

Abstract
Autophagy is an evolutionarily conserved intracellular mechanism for degradation of long-lived proteins and organelles. Accumulating lines of evidence indicate that autophagy is deeply involved in the development of cardiac disease. Autophagy is upregulated in almost all cardiac pathological states, exerting both protective and detrimental functions. Whether autophagy activation is an adaptive or maladaptive mechanism during cardiac stress seems to depend upon the pathological context in which it is upregulated, the extent of its activation, and the signaling mechanisms promoting its enhancement. Pharmacological modulation of autophagy may therefore represent a potential therapeutic strategy to limit myocardial damage during cardiac stress. Several pharmacological agents that are able to modulate autophagy have been identified, such as mTOR inhibitors, AMPK modulators, sirtuin activators, IP3 and calcium lowering agents, and lysosome inhibitors. Although few of these modulators of autophagy have been directly tested during cardiac stress, many of them appear to have high potential to be efficient in the treatment of cardiac disease. We will discuss the potential usefulness of different pharmacological activators and inhibitors of autophagy in the treatment of cardiac diseases.
Introduction
Macroautophagy is an intracellular bulk degradation process, in which long-lived proteins and organelles are sequestered by double-membrane vacuoles, termed autophagosomes, and delivered to lysosomes for degradation 1, 2.
Macroautophagy (hereafter autophagy) occurs under basal conditions and mediates homeostatic functions in cells. However, autophagy is also induced by stress, such as energy deprivation, endoplasmic reticulum stress and oxidative stress. When autophagy is upregulated to moderate levels, it exerts protective cellular functions, such as ATP production and clearance of oxidized proteins and damaged organelles. Therefore, defects in protective autophagy would exacerbate energy stress, ER stress and mitochondrial dysfunction, thus promoting necrotic or apoptotic cell death. On the other hand, when it is activated excessively, autophagy can induce cell death, possibly through depletion of essential proteins and organelles. This form of cell death is defined as type II programmed cell death or autophagic cell death 1, 2. Recent evidence also indicates that exaggerated activation of autophagy may promote other forms of cell death as well. It has been shown in the context of autophagy activation, cleavage of Atg5, Atg4D and Beclin-1, or Bcl-2 sequestration by Beclin-1 may also promote or enhance apoptosis 3. In addition, autophagy can induce necrosis in cells with defective apoptosis 4.
Accumulating lines of evidence indicate that autophagy is activated at baseline and in response to stress, including myocardial ischemia, reperfusion and heart failure, in the heart and the cardiomyocytes therein 5. In experimental animals, modulation of autophagy in the heart affects LV function, the extent of myocardial injury, and even survival of the animals both at baseline and in response to stress. These findings suggest that autophagy is intimately involved in the pathogenesis of heart disease and that modulation of autophagy may be considered as a novel modality of treatment for heart disease. This review discusses the potential usefulness of different pharmacological activators and inhibitors of autophagy in the treatment of cardiac diseases.
Mechanisms regulating autophagy
Autophagosomes are vesicles limited by a lipid bilayer membrane, representing the functional unit of autophagy. Autophagosome formation, consisting of induction, nucleation, expansion and maturation/retrieval of autophagosomes, is finely regulated by the autophagy-related proteins. Induction and nucleation are characterized by the formation of an isolated membrane, known as the phagophore, located at the phagophore assembly site. The source site of the phagophore has been advocated to be in the endoplasmic reticulum, in the mitochondrion, or in other unknown sites 1, 2. Phagophore formation is regulated by two multiprotein complexes. The first complex, composed of Atg13, Unc-51-like kinase 1 (ULK-1, a mammalian homolog of Atg1) and ULK-2, phosphorylates the focal adhesion kinase-family interacting protein 200 (FIP200) and promotes phagophore initiation. The second functional complex is composed of the class-III phosphoinositide 3-kinase, Vps34, which binds to Beclin-1, Atg14 and Vps150, constituting a macromolecular structure which generates phosphatidylinositol 3-phosphate. Phosphatidylinositol 3-phosphate is required for the recruitment of other regulatory proteins, such as Atg18, Atg20, Atg21 and Atg24, at the phagophore assembly site, thus allowing phagophore expansion. Phagophore expansion is associated with the inclusion of cytoplasmic elements and is regulated by two ubiquitylation-like reactions. Firstly, Atg12 is conjugated to Atg5 through a reaction catalyzed by Atg7 and Atg10. The Atg12-Atg5 complex interacts with Atg16 and binds the phagophore membrane, promoting its elongation. Subsequently, the cytosolic form of LC3 (Atg8), known as LC3-I, which has been cleaved by Atg4 and activated by Atg7 and Atg3, is conjugated to the lipid phosphatidylethanolamine in a reaction that is regulated by the Atg12-5 complex. The lipidated form of LC3, known as LC3-II, is crucial for autophagosome expansion. The final step of the autophagic machinery is characterized by the fusion of mature autophagosomes with lysosomes, through a process which necessitates the lysosome membrane protein LAMP-2 and the small GTPase Rab7 1, 2.
Mammalian target of rapamycin (mTOR) represents a critical regulator of autophagy in lower organisms and mammalian cells. mTOR inhibits autophagy through phosphorylation of Ulk1/2 and inhibition of phagophore initiation 1, 2. However, autophagy is also regulated through mTOR-independent mechanisms. AMP-dependent kinase (AMPK) activates autophagy, not only through the inhibition of mTOR, but also by directly phosphorylating/activating Ulk1. Sirt1, which is a member of the class III histone deacetylases, upregulates autophagy independently of mTOR by deacetylating proteins regulating autophagy, such as Foxo1. Myo-inositol-1,4,5-triphosphate (IP3) inhibits autophagy through increases in intracellular calcium concentrations. Intracellular cyclic AMP also inhibits autophagy through activation of protein kinase A, which inhibits phagophore initiation, and through Epac/Rap2B/phospholipase C-ε 1, 2.
Recent evidence indicates that cargo-specific forms of autophagy exist. Among them, mitophagy has been the most studied. Mitophagy is a form of autophagy which selectively sequesters mitochondria for degradation 2, 6. Mitophagy is required for turnover of mitochondria at baseline and is a mitochondrial quality control mechanism that allows selective degradation of damaged mitochondria during stress 2, 6. During stress, mitophagy is mainly regulated by two proteins: parkin and PTEN-induced putative kinase protein 1 (PINK1) 2, 6. Parkin is a cytosolic protein that specifically translocates to dysfunctioning mitochondria during stress. Parkin ubiquitylates several mitochondrial substrates and subsequently mediates the association of ubiquitylated cargo to LC3, which allows mitochondrial sequestration into autophagosomes 2, 6. Parkin translocation to mitochondria during stress is mediated by PINK1, which selectively labels damaged mitochondria during stress 2, 6.
Potential use of pharmacological modulators of autophagy in the treatment of cardiac disease
Several pharmacological compounds and classes of drugs have been shown to either enhance or inhibit autophagy (Figure 1). Some of them promote or inhibit autophagosome formation (i.e. rapamycin and 3-metyladenine respectively), whereas others affect autophagosome/lysosome fusion (bafilomycin) or lysosomal enzyme activity (chloroquine). Of note, the majority of these drugs modulate autophagy indirectly (i.e. AMPK activators or mTORC1 inhibitors), and they do not directly interfere with the autophagic machinery 7.
Figure 1.
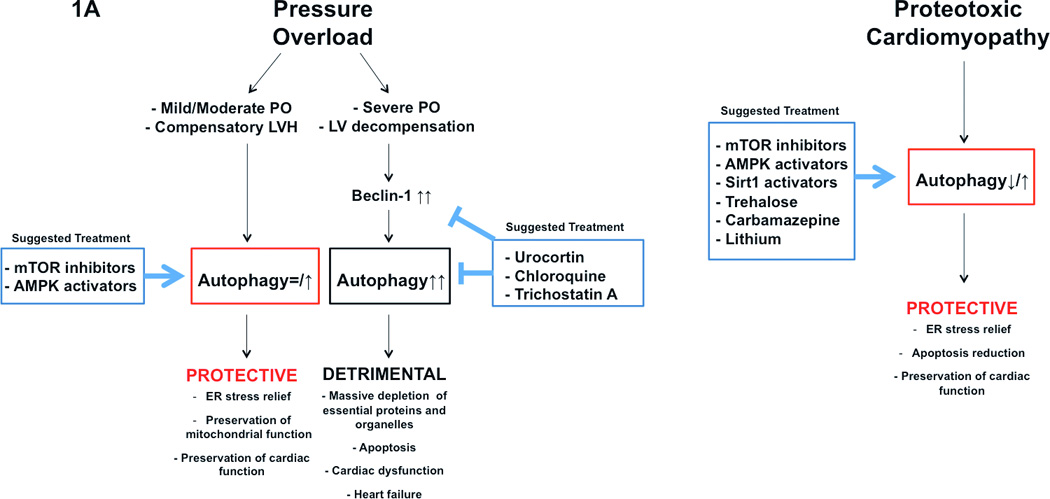
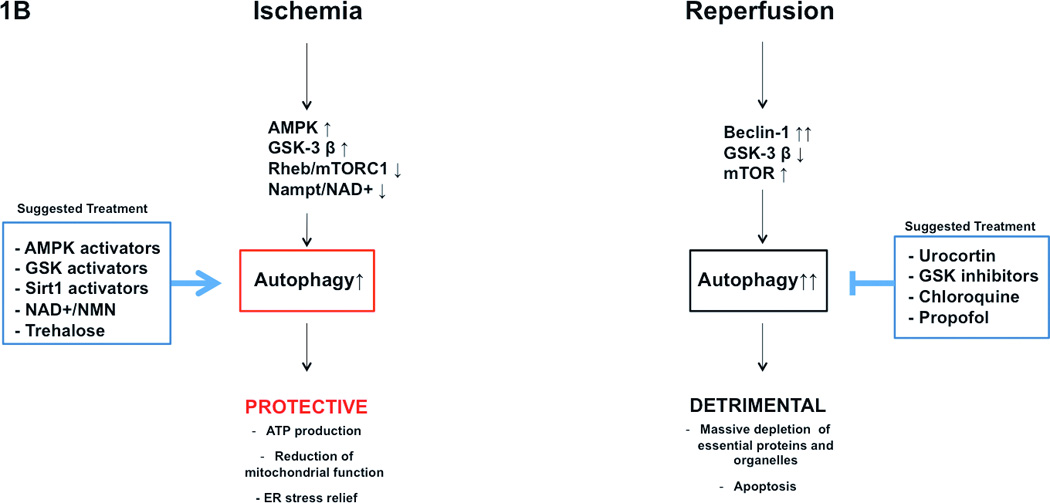
Pathophysiological significance of autophagy in the development of cardiomyopathy (A), and ischemia/reperfusion (B) is represented. Potential usefulness of pharmacological modulation of autophagy is also schematized.
mTOR inhibitors are the most studied autophagy inducers, and among them, rapamycin was the first to be identified. Rapamycin and its analogues bind to a cytosolic protein FK-binding protein12, thereby inhibiting mTOR, particularly the complex 1 of mTOR. Recently, novel mTOR inhibitors have been identified, such as Torin1, perhexiline, niclosamide and rottlerin. Among these, Torin1, an ATP-competitive small molecule, strongly activates autophagy 7. Activators of AMPK, including AICAR and metformin, are also important pharmacological inducers of autophagy, although the role of AICAR in the regulation of mammalian autophagy is still debated 8, 9. AMPK activates autophagy through mTOR inhibition and direct activation of Ulk1 8. On the other hand, AMPK inhibitors (i.e. ARA-A and compound C) inhibit autophagy 8. Akt inhibitors are also important activators of autophagy, acting through mTOR inhibition and FOXO activation 10. On the other hand, GSK-3 β inhibitors inhibit autophagy through mTOR activation 11. Chloroquine and analogous agents are strong inhibitors of autophagic flux, targeting lysosomes and inhibiting lysosomal enzyme activity 12. In addition, several drugs with well-established pharmacological actions upon other cellular functions modulate autophagy independently of mTOR. Carbamazepine, valproic acid and lithium activate autophagy by reducing the intracellular levels of IP3 7, 13. Calcium channel blockers and antiarrhythmic drugs, such as verapamil, loperamide, amiodarone, nimodipine, nitrendipine, niguldipine and pimozide, activate autophagy by inhibiting intracellular levels of calcium 7, 14. Dideoxyadenosine and β-adrenergic antagonists induce autophagy by reducing intracellular cyclic AMP, whereas fluspirilene and trifluoperazine promote autophagy by antagonizing dopamine 7, 14. Tamoxifen promotes autophagy by increasing the intracellular levels of ceramide 15. Finally, newly discovered compounds, including trehalose, a disaccharide 16, and the Small Molecules Enhancer of Rapamycin 17, stimulate autophagy through unknown mechanisms.
Although only a few of the aforementioned pharmacological modulators have been tested for their direct effects upon autophagy thus far, many of them have a high potential to be effective in treating cardiac disease. Several factors should be considered when a certain drug is chosen to modulate autophagy under certain pathological conditions in the heart. First, activators or inhibitors of autophagy should be chosen based on the pathological context in which they are used. For example, autophagy should be enhanced only when it is protective. Although it is still under debate, autophagy promotes cell death under some conditions. Second, the extent of autophagy modulation should be taken into account. Supraphysiological activation and complete inhibition of autophagy are both detrimental and can trigger cell death. Third, autophagy is activated through different signaling mechanisms in different cardiac diseases. Ideally, pharmacological modulation of autophagy under certain pathological conditions should be achieved by modulation of specific underlying signaling mechanisms. Finally, almost all of the pharmacological modulators of autophagy exert autophagy-independent functions as well, which should be taken into account when the most suitable drug must be chosen under a specific condition.
Based on these four considerations, in the following we will discuss which pharmacological modulators of autophagy have the potential to be specifically used under different cardiac pathological conditions (Figure 1).
It should be pointed out that to date no clinical data exists regarding the efficacy of pharmacological modulation of autophagy in cardiac diseases. Therefore these recommendations are based exclusively on the current experimental evidence. Further clinical studies are encouraged to establish whether they can really be translated into the clinical context.
Of note, the efficacy of clinical pharmacological inhibition of autophagy is currently being tested in patients affected by cancer. In fact, although a defect in autophagy has been proposed as a factor favoring cancer development 18, many lines of evidence indicate that several typologies of cancer are resistant to external stresses, such as hypoxia, nutrient deprivation, oxidative stress and anticancer therapy, through a marked activation of autophagy, which is protective under these conditions. Thus, inhibition of autophagy may represent a promising therapeutic intervention in cancer treatment 19.
Pharmacological modulation of autophagy in cardiomyopathy and heart failure
In the heart subjected to pressure overload, a modest activation of autophagy appears to be required to increase the clearance of misfolded proteins and maintain cardiac function. Complete inhibition of autophagy in this context rapidly causes heart failure 20. On the other hand, a strong activation of autophagy promoted by Beclin-1 upregulation is observed in response to severe transverse aortic constriction, and it mediates the transition from compensatory left ventricular hypertrophy to heart failure 21. It seems reasonable to postulate that pharmacological modulation of autophagy during pressure overload should maintain the activity of autophagy within physiological levels. Autophagy should be modestly activated during compensated hypertrophy in response to moderate pressure overload. Low-dose mTOR inhibitors or AMPK activators appear to be beneficial for this purpose, since they activate autophagy while limiting the progression of hypertrophy. This idea is also supported by studies demonstrating that rapamycin, and both AICAR and metformin, improve cardiac function, reduce cardiac hypertrophy and delay the onset of heart failure during pressure overload, although the involvement of autophagy activation in such beneficial effects was not specifically addressed in these studies 22–24. Of note, metformin, a glucose-lowering agent, is prescribed to patients with diabetes who display impaired cardiac activity of AMPK 25.
On the other hand, autophagy activation should be reduced in the presence of severe pressure overload. Specific inhibitors of Beclin-1 may be used in this context. Urocortin, a protein that belongs to the family of corticotropin-releasing factors, was shown to be able to inhibit Beclin-1 and autophagy, and thus, it could be a valid therapeutic option 26. In addition, since exaggerated autophagy may trigger cell death through massive depletion of essential proteins and organelles, lysosomal enzyme inhibitors, such as chloroquine, could be appropriate in this condition to delay autophagy-induced protein degradation and to reduce cell death. Finally, a recent study demonstrated that administration of Trichosatin A, a class I–II histone deacetylase inhibitor, inhibits autophagy and blunts pathological left ventricular remodeling during severe pressure overload, thus supporting the use of this drug. 27.
Autophagy is protective in cardiomyopathy caused by accumulation of misfolded proteins. In proteotoxic cardiomyopathy caused by aggregation of the alphaB-crystallin mutant, Atg7-dependent activation of autophagy reduces accumulation of amyloid oligomers in cardiomyocytes, suggesting that stimulation of autophagy may improve cardiac function and reduce ventricular remodeling 28. On the other hand, inhibition of autophagy through beclin-1 partial genetic deletion accelerates ventricular dysfunction in the alphaB-crystallin mutant mice 29.
mTOR inhibitors, AMPK activators, Sirt1 activators and trehalose have been shown to be effective in the treatment of neurodegenerative disorders, such as Alzheimer's disease, Parkinson's disease and Huntington's disease, through activation of autophagy and clearance of aberrant intracellular protein aggregates 7. These classes of drugs also appear appropriate for the treatment of proteotoxic cardiomyopathies.
Carbamazepine and lithium salt would also represent valid therapeutic tools to induce autophagy and to reduce intracellular aggregates in proteotoxic cardiomyopathy. In fact, both carbamazepine and lithium strongly increase intracellular clearance of misfolded protein accumulation through induction of autophagy 7. Unfortunately, serious adverse side events, which accompany carbamazepine and lithium treatments, should discourage the use of these drugs. In this regard, since lithium is an inhibitor of GSK-3, it may inhibit autophagy and stimulate hypertrophy in cardiomyocytes.
Pharmacological modulation of autophagy during ischemia and reperfusion
Autophagy is significantly enhanced in the heart during ischemia and reperfusion. However, the functional significance of autophagy activation appears to be different in one condition with respect to the other. Autophagy activation is protective during chronic/prolonged ischemia. Autophagy is significantly upregulated in the swine hibernated myocardium, and the level of autophagy inversely correlates with that of apoptosis in the ischemic area, suggesting that activation of autophagy prevents cell death 30. Autophagy is moderately activated during prolonged ischemia through activation of AMPK and GSK-3 β and inhibition of Rheb, which inhibit mTOR activity 11, 31, 32. Inhibition of endogenous AMPK and GSK-3 β or activation of Rheb suppresses autophagy and enhances myocardial ischemic injury. Autophagy may compensate for the loss of energy through regeneration of amino acids and fatty acids, which are used for ATP synthesis. Alternatively, autophagy removes damaged mitochondria and eliminates protein aggregates, which accumulate during ischemia and interfere with cellular functions 1.
On the other hand, autophagy activation appears to exert detrimental effects during reperfusion. We found that both autophagosome formation and autophagic flux are enhanced by oxidative stress during reperfusion injury 33. Mechanistically, autophagy activation during reperfusion is mediated by marked upregulation of Beclin-1. Interestingly, downregulation of Beclin-1 blocked autophagy and reduced reperfusion injury, thus suggesting that exaggerated autophagy activation is detrimental under this condition 31. Accordingly, we recently showed that inhibition of GSK-3 β during myocardial reperfusion is protective through activation of mTOR and inhibition of autophagy, thus further indicating that autophagy activation during reperfusion may promote cell death 11. Our observations are also supported by a study conducted in vitro, showing that Beclin-1 inhibition by urocortin inhibits autophagy and reduces death of cardiomyocytes subjected to hypoxia-reoxygenation 26.
It should be noted that activation of autophagy during reperfusion is protective under some experimental conditions 34, 35. The exact reason for this discrepancy has not yet been resolved. We have shown recently that the beneficial effect of autophagy suppression is attenuated by prolonging the time for ischemia before reperfusion 11. Thus, it is possible that the overall effect of autophagy suppression is determined by the balance between its effect upon ischemia and that upon reperfusion.
Then, how should autophagy be modulated during an ischemic episode in order to reduce myocardial loss? In patients with chronic stable coronary artery disease or in patients with stable subendocardial ischemia, which does not require an urgent mechanical coronary recanalization, autophagy should be pharmacologically enhanced. On the other hand, in patients with acute and total coronary occlusion, autophagy should be enhanced during the ischemic phase, though it should be reduced later on, during the reperfusion phase. Unfortunately, based on the current available evidence, it is impossible to establish what the optimal timing for these interventions is and whether these interventions are actually possible. Future studies are required to specifically address this important issue. Of note, autophagy could be activated during ischemia through modulation of the signaling pathways, including AMPK, that are not involved in the regulation of autophagy during reperfusion. This strategy, namely targeting the phase-specific signaling mechanism, would permit one to activate autophagy during ischemia without further activating autophagy during reperfusion.
Treatments with activators of AMPK or GSK-3 β represent valid therapeutic options to enhance autophagy during ischemia 11, 31. Alternatively, mTORC1 inhibition would also represent an efficacious intervention during ischemia, particularly in subjects with obesity and metabolic syndrome who have defective cardiac autophagy through deregulated Rheb/mTORC1 activation 32. We have shown that GSK-3 β promotes autophagy during ischemia through mTOR inhibition 11. However, the beneficial effect of a direct and selective modulation of mTORC1 vs. mTORC2 in this model is still under investigation.
We have recently shown that nicotinamide phosphoribosyltransferase (Nampt), a rate-limiting enzyme in the NAD+ salvage pathway, critically increases cardiomyocyte NAD+ and ATP content, inhibits apoptosis and stimulates autophagic flux in cardiomyocytes. Activation of Nampt during myocardial ischemia activates autophagy and inhibits myocardial injury 36. Since Nampt modulates autophagy by regulating NAD+ content, supplementation of NAD+ or its precursor, nicotinamide mononucleotide (NMN), could be used to stimulate autophagy during myocardial ischemia. In addition, Nampt and NAD+ activate Sirt1, which in turn enhances nuclear localization of FoxO1 and stimulates autophagy 37. Therefore, pharmacological activation of Sirt1 could also be efficient in the activation of autophagy and reduction of ischemic injury.
Trehalose significantly activates autophagy in cardiomyocytes and reduces cardiomyocyte death during glucose deprivation, a model mimicking ischemia 32. This suggests that trehalose may also be considered as an alternative autophagy inducer. Interestingly, pharmacological agents known to ameliorate ischemic tolerance and reduce blood pressure, such as propranolol, verapamil, nicardipine and nimodipine, can also stimulate cardiomyocyte autophagy 1, 7. These drugs may therefore confer additional cardioprotection if administered during ischemia.
During reperfusion, inhibition of autophagy with urocortin or GSK-3 β inhibitors would be the most appropriate treatment 11, 26. In addition, administration of chloroquine is indicated to delay autophagy-induced degradation of proteins, such as catalases, that are essential for the myocardial response to reperfusion injury 5. Propofol, a common drug used for induction of anesthesia, which has antioxidant properties, has also been shown to inhibit autophagy and limit myocardial damage during reperfusion injury. Notably, propofol inhibits autophagy through inhibition of Beclin-1 and activation of mTOR 38.
Interestingly, two antimicrobial agents, chloramphenicol and sulfaphenazole, have recently been shown to activate autophagy and reduce myocardial damage during ischemia/reperfusion 34, 35. However, these drugs are also strong reactive oxygen species (ROS) production inhibitors 39. Therefore, whether autophagy directly mediates the protective effects exerted by chloramphenicol and sulfaphenazole needs to be clarified.
Perspectives
Pharmacological modulation of autophagy appears to be a novel and promising strategy for treating heart disease. However, there are many unresolved issues that need to be addressed.
First, the role of autophagy during cardiac stress remains to be fully understood in vivo. Since some autophagy genes are involved in other cellular functions besides autophagy, evaluating the role of autophagy from only a single line of a genetically altered mouse model could be misleading. Studies using more lines of genetically altered mice, with loss- and gain-of-functions of autophagy, would help clarify whether, and in what condition, autophagy is protective or detrimental in the heart. In addition, the development of small molecule compounds targeting the autophagic machinery, which either selectively stimulate or inhibit autophagy, would be useful to further address this issue.
Second, whether autophagy can induce programmed cell death is still under debate and requires more investigation. It is unknown whether autophagic cell death is simply caused by excessive autophagy or by a distinct form of autophagy. If a protective form and a detrimental form of autophagy are mediated by distinct signaling mechanisms, pharmacological therapy should focus on enhancing “good” autophagy or attenuating “bad” autophagy. Recent evidence suggests that distinct forms of autophagy exist in mammalian cells, namely Atg5/Atg7-dependent and independent ones 40. Cargo-specific forms of autophagy, such as mitophagy, could be mediated by a distinct set of signaling mechanisms 41. Pharmacological agents that can selectively modulate these subtypes of autophagy may also represent promising tools for cardiac disease treatment. If activation of autophagy can induce cell death, elucidating the underlying molecular mechanism of cell death may allow us to inhibit cell death without affecting physiological autophagy.
In conclusion, increasing lines of evidence support the involvement of altered autophagy in cardiac diseases. The function of autophagy in the heart during stress is complex and appears stimulus-, dose- and time-dependent. Many signaling mechanisms involved in autophagy have cross-talk with other cellular functions, such as ubiquitin proteasome degradation and apoptotic cell death. These make modulation of autophagy through drug therapy challenging. However, judging from the fundamental importance of autophagy in a wide variety of cellular functions, we expect that medical therapy targeting autophagy should become an effective modality to protect the heart from stress in the near future.
Acknowledgements
The authors thank Christopher D. Brady for critical reading of the manuscript.
Funding support:
This work was supported in part by U.S. Public Health Service Grants HL102738, HL67724, HL69020, HL91469, AG23039, and AG27211. This work was also supported by the Foundation Leducq Transatlantic Networks of Excellence. SS is supported by a Postdoctoral Fellowship from the Founders Affiliate, American Heart Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev. 2010;90:1383–1435. doi: 10.1152/physrev.00030.2009. [DOI] [PubMed] [Google Scholar]
- 3.Djavaheri-Mergny M, Maiuri MC, Kroemer G. Cross talk between apoptosis and autophagy by caspase-mediated cleavage of beclin 1. Oncogene. 2010;29:1717–1719. doi: 10.1038/onc.2009.519. [DOI] [PubMed] [Google Scholar]
- 4.Ullman E, Fan Y, Stawowczyk M, Chen HM, Yue Z, Zong WX. Autophagy promotes necrosis in apoptosis-deficient cells in response to er stress. Cell Death Differ. 2008;15:422–425. doi: 10.1038/sj.cdd.4402234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy in the heart. Cell Death Differ. 2009;16:31–38. doi: 10.1038/cdd.2008.163. [DOI] [PubMed] [Google Scholar]
- 6.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fleming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol. 2011;7:9–17. doi: 10.1038/nchembio.500. [DOI] [PubMed] [Google Scholar]
- 8.Mihaylova MM, Shaw RJ. The ampk signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meijer AJ, Codogno P. Amp-activated protein kinase and autophagy. Autophagy. 2007;3:238–240. doi: 10.4161/auto.3710. [DOI] [PubMed] [Google Scholar]
- 10.Mammucari C, Schiaffino S, Sandri M. Downstream of akt: Foxo3 and mtor in the regulation of autophagy in skeletal muscle. Autophagy. 2008;4:524–526. doi: 10.4161/auto.5905. [DOI] [PubMed] [Google Scholar]
- 11.Zhai P, Sciarretta S, Galeotti J, Volpe M, Sadoshima J. Differential roles of gsk-3{beta} during myocardial ischemia and ischemia/reperfusion. Circ Res. 2011;109:502–511. doi: 10.1161/CIRCRESAHA.111.249532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carew JS, Nawrocki ST, Giles FJ, Cleveland JL. Targeting autophagy: A novel anticancer strategy with therapeutic implications for imatinib resistance. Biologics. 2008;2:201–204. doi: 10.2147/btt.s1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S, Michalopoulos G, Perlmutter DH. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin z and reduces hepatic fibrosis. Science. 2010;329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 14.Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P, O'Kane CJ, Floto RA, Rubinsztein DC. Novel targets for huntington's disease in an mtor-independent autophagy pathway. Nat Chem Biol. 2008;4:295–305. doi: 10.1038/nchembio.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Medina P, Silvente-Poirot S, Poirot M. Tamoxifen and aebs ligands induced apoptosis and autophagy in breast cancer cells through the stimulation of sterol accumulation. Autophagy. 2009;5:1066–1067. doi: 10.4161/auto.5.7.9820. [DOI] [PubMed] [Google Scholar]
- 16.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mtor-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–5652. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 17.Floto RA, Sarkar S, Perlstein EO, Kampmann B, Schreiber SL, Rubinsztein DC. Small molecule enhancers of rapamycin-induced tor inhibition promote autophagy, reduce toxicity in huntington's disease models and enhance killing of mycobacteria by macrophages. Autophagy. 2007;3:620–622. doi: 10.4161/auto.4898. [DOI] [PubMed] [Google Scholar]
- 18.Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kimmelman AC. The dynamic nature of autophagy in cancer. Genes Dev. 2011;25:1999–2010. doi: 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 21.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, Izumo S. Inhibition of mtor signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. 2004;109:3050–3055. doi: 10.1161/01.CIR.0000130641.08705.45. [DOI] [PubMed] [Google Scholar]
- 23.Zhang CX, Pan SN, Meng RS, Peng CQ, Xiong ZJ, Chen BL, Chen GQ, Yao FJ, Chen YL, Ma YD, Dong YG. Metformin attenuates ventricular hypertrophy by activating the amp-activated protein kinase-endothelial nitric oxide synthase pathway in rats. Clin Exp Pharmacol Physiol. 2011;38:55–62. doi: 10.1111/j.1440-1681.2010.05461.x. [DOI] [PubMed] [Google Scholar]
- 24.Li HL, Yin R, Chen D, Liu D, Wang D, Yang Q, Dong YG. Long-term activation of adenosine monophosphate-activated protein kinase attenuates pressure-overload-induced cardiac hypertrophy. J Cell Biochem. 2007;100:1086–1099. doi: 10.1002/jcb.21197. [DOI] [PubMed] [Google Scholar]
- 25.Shibata R, Ouchi N, Ito M, Kihara S, Shiojima I, Pimentel DR, Kumada M, Sato K, Schiekofer S, Ohashi K, Funahashi T, Colucci WS, Walsh K. Adiponectin-mediated modulation of hypertrophic signals in the heart. Nat Med. 2004;10:1384–1389. doi: 10.1038/nm1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valentim L, Laurence KM, Townsend PA, Carroll CJ, Soond S, Scarabelli TM, Knight RA, Latchman DS, Stephanou A. Urocortin inhibits beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J Mol Cell Cardiol. 2006;40:846–852. doi: 10.1016/j.yjmcc.2006.03.428. [DOI] [PubMed] [Google Scholar]
- 27.Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG, Hill JA. Histone deacetylase (hdac) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108:4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in cryabr120g cardiomyocytes. Circ Res. 2011;109:151–160. doi: 10.1161/CIRCRESAHA.110.237339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tannous P, Zhu H, Johnstone JL, Shelton JM, Rajasekaran NS, Benjamin IJ, Nguyen L, Gerard RD, Levine B, Rothermel BA, Hill JA. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. 2008;105:9745–9750. doi: 10.1073/pnas.0706802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan L, Sadoshima J, Vatner DE, Vatner SF. Autophagy in ischemic preconditioning and hibernating myocardium. Autophagy. 2009;5:709–712. doi: 10.4161/auto.5.5.8510. [DOI] [PubMed] [Google Scholar]
- 31.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of amp-activated protein kinase and beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 32.Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, Condorelli G, Sadoshima J. Rheb is a critical regulator of autophagy during myocardial ischemia: Pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012 doi: 10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal. 2011;14:2179–2190. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sala-Mercado JA, Wider J, Undyala VV, Jahania S, Yoo W, Mentzer RM, Jr, Gottlieb RA, Przyklenk K. Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia-reperfusion injury. Circulation. 2010;122:S179–S184. doi: 10.1161/CIRCULATIONAHA.109.928242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang C, Liu W, Perry CN, Yitzhaki S, Lee Y, Yuan H, Tsukada YT, Hamacher-Brady A, Mentzer RM, Jr, Gottlieb RA. Autophagy and protein kinase c are required for cardioprotection by sulfaphenazole. Am J Physiol Heart Circ Physiol. 2010;298:H570–H579. doi: 10.1152/ajpheart.00716.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsu CP, Hariharan N, Alcendor RR, Oka S, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through autophagy in cardiomyocytes. Autophagy. 2009;5:1229–1231. doi: 10.4161/auto.5.8.10275. [DOI] [PubMed] [Google Scholar]
- 37.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of foxo by sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res. 2010;107:1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Noh HS, Shin IW, Ha JH, Hah YS, Baek SM, Kim DR. Propofol protects the autophagic cell death induced by the ischemia/reperfusion injury in rats. Mol Cells. 2010;30:455–460. doi: 10.1007/s10059-010-0130-z. [DOI] [PubMed] [Google Scholar]
- 39.Granville DJ, Tashakkor B, Takeuchi C, Gustafsson AB, Huang C, Sayen MR, Wentworth P, Jr, Yeager M, Gottlieb RA. Reduction of ischemia and reperfusion-induced myocardial damage by cytochrome p450 inhibitors. Proc Natl Acad Sci U S A. 2004;101:1321–1326. doi: 10.1073/pnas.0308185100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of atg5/atg7-independent alternative macroautophagy. Nature. 2009;461:654–658. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 41.Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by parkin and p62/sqstm1. PLoS One. 2011;6:e20975. doi: 10.1371/journal.pone.0020975. [DOI] [PMC free article] [PubMed] [Google Scholar]