimulation of the globus pallidus internus for treatment of non-dYT1 generalized dystonia and choreoathetosis: 2-year follow up. J Neurosurg. 2003;98:785–92. doi: 10.3171/jns.2003.98.4.0785. [DOI] [PubMed] [Google Scholar]
- 123.Capelle HH, Blahak C, Schrader C, et al. Chronic deep brain stimulation in patients with tardive dystonia without a history of major psychosis. Mov Disord. 2010;25:1477–81. doi: 10.1002/mds.23123. [DOI] [PubMed] [Google Scholar]
- 124.Chang EF, Schrock LE, Starr PA, et al. Long-term benefit sustained after bilateral pallidal deep brain stimulation in patients with refractory tardive dystonia. Stereotact Funct Neurosurg. 2010;88:304–10. doi: 10.1159/000316763. [DOI] [PubMed] [Google Scholar]
- 125.Gruber D, Trottenberg T, Kivi A, et al. Long-term effects of pallidal deep brain stimulation in tardive dystonia. Neurology. 2009;73:53–8. doi: 10.1212/WNL.0b013e3181aaea01. [DOI] [PubMed] [Google Scholar]
- 126.Usmani N, Bedi GS, Sengun C, et al. Late onset of cervical dystonia in a 39-year-old patient following cerebellar hemorrhage. J Neurol. 2011;258:149–51. doi: 10.1007/s00415-010-5685-2. [DOI] [PubMed] [Google Scholar]
- 127.Waln O, LeDoux MS. Delayed-onset oromandibular dystonia after a cerebellar hemorrhagic stroke. Parkinsonism Relat Disord. 2010;16:623–5. doi: 10.1016/j.parkreldis.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 128.Zadro I, Brinar VV, Barun B, et al. Cervical dystonia due to cerebellar stroke. Mov Disord. 2008;23:919–20. doi: 10.1002/mds.21981. [DOI] [PubMed] [Google Scholar]
- 129.O'Rourke K, O'Riordan S, Gallagher J, et al. Paroxysmal torticollis and blepharospasm following bilateral cerebellar infarction. J Neurol. 2006;253:1644–5. doi: 10.1007/s00415-006-0202-3. [DOI] [PubMed] [Google Scholar]
- 130.Rumbach L, Barth P, Costaz A, et al. Hemidystonia consequent upon ipsilateral vertebral artery occlusion and cerebellar infarction. Mov Disord. 1995;10:522–5. doi: 10.1002/mds.870100424. [DOI] [PubMed] [Google Scholar]
- 131.Krauss JK, Seeger W, Jankovic J. Cervical dystonia associated with tumors of the posterior fossa. Mov Disord. 1997;12:443–7. doi: 10.1002/mds.870120329. [DOI] [PubMed] [Google Scholar]
- 132.LeDoux MS, Brady KA. Secondary cervical dystonia associated with structural lesions of the central nervous system. Mov Disord. 2003;18:60–9. doi: 10.1002/mds.10301. [DOI] [PubMed] [Google Scholar]
- 133.Caress JB, Nohria V, Fuchs H, et al. Torticollis acquired in late infancy due to a cerebellar gangliocytoma. Int J Pediatr Otorhinolaryngol. 1996;36:39–44. doi: 10.1016/0165-5876(95)01320-2. [DOI] [PubMed] [Google Scholar]
- 134.Alarcon F, Tolosa E, Munoz E. Focal limb dystonia in a patient with a cerebellar mass. Arch Neurol. 2001;58:1125–7. doi: 10.1001/archneur.58.7.1125. [DOI] [PubMed] [Google Scholar]
- 135.Draganski B, Thun-Hohenstein C, Bogdahn U, et al. “Motor circuit” gray matter changes in idiopathic cervical dystonia. Neurology. 2003;61:1228–31. doi: 10.1212/01.wnl.0000094240.93745.83. [DOI] [PubMed] [Google Scholar]
- 136.Obermann M, Yaldizli O, De Greiff A, et al. Morphometric changes of sensorimotor structures in focal dystonia. Mov Disord. 2007;22:1117–23. doi: 10.1002/mds.21495. [DOI] [PubMed] [Google Scholar]
- 137.Delmaire C, Vidailhet M, Elbaz A, et al. Structural abnormalities in the cerebellum and sensorimotor circuit in writer's cramp. Neurology. 2007;69:376–80. doi: 10.1212/01.wnl.0000266591.49624.1a. [DOI] [PubMed] [Google Scholar]
- 138.Kluge A, Kettner B, Zschenderlein R, et al. Changes in perfusion pattern using ECD-SPECT indicate frontal lobe and cerebellar involvement in exercise-induced paroxysmal dystonia. Mov Disord. 1998;13:125–34. doi: 10.1002/mds.870130124. [DOI] [PubMed] [Google Scholar]
- 139.Yoon JH, Lee PH, Yoon SN. Subtraction brain SPECT imaging in a patient with paroxysmal exercise-induced dystonia: role of the primary somatosensory cortex. Arch Neurol. 2007;64:1652–6. doi: 10.1001/archneur.64.11.1652. [DOI] [PubMed] [Google Scholar]
- 140.LeDoux MS, Rutledge SL, Mountz JM, et al. SPECT abnormalities in generalized dystonia. Pediatr Neurol. 1995;13:5–10. doi: 10.1016/0887-8994(95)00109-s. [DOI] [PubMed] [Google Scholar]
- 141.Preibisch C, Berg D, Hofmann E, et al. Cerebral activation patterns in patients with writer's cramp: a functional magnetic resonance imaging study. J Neurol. 2001;248:10–7. doi: 10.1007/s004150170263. [DOI] [PubMed] [Google Scholar]
- 142.Hutchinson M, Nakamura T, Moeller JR, et al. The metabolic topography of essential blepharospasm: a focal dystonia with general implications. Neurology. 2000;55:673–7. doi: 10.1212/wnl.55.5.673. [DOI] [PubMed] [Google Scholar]
- 143.Argyelan M, Carbon M, Niethammer M, et al. Cerebellothalamocortical connectivity regulates penetrance in dystonia. J Neurosci. 2009;29:9740–7. doi: 10.1523/JNEUROSCI.2300-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Campbell DB, Hess EJ. Cerebellar circuitry is activated during convulsive episodes in the tottering (tg/tg) mutant mouse. Neuroscience. 1998;85:773–83. doi: 10.1016/s0306-4522(97)00672-6. [DOI] [PubMed] [Google Scholar]
- 145.Chen G, Popa LS, Wang X, et al. Low-frequency oscillations in the cerebellar cortex of the tottering mouse. J Neurophysiol. 2009;101:234–45. doi: 10.1152/jn.90829.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Brown LL, Lorden JF. Regional cerebral glucose utilization reveals widespread abnormalities in the motor system of the rat mutant dystonic. J Neurosci. 1989;9:4033–41. doi: 10.1523/JNEUROSCI.09-11-04033.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Richter A, Brotchie JM, Crossman AR, et al. [3H]-2-deoxyglucose uptake study in mutant dystonic hamsters: abnormalities in discrete brain regions of the motor system. Mov Disord. 1998;13:718–25. doi: 10.1002/mds.870130419. [DOI] [PubMed] [Google Scholar]
- 148.Pizoli CE, Jinnah HA, Billingsley ML, et al. Abnormal cerebellar signaling induces dystonia in mice. J Neurosci. 2002;22:7825–33. doi: 10.1523/JNEUROSCI.22-17-07825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.LeDoux MS, Lorden JF, Meinzen-Derr J. Selective elimination of cerebellar output in the genetically dystonic rat. Brain Res. 1995;697:91–103. doi: 10.1016/0006-8993(95)00792-o. [DOI] [PubMed] [Google Scholar]
- 150.Devanagondi R, Egami K, LeDoux MS, et al. Neuroanatomical substrates for paroxysmal dyskinesia in lethargic mice. Neurobiol Dis. 2007;27:249–57. doi: 10.1016/j.nbd.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Campbell DB, North JB, Hess EJ. Tottering mouse motor dysfunction is abolished on the Purkinje cell degeneration (pcd) mutant background. Exp Neurol. 1999;160:268–78. doi: 10.1006/exnr.1999.7171. [DOI] [PubMed] [Google Scholar]
- 152.Calderon DP, Fremont R, Kraenzlin F, et al. The neural substrates of rapid-onset Dystonia-Parkinsonism. Nat Neurosci. 2011;14:357–65. doi: 10.1038/nn.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Baumer T, Demiralay C, Hidding U, et al. Abnormal plasticity of the sensorimotor cortex to slow repetitive transcranial magnetic stimulation in patients with writer's cramp. Mov Disord. 2007;22:81–90. doi: 10.1002/mds.21219. [DOI] [PubMed] [Google Scholar]
- 154.Braun C, Schweizer R, Heinz U, et al. Task-specific plasticity of somatosensory cortex in patients with writer's cramp. Neuroimage. 2003;20:1329–38. doi: 10.1016/S1053-8119(03)00375-6. [DOI] [PubMed] [Google Scholar]
- 155.Edwards MJ, Huang YZ, Mir P, et al. Abnormalities in motor cortical plasticity differentiate manifesting and nonmanifesting DYT1 carriers. Mov Disord. 2006;21:2181–6. doi: 10.1002/mds.21160. [DOI] [PubMed] [Google Scholar]
- 156.Quartarone A, Morgante F, Sant'angelo A, et al. Abnormal plasticity of sensorimotor circuits extends beyond the affected body part in focal dystonia. J Neurol Neurosurg Psychiatry. 2008;79:985–90. doi: 10.1136/jnnp.2007.121632. [DOI] [PubMed] [Google Scholar]
- 157.Tamura Y, Ueki Y, Lin P, et al. Disordered plasticity in the primary somatosensory cortex in focal hand dystonia. Brain. 2009;132:749–55. doi: 10.1093/brain/awn348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mehrkens JH, Botzel K, Steude U, et al. Long-term efficacy and safety of chronic globus pallidus internus stimulation in different types of primary dystonia. Stereotact Funct Neurosurg. 2009;87:8–17. doi: 10.1159/000177623. [DOI] [PubMed] [Google Scholar]
- 159.Kohling R, Koch UR, Hamann M, et al. Increased excitability in cortico-striatal synaptic pathway in a model of paroxysmal dystonia. Neurobiol Dis. 2004;16:236–45. doi: 10.1016/j.nbd.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 160.Martella G, Tassone A, Sciamanna G, et al. Impairment of bidirectional synaptic plasticity in the striatum of a mouse model of DYT1 dystonia: role of endogenous acetylcholine. Brain. 2009;132:2336–49. doi: 10.1093/brain/awp194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pisani A, Martella G, Tscherter A, et al. Altered responses to dopaminergic D2 receptor activation and N-type calcium currents in striatal cholinergic interneurons in a mouse model of DYT1 dystonia. Neurobiol Dis. 2006;24:318–25. doi: 10.1016/j.nbd.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 162.Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson's disease models. Nature. 2007;445:643–7. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- 163.Ferre S. Adenosine-dopamine interactions in the ventral striatum. Implications for the treatment of schizophrenia. Psychopharmacology (Berl) 1997;133:107–20. doi: 10.1007/s002130050380. [DOI] [PubMed] [Google Scholar]
- 164.Elbert T, Candia V, Altenmuller E, et al. Alteration of digital representations in somatosensory cortex in focal hand dystonia. Neuroreport. 1998;9:3571–5. doi: 10.1097/00001756-199811160-00006. [DOI] [PubMed] [Google Scholar]
- 165.Granert O, Peller M, Gaser C, et al. Manual activity shapes structure and function in contralateral human motor hand area. Neuroimage. 2011;54:32–41. doi: 10.1016/j.neuroimage.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 166.Kang JS, Terranova C, Hilker R, et al. Deficient homeostatic regulation of practice-dependent plasticity in writer's cramp. Cereb Cortex. 2011;21:1203–12. doi: 10.1093/cercor/bhq204. [DOI] [PubMed] [Google Scholar]
- 167.Byl NN, Merzenich MM, Jenkins WM. A primate genesis model of focal dystonia and repetitive strain injury: I. Learning-induced dedifferentiation of the representation of the hand in the primary somatosensory cortex in adult monkeys. Neurology. 1996;47:508–20. doi: 10.1212/wnl.47.2.508. [DOI] [PubMed] [Google Scholar]
- 168.Alterman RL, Miravite J, Weisz D, et al. Sixty hertz pallidal deep brain stimulation for primary torsion dystonia. Neurology. 2007;69:681–8. doi: 10.1212/01.wnl.0000267430.95106.ff. [DOI] [PubMed] [Google Scholar]
- 169.Sun B, Chen S, Zhan S, et al. Subthalamic nucleus stimulation for primary dystonia and tardive dystonia. Acta Neurochir Suppl. 2007;97:207–14. doi: 10.1007/978-3-211-33081-4_23. [DOI] [PubMed] [Google Scholar]
- 170.Bressman SB. Dystonia genotypes, phenotypes, and classification. Adv Neurol. 2004;94:101–7. [PubMed] [Google Scholar]
- 171.Kojovic M, Caronni A, Bologna M, et al. Botulinum toxin injections reduce associative plasticity in patients with primary dystonia. Mov Disord. 2011;26:1282–9. doi: 10.1002/mds.23681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Blomstedt P, Tisch S, Hariz MI. Pallidal deep brain stimulation in the treatment of Meige syndrome. Acta Neurol Scand. 2008;118:198–202. doi: 10.1111/j.1600-0404.2008.00999.x. [DOI] [PubMed] [Google Scholar]
- 173.Fukaya C, Katayama Y, Kano T, et al. Thalamic deep brain stimulation for writer's cramp. J Neurosurg. 2007;107:977–82. doi: 10.3171/JNS-07/11/0977. [DOI] [PubMed] [Google Scholar]
- 174.Reese R, Charron G, Nadjar A, et al. High frequency stimulation of the entopeduncular nucleus sets the cortico-basal ganglia network to a new functional state in the dystonic hamster. Neurobiol Dis. 2009;35:399–405. doi: 10.1016/j.nbd.2009.05.022. [DOI] [PubMed] [Google Scholar]
- 175.Tisch S, Rothwell JC, Bhatia KP, et al. Pallidal stimulation modifies after-effects of paired associative stimulation on motor cortex excitability in primary generalised dystonia. Exp Neurol. 2007;206:80–5. doi: 10.1016/j.expneurol.2007.03.027. [DOI] [PubMed] [Google Scholar]
- 176.Kuncel AM, Turner DA, Ozelius LJ, et al. Myoclonus and tremor response to thalamic deep brain stimulation parameters in a patient with inherited myoclonus-dystonia syndrome. Clin Neurol Neurosurg. 2009;111:303–6. doi: 10.1016/j.clineuro.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Panizza M, Lelli S, Nilsson J, et al. H-reflex recovery curve and reciprocal inhibition of H-reflex in different kinds of dystonia. Neurology. 1990;40:824–8. doi: 10.1212/wnl.40.5.824. [DOI] [PubMed] [Google Scholar]
- 178.Nakashima K, Rothwell JC, Day BL, et al. Reciprocal inhibition between forearm muscles in patients with writer's cramp and other occupational cramps, symptomatic hemidystonia and hemiparesis due to stroke. Brain. 1989;112:681–97. doi: 10.1093/brain/112.3.681. [DOI] [PubMed] [Google Scholar]
- 179.Tisch S, Limousin P, Rothwell JC, et al. Changes in forearm reciprocal inhibition following pallidal stimulation for dystonia. Neurology. 2006;66:1091–3. doi: 10.1212/01.wnl.0000204649.36458.8f. [DOI] [PubMed] [Google Scholar]
- 180.Priori A, Berardelli A, Mercuri B, et al. Physiological effects produced by botulinum toxin treatment of upper limb dystonia. Changes in reciprocal inhibition between forearm muscles Brain. 1995;118:801–7. doi: 10.1093/brain/118.3.801. [DOI] [PubMed] [Google Scholar]
- 181.Ridding MC, Sheean G, Rothwell JC, et al. Changes in the balance between motor cortical excitation and inhibition in focal, task specific dystonia. J Neurol Neurosurg Psychiatry. 1995;59:493–8. doi: 10.1136/jnnp.59.5.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Beck S, Shamim EA, Richardson SP, et al. Inter-hemispheric inhibition is impaired in mirror dystonia. Eur J Neurosci. 2009;29:1634–40. doi: 10.1111/j.1460-9568.2009.06710.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Sohn YH, Hallett M. Disturbed surround inhibition in focal hand dystonia. Ann Neurol. 2004;56:595–9. doi: 10.1002/ana.20270. [DOI] [PubMed] [Google Scholar]
- 184.Beck S, Houdayer E, Richardson SP, et al. The role of inhibition from the left dorsal premotor cortex in right-sided focal hand dystonia. Brain Stimul. 2009;2:208–14. doi: 10.1016/j.brs.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Matsumura M, Sawaguchi T, Oishi T, et al. Behavioral deficits induced by local injection of bicuculline and muscimol into the primate motor and premotor cortex. J Neurophysiol. 1991;65:1542–53. doi: 10.1152/jn.1991.65.6.1542. [DOI] [PubMed] [Google Scholar]
- 186.Matsumura M, Sawaguchi T, Kubota K. GABAergic inhibition of neuronal activity in the primate motor and premotor cortex during voluntary movement. J Neurophysiol. 1992;68:692–702. doi: 10.1152/jn.1992.68.3.692. [DOI] [PubMed] [Google Scholar]
- 187.Deuschl G, Heinen F, Kleedorfer B, et al. Clinical and polymyographic investigation of spasmodic torticollis. J Neurol. 1992;239:9–15. doi: 10.1007/BF00839204. [DOI] [PubMed] [Google Scholar]
- 188.Muller J, Wissel J, Masuhr F, et al. Clinical characteristics of the geste antagoniste in cervical dystonia. J Neurol. 2001;248:478–82. doi: 10.1007/s004150170156. [DOI] [PubMed] [Google Scholar]
- 189.Wissel J, Muller J, Ebersbach G, et al. Trick maneuvers in cervical dystonia: investigation of movement- and touch-related changes in polymyographic activity. Mov Disord. 1999;14:994–9. doi: 10.1002/1531-8257(199911)14:6<994::aid-mds1013>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 190.Naumann M, Magyar-Lehmann S, Reiners K, et al. Sensory tricks in cervical dystonia: perceptual dysbalance of parietal cortex modulates frontal motor programming. Ann Neurol. 2000;47:322–8. [PubMed] [Google Scholar]
- 191.Schramm A, Classen J, Reiners K, et al. Characteristics of sensory trick-like manoeuvres in jaw-opening dystonia. Mov Disord. 2007;22:430–3. doi: 10.1002/mds.21354. [DOI] [PubMed] [Google Scholar]
- 192.Sheehy M, Marsden C. Writers' cramp- a focal dystonia. Brain. 1982;105:465–80. doi: 10.1093/brain/105.3.461. [DOI] [PubMed] [Google Scholar]
- 193.Gomez-Wong E, Marti MJ, Tolosa E, et al. Sensory modulation of the blink reflex in patients with blepharospasm. Arch Neurol. 1998;55:1233–7. doi: 10.1001/archneur.55.9.1233. [DOI] [PubMed] [Google Scholar]
- 194.Yoneda Y, Rome S, Sagar HJ, et al. Abnormal perception of the tonic vibration reflex in idiopathic focal dystonia. Eur J Neurol. 2000;7:529–33. doi: 10.1046/j.1468-1331.2000.t01-1-00102.x. [DOI] [PubMed] [Google Scholar]
- 195.Frima N, Nasir J, Grunewald RA. Abnormal vibration-induced illusion of movement in idiopathic focal dystonia: an endophenotypic marker? Mov Disord. 2008;23:373–7. doi: 10.1002/mds.21838. [DOI] [PubMed] [Google Scholar]
- 196.Kaji R, Rothwell JC, Katayama M, et al. Tonic vibration reflex and muscle afferent block in writer's cramp. Ann Neurol. 1995;38:155–62. doi: 10.1002/ana.410380206. [DOI] [PubMed] [Google Scholar]
- 197.Rosenkranz K, Williamon A, Butler K, et al. Pathophysiological differences between musician's dystonia and writer's cramp. Brain. 2005;128:918–31. doi: 10.1093/brain/awh402. [DOI] [PubMed] [Google Scholar]
- 198.Tamburin S, Manganotti P, Marzi CA, et al. Abnormal somatotopic arrangement of sensorimotor interactions in dystonic patients. Brain. 2002;125:2719–30. doi: 10.1093/brain/awf279. [DOI] [PubMed] [Google Scholar]
- 199.Bara-Jimenez W, Catalan MJ, Hallett M, et al. Abnormal somatosensory homunculus in dystonia of the hand. Ann Neurol. 1998;44:828–31. doi: 10.1002/ana.410440520. [DOI] [PubMed] [Google Scholar]
- 200.Nelson AJ, Blake DT, Chen R. Digit-specific aberrations in the primary somatosensory cortex in Writer's cramp. Ann Neurol. 2009;66:146–54. doi: 10.1002/ana.21626. [DOI] [PubMed] [Google Scholar]
- 201.Lim J, Hao T, Shaw C, et al. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–14. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 202.Vila M, Przedborski S. Genetic clues to the pathogenesis of Parkinson's disease. Nat Med. 2004;(10):S58–62. doi: 10.1038/nm1068. [DOI] [PubMed] [Google Scholar]
- 203.Stone EA, Lin Y, Quartermain D. A final common pathway for depression? Progress toward a general conceptual framework. Neurosci Biobehav Rev. 2008;32:508–24. doi: 10.1016/j.neubiorev.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Benatar M. Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol Dis. 2007;26:1–13. doi: 10.1016/j.nbd.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 205.Friese MA, Montalban X, Willcox N, et al. The value of animal models for drug development in multiple sclerosis. Brain. 2006;129:1940–52. doi: 10.1093/brain/awl083. [DOI] [PubMed] [Google Scholar]